

Abstract
A series of 1,5-disubstituted pyridones was identified as positive allosteric modulators (PAMs) of the metabotropic glutamate receptor 2 (mGluR2) via high throughput screening (HTS). Subsequent SAR exploration led to the identification of several compounds with improved in vitro activity. Lead compound 8 was further profiled and found to attenuate the increase in PCP induced locomotor activity in mice.
Keywords: mGluR2, metabotropic, glutamate, allosteric, PAM, modulators
Metabotropic glutamate receptors (mGluRs) represent a family of G protein-coupled receptors that are activated by the excitatory neurotransmitter glutamate. Activation of group II metabotropic glutamate receptors (mGluR2 and mGluR3) may provide anxiolytic and/or antipsychotic effects (1−3). Mixed mGluR2/mGluR3 agonists such as LY354740 (1, Figure 1) have shown activity in a range of preclinical animal models of anxiety and schizophrenia. The anxiolytic potential of LY354740 was also confirmed in healthy human volunteers, showing activity in fear-potentiated startle and panic induction after a CO2 challenge (4,5). A related prodrug, LY2140023 (2), demonstrated improvements in positive and negative symptoms compared to that by the placebo in schizophrenic patients (6). In addition, evidence exists from knockout studies that the preclinical antipsychotic effects may be mediated via the mGluR2 receptor (7).
Figure 1.

Structures of mixed mGluR2/3 agonists LY354740 and LY2140023, and of the first reported mGluR2 PAM LY487379.
Over recent years, there has been increasing interest in trying to identify positive allosteric modulators (PAMs) of mGluR2, which bind at an alternative site to the orthosteric endogenous agonist (8). The first mGluR2 PAMs were a series of sulfonamide derivatives discovered by scientists at Lilly, including LY487379 (3, Figure 1) (9,10). We have recently conducted a review of the currently reported mGluR2 PAMs (11), and new reports are appearing on a frequent basis (12). Recent reports from our laboratories described a series of imidazopyridines identified with the help of computational shape and electrostatic field similarity (13). In this article, the discovery of 1,5-disubstituted pyridones as a new series of mGluR2 PAMs is presented. Initial SAR exploration is given along with in vivo profiling of a lead compound.
Results and Discussion
High throughput screening (HTS) of the compound collection from Addex Pharmaceuticals in an mGluR2 PAM FLIPR (fluorometric imaging plate reader) assay led to the discovery of pyridones 4 and 5. Their potentiating activity was also confirmed in a mGluR2 [35S]-GTPγS assay (Figure 2). The in vitro GTPγS assay provided two measures of compound activity, the EC50 for the potentiation of glutamate, and the maximal increase in observed glutamate response, the % Emax(14). Compound 4 displayed an EC50 of 4.47 μM and a 133% increase in the maximal glutamate effect, while compound 5 had an EC50 of 6.29 μM and an Emax of 138%. Despite their low activity, compounds 4 and 5 were selected as viable starting points for a CNS focused exploration given their relatively low molecular weight, 374 and 291, respectively, and polar surface area, 50 and 30, respectively. Our initial efforts focused on building structure−activity relationships (SAR) by sequential modification of both R1 and R2 substituents of the pyridone ring.
Figure 2.

Pyridone hits 4 and 5 identified from an mGluR2 PAM high throughput screen.
The initial set of compounds 6−31 covered the variation of the R1 group at position C-5 of the pyridone ring while maintaining the 4-chloro-2-fluoro benzyl substituent in compound 4 constant. The target compounds 6−31 were prepared following the synthetic strategies shown in Schemes 1−3.
Scheme 1.

Reagents and conditions: (i) 4-chloro-2fluorobenzylbromide, K2CO3, THF, 60 °C, 17 h, 79%. (ii) ArB(OH)2, 10% Pd(PPh3)4, NaHCO3 (aq.)/1,4-dioxane, 140 °C, 5 min at μW, 30−75%. (iii) 4-Phenylpiperidine, Pd(OAc)2, BINAP, t-BuOK, toluene, 100 °C, 16 h, 77%. (iv) BBr3, CH2Cl2, −40 °C to rt, 24 h, 83%. (v) 2-chloroacetophenone, K2CO3, THF, 110 °C, 30 min at μW, 20%. (vi) 2-Chloropropiophenone, K2CO3, THF, 110 °C, 30 min at μW, 34%. (vii) (a) N-BOC-2-hydroxyethanol, Ph3P, DEAD, 0 °C to rt, 16 h, 66%; (b) HCl/1,4-dioxane, MeOH, 80 °C, 2 days, 100%; (c) AcCl (12) or MeSO2Cl (13), Et3N, CH2Cl2, rt, 1 h, 47−65%, respectively. (viii) 2-Bromoacetonitrile, K2CO3, acetonitrile, 180 °C, 5 min at μW, 42%.
Scheme 3.

Reagents and conditions: (i) 4-Methoxybenzylmagnesium bromide, THF, −78 °C to rt, 14 h, 68%. (ii) 4-Chloro-2fluorobenzylbromide, acetonitrile, NaI, 90 °C, 14 h, 48−58%. (iii) Et3SiH, TFA, rt, 1 h, 79%.
The majority of the analogues were prepared following the synthetic routes depicted in Scheme 1. N-Alkylation of 5-bromo-2-hydroxypyridine (32) with 4-chloro-2-fluorobenzyl bromide afforded compound 33 in 79% yield. Subsequent Suzuki coupling of the 5-bromopyridone 33 with several aryl boronic acids under microwave irradiation led to the corresponding target compounds 4, 6, 8, 9, 14−17, and 29−31 in moderate to good yields. Additionally, 33 was coupled with 4-phenylpiperidine under standard Buchwald−Hartwig coupling conditions to yield the pyridone derivative 26. Compound 8 was further modified by cleavage of the methoxy ether with BBr3 to give the phenolic derivative 7, which was then coupled with different alkylbromides under microwave irradiation resulting in the corresponding O-alkylated products 10−11 and 19. Compound 7 was also treated with the commercially available N-BOC-hydroxyethanolamine under Mitsunobu reaction conditions followed by cleavage of the BOC group. The subsequent acetylation or sulfonylation of the amino group afforded compounds 12 or 13, respectively.
Compound 25, in which position C-5 of the pyridone ring is substituted by a 3-phenylpropyl, was prepared as shown in Scheme 2. Halogen−lithium exchange on commercially available 3-bromo-2-methoxypyridine (34) with n-butyllithium followed by treatment with 1-bromo-3-phenylpropane led to the methoxypyridine derivative 35 in 17% yield. Treatment of 35 with 4-chloro-2-fluorobenzyl bromide in the presence of NaI afforded the target compound 25 in moderate yield (48%).
Scheme 2.

Reagents and conditions: (i) 1-(3-Bromopropyl)benzene, n-BuLi, THF, −78 °C to rt, 1 h, 17%. (ii) 4-Chloro-2fluorobenzyl bromide, acetonitrile, NaI, 90 °C, 14 h, 48−58%. (iii) Phenol, CuI, Cs2CO3, Me2NCH2COOH, 1,4-dioxane, 150 °C, 25 min at μW, 44%.
The introduction of an oxygen linker between the pyridine core and the aryl substituent at position C-5 proceeded via the reaction of the bromopyridine 34 with phenol under copper-catalyzed Ullmann coupling conditions to afford 2-methoxy-4-phenoxypyridine 36, which was converted into the final compound 28 under standard N-benzylation conditions (Scheme 2).
Finally, compound 27, an analogue of 8 which contains a methylene spacer, was synthesized as shown in Scheme 3. In the first step, the addition of 4-methoxybenzylmagnesium bromide to 37 led to the corresponding alcohol 38, which was then subjected to N-benzylation to give pyridone 39 that was finally transformed into 27 by treatment with Et3SiH in trifluoroacetic acid.
The variations around the R1 group and the functional activity of N-(4-chloro-2-fluorobenzyl)pyridones 4 and 6−31 are listed in Table 1.
Table 1. Functional Activity of Representative mGluR2 PAMs 4 and 6−31a.

| compound | R1 | mGlu2 EC50 (μM)a | mGlu2 Emax (%) |
|---|---|---|---|
| 4 | HOCH2CH2O-Ph- | 4.47 | 133 |
| 6 | Ph- | 6.31 | 138 |
| 7 | 4-OH-Ph- | 5.01 | 124 |
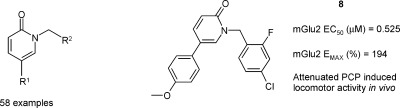
| 8 | 4-MeO-Ph- | 0.53 | 194 |
| 9 | 4-CF3OPh- | >10 | |
| 10 | 4-(PhC(O)CH2O)-Ph- | 11.5 | 182 |
| 11 | 4-(PhC(O)CH2CH2O)-Ph- | 1.26 | 196 |
| 12 | 4-(AcNHCH2CH2O)-Ph- | 0.79 | 102 |
| 13 | 4-(MeSO2NHCH2CH2O)-Ph- | 0.68 | 120 |
| 14 | 3-NH2-Ph- | 3.98 | 67 |
| 15 | 4-NH2-Ph- | 9.33 | 102 |
| 16 | 4-AcNH-Ph- | 0.81 | 48 |
| 17 | 4-(pyrrolidinone-1-yl)-Ph- | 1.74 | 112 |
| 18 | HOCH2Ph- | 5.25 | 145 |
| 19 | 4-(CNCH2O)-Ph- | 3.39 | 184 |
| 20 | 3-F-4-MeO-Ph- | 0.71 | 241 |
| 21 | 3,4-diMeO-Ph- | 3.31 | 152 |
| 22 | 3-Me-4-MeO-Ph- | 1.05 | 185 |
| 23 | 5,3-diMe-4-MeO-Ph- | 1.00 | 116 |
| 24 | 3-(6-MeO)pyridyl- | 9.33 | 149 |
| 25 | PhCH2CH2CH2- | 0.56 | 33 |
| 26 | 4-phenylpiperidinyl- | 3.24 | 101 |
| 27 | 4-MeO-PhCH2- | 1.93 | 70 |
| 28 | Ph-O- | >10 | -- |
| 29 | 3-(5-tetrazolyl)-Ph- | 7.59 | 65 |
| 30 | 4-(5-tetrazolyl)-Ph- | 1.00 | 32 |
| 31 | 4-(4-pyridylthiomethyl)-Ph- | 0.18 | 168 |
Values are means of three experiments.
The effect of substituents on the C-5 aromatic ring present in 4 was explored with compounds 6−24. The unsubstituted compound 6 had the same level of in vitro potency, EC50 of 6.31 μM and Emax 138%, compared to that of 4. The 4-hydroxyphenyl derivative 7 and its elongated analogue 18 also displayed similar activities, EC50 of 5.01 μM and 5.25 μM and Emax of 124 and 145%, respectively. However, a 10-fold increase in potency was obtained with the simple 4-methoxy substituent in compound 8, which had an EC50 of 0.53 μM and an Emax of 194%. This compound is the direct analogue of the initial hit compound 5 and demonstrated the beneficial effect of the 4-chloro-2-fluoro decoration on the benzyl R2 substituent. A decrease in potency was observed with the pyridyl analogue 24, and the activity was completely lost with the 4-trifluoromethoxy derivative 9, which indicates that an electron-rich phenyl ring at position C-5 of the pyridone core might be preferred. The effect of a H-bond acceptor was also explored with the introduction of several alkyl chains at the para-position of the C-5 phenyl ring (10, 11, and 19). The results obtained suggested that although an acceptor function is tolerated it does not result in an increase in activity. Amino substitution either at the meta- or para-position (14, 15) was detrimental for the Emax activity, with values of 67% and 102%, respectively. However, improved potency was observed with amide derivatives 16 and 17, which had EC50 values of 0.81 μM and 1.74 μM, respectively. The interesting activity seen for compound 8 prompted us to study the 4-methoxy substitution in combination with other small substituents on the same phenyl ring (20−23). Unfortunately, none of these analogues showed better activity than 8.
Several different spacers were introduced between the pyridone core and the phenyl ring (compounds 25−28), although only the 4-piperidinyl spacer (26) was somewhat tolerated. The introduction of tetrazole substituents commonly found in series of mGluR2 PAM indanones from Merck (15) resulted in weakly active compounds (29, 30) in this class. In contrast, the 4-pyridylthiomethyl (31), also found in Merck’s indanone series (16), gave rise to the most potent compound identified in this R1 exploration. This finding supports the hypothesis that an electron-rich phenyl ring in combination with groups that introduce moderate polarity such as a H-bond acceptor is beneficial for activity.
We then focused our attention on exploring substitution at the pyridone nitrogen of the initial hit 5 while maintaining the 4-methoxyphenyl substituent constant at the C-5 position of the pyridone ring. A straightforward synthesis route that allowed the rapid preparation of a set of 27 compounds (41−68) is shown in Scheme 4. Suzuki coupling between 4-methoxyphenylboronic acid and 4-bromo-2-methoxypyridine 34 afforded the corresponding coupling product 40, which was transformed into the final compounds 41−68 by treatment with the appropriate alkylating agent in the presence of NaI.
Scheme 4.

Reagents and conditions: (i) 4-Methoxyphenylboronic acid, 10% Pd(PPh3)4, NaHCO3 (aq.)/1,4-dioxane, 140 °C, 5 min at μW, 75%. (ii) R1CH2X, acetonitrile, NaI, 150 °C, 30 min at μW, 30−65%.
As shown in Table 2, lipophilic decoration on the aryl via halogen atoms or halogenated alkyl groups (41−43, 45−49) was beneficial for potency. Compound 45 was the most active one identified during this R2 exploration, with an EC50 of 0.24 μM and an Emax of 228%. The increase in potency is obvious in comparison with those of analogues having hydrophilic substitutions such as the phenol analogue 44, which was inactive up to 10 μM. The same trend in SAR was seen with analogues 51−56, where the aryl ring is replaced by different heterocycles such as pyridyl, thiazolyl, benzimidazolyl, or benzothiazolyl, which only showed weak activity. The unsubstituted pyridyl 51 was inactive; however, the introduction of additional lipophilic substituents such as trifluoromethyl or methoxy resulted in weakly active compounds (52 and 53). Lengthening the distance between the pyridone and the aryl through an ethyl- or methyloxy-chain (57, 58) did not have an effect on activity. Lipophilic mapping on these elongated analogues again increased activity (59, 60). Finally, the importance of having an aromatic ring as a substituent on the pyridone nitrogen was investigated with several analogues having N-aliphatic groups (61−68). Compounds showed comparable potency indicating that aromaticity played no particular role in potency. However, these data confirm the importance of having lipophilic substituents in this area of the molecule, with the most lipophilic alkyl groups displaying better activities (67, 68). In summary, the exploration of the R2 substituents revealed that neither polarity nor weak basicity are well tolerated at R2, while hydrophobicity is essential to maintain reasonable potency levels.
Table 2. Functional Activity of Representative mGluR2 PAMs 5, 41−68a.

| compound | R2 | mGlu2 EC50 (μM)a | mGlu2 Emax (%) |
|---|---|---|---|
| 5 | Ph- | 6.29 | 138 |
| 41 | 2-F-Ph- | 1.00 | 44 |
| 42 | 3-F-Ph- | 1.91 | 96 |
| 43 | 4-Cl-Ph- | 1.41 | 189 |
| 44 | 4-OH-Ph- | >10 | |
| 45 | 4-CF3O-Ph- | 0.24 | 228 |
| 46 | 2,3-diF-Ph- | 10 | 131 |
| 47 | 2,4-diF-Ph- | 4.79 | 128 |
| 48 | 2,4,6-triF-Ph- | 2.96 | 118 |
| 49 | 3-F-4-Cl-Ph- | 0.48 | 180 |
| 50 | 2-F-4-CF3−Ph- | 3.16 | 124 |
| 51 | 3-Py- | >10 | |
| 52 | 3-(6-MeO)Py- | 6.03 | 66 |
| 53 | 3-(6-CF3)Py- | 6.76 | 144 |
| 54 | 4-(2-Me)-triazolyl- | >10 | |
| 55 | 2-benzimidazolyl- | >10 | |
| 56 | 2-benzothiazolyl- | 5.62 | 79 |
| 57 | PhCH2CH2- | 3.71 | 117 |
| 58 | PhOCH2- | 3.98 | 87 |
| 59 | 3-Cl-PhOCH2- | 3.16 | 124 |
| 60 | 4-Cl-PhOCH2- | 1.91 | 115 |
| 61 | n-propyl- | 7.24 | 53 |
| 62 | n-butyl- | 9.28 | 122 |
| 63 | i-butyl- | 0.54 | 75 |
| 64 | s-butyl- | 12.88 | 97 |
| 65 | CF3CH2CH2- | 8.71 | 97 |
| 66 | cyclopentyl- | 9.33 | 122 |
| 67 | cyclohexyl- | 0.43 | 120 |
| 68 | cyclohexylmethyl- | 1.08 | 161 |
Values are means of three experiments.
To further profile the series, a representative set of compounds was tested for single point microsomal stability in human liver microsomes (HLM). The results are shown in Table 3, with most of the compounds suffering from extensive metabolism. The 4-methoxyphenyl compound 8 was completely metabolized after 15 min of incubation with HLM, with the major metabolite identified as the corresponding des-methyl analogue (7). Somewhat decreased metabolism was found with the 4-hydroxy analogue 7 (70%) and its elongated version 18 (82%). Aniline derivative 15 and its N-acetyl analogue 16 were also somewhat more stable than 8 (64% and 83% metabolized, respectively). A remarkable improvement was found for compounds 12 and 13 where the 4-methoxy group was replaced by bulkier alkoxy groups (24% and 35%, respectively). The presence of either electron donating or withdrawing ortho-substituents to the 4-methoxy group (compounds 20, 22) did not prevent extensive degradation. Improved stability was observed with the tetrazole derivative 30 (59%), which suggests that introduction of polar functional groups may increase metabolic stability in this series. Finally, the two most interesting examples with respect to their primary activity, compounds 31 and 45, suffered from extensive metabolism with 73% and 98% metabolized in the HLM assay, respectively.
Table 3. Metabolic Stability in Human Liver Microsomes (HLM) of Some Representative Compounds.
| compound | HLMa (%) |
|---|---|
| 7 | 70 |
| 8 | 100 |
| 12 | 24 |
| 13 | 35 |
| 15 | 64 |
| 16 | 83 |
| 18 | 82 |
| 20 | 100 |
| 22 | 87 |
| 30 | 59 |
| 31 | 73 |
| 45 | 98 |
HLM data refer to the percentage of compound metabolized after 15 min at 5 μM concentration.
Despite its high metabolic turnover, compound 8 was chosen as a prototype to further evaluate the potential of this novel mGluR2 PAM series.
The ability of compound 8 to shift the in vitro concentration−response curve (CRC) of glutamate was investigated further. As shown in Figure 3, the CRC of glutamate shifts to the left and upward with increasing concentrations of compound 8. A 7-fold shift in the glutamate EC50 was seen in the presence of 3 μM of 8, with the glutamate pEC50 shifted from 4.9 to 5.8. These results are in line with a positive allosteric interaction between glutamate and compound 8.
Figure 3.

Glutamate (Glu) concentration response curve in presence of varying concentration of compound 8, demonstrating an approximate 7-fold-shift in Glu EC50 at 3 μM concentration of 8 (experiment performed with CHO cells expressing cloned human mGluR2).
Targeting the allosteric site of mGluRs is expected to improve the chance of identifying selective ligands. Possible agonist or antagonist effects of compound 8 against mGluR3, mGluR5, mGluR7, and mGluR8 were first evaluated (17). Compound 8 was inactive in all assays except for mGluR7 antagonism where it had an EC50 of 4.94 μM and an Emax of 78%. In addition, compound 8 was profiled against 50 targets in a selectivity screen performed at CEREP (18). Selectivity was good overall, and the only interaction greater than 50% effect at a 10 μM concentration was with the norepinephrine transporter.
Compound 8 was further evaluated in vivo in the phencyclidine (PCP)-induced hyperlocomotion model in mice. This model is based on the evidence that the noncompetitive NMDA receptor channel blocker PCP induces schizophrenia-like behavior in humans (19). Hence, PCP is widely used in animal models of psychosis. In rodents, PCP causes an increase in motor activity. As this induced behavior may be linked to increased glutamate release, it is speculated that mGlu2 receptor activation may block these specific PCP-mediated effects (20). As shown in Figure 4A, no alteration in locomotor activity was observed in the absence of PCP indicating that compound 8 has no effect on locomotion per se. However, from Figure 4B it can be seen that compound 8 (200 mg/kg intraperitoneal (ip)) significantly attenuated the increase in locomotor activity induced by PCP (5 mg/kg ip). The effect level was comparable to that of the Lilly reference PAM, LY487379, 3, administered at the same dose and by the same route (20). Monitoring of plasma and brain levels of 8 showed concentrations of 3270 ng/mL and 4478 ng/g, respectively, 30 min after dosing, resulting in a brain to plasma ratio of 1.4.
Figure 4.

Effect of compound 8 (200 mg/kg ip) on PCP-induced (5 mg/kg sc) locomotor activity in mice. (A) Compound 8 shows no effect on locomotor activity in the absence of PCP. (B) Compound 8 and LY487379 (3) both reduce PCP-induced activity (same dose for both compounds). * indicates p < 0.001.
In summary, a novel series of 1,5-disubstituted pyridones with mGluR2 PAM activity have been presented. Systematic SAR studies from the initially available hits 4 and 5 have resulted in compounds with better potency in a GTPγS assay. Although those analogues showed poor in vitro metabolic stability in HLM, alternative compounds were identified with reduced metabolic turnover. Compound 8 displayed good brain levels after IP administration and comparable activity to a reference PAM, LY487379, in the in vivo PCP induced locomotor activity test. Taken together, these results suggest promise for the 1,5-pyridone series. However, the challenge remains to combine potency with metabolic stability and identify compounds suitable for in vivo administration by alternative routes. These studies are underway and will be the subject of future reports from our laboratories.
Acknowledgments
We thank members of the Purification and Analysis group and the In Vitro Metabolism team from the Discovery ADME/Tox group.
Note Added after ASAP Publication
This paper was published on the Web on August 23, 2010. There was a change in the authorship for the paper, and the corrected version was reposted on November 4, 2010.
Present address: Experimental Therapeutics Programme, Spanish National Cancer Research Centre (CNIO), Melchor Fernandez Almagro 3,28029 Madrid, Spain.
References
- Swanson C. J.; Bures M.; Johnson M. P.; Linden A. M.; Monn J. A.; Schoepp D. D. (2005) Glutamate Receptors as novel targets for anxiety and stress disorders. Nat. Rev. Drug Discovery 4, 131–144. [DOI] [PubMed] [Google Scholar]
- Schoepp D. D.; Marek G. J. (2002) Preclinical pharmacology of mGluR2/3 receptor agonists: novel agents for schizophrenia?. Curr. Drug Targets: CNS Neurol. Disord. 1, 215–225. [DOI] [PubMed] [Google Scholar]
- Conn P. J.; Lindsley C. W.; Jones C. K. (2009) Activation of the metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol. Sci. 30, 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine L.; Gaydos B.; Sheehan D.; Goddard A.; Feighner J.; Potter W.; Schoepp D. D. (2002) The mGlu2/3 Receptor agonist, LY354740, reduces panic anxiety induced by a CO2 challenge in patients diagnosed with panic disorder. Neuropharmacology 43, 294–295. [Google Scholar]
- Schoepp D. D.; Wright R. A.; Levine L. R.; Gaydos B.; Potter W. Z. (2003) LY354740, an mGlu2/3 receptor agonist as a novel approach to treat anxiety/stress. Stress 6, 189–197. [DOI] [PubMed] [Google Scholar]
- Patil S. T.; Zhang L.; Martenyi F.; Lowe S. L.; Jackson K. A.; Andreev B. V.; Avedisova A. S.; Bardenstein L. M.; Gurovich I. Y.; Morozova M. A.; Mosolov S. N.; Neznanov N. G.; Reznik A. M.; Smulevich A. B.; Tochilov V. A.; Johnson B. G.; Monn J. A.; Schoepp D. D. (2007) Activation of mGlu2/3 receptors as a new approach totreat schizophrenia: a randomized Phase 2 clinical trial. Nat. Med. 13, 1102–1107. [DOI] [PubMed] [Google Scholar]
- Fell M. J.; Svensson K. A.; Johnson B. G.; Schoepp D. D. (2008) Evidence for the role of metabotropic glutamate (mGlu)2 not mGlu3 receptors in the preclinical antipsychotic pharmacology of the mGlu2/3 receptor agonist (−)-(1R,4S,5S,6S)-4-amino-2-sulfonylbicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY404039). J. Pharmacol. Exp. Ther. 326, 209–217. [DOI] [PubMed] [Google Scholar]
- Jeffrey Conn P.; Christopoulos A.; Lindsley C. W. (2009) Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug. Discovery 8, 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. P.; Baez M.; Jagdmann G. E.; Britton T. C.; Large T. H.; Callagaro D. O.; Tizzano J. P.; Monn J. A.; Schoepp D. D. (2003) Discovery of allosteric potentiators for the metabotropic glutamate 2 receptor: synthesis and subtype selectivity of N-(4-(2-methoxyphenoxy)phenyl)-N-(2,2,2-trifluoroethylsulfonyl)pyrid-3-ylmethylamine. J. Med. Chem. 46, 3189–3192. [DOI] [PubMed] [Google Scholar]
- In our in house GTPγS assay, compound 3 displayed an mGluR2 PAM Emax of 169%; however, a definitive EC50 could not be determined because, prior to reaching a plateau, the GTPγS response gradually decreased, likely due to the solubility limits for the compound.
- Trabanco A. A., Cid J. M., Lavreysen H., Macdonald G. J., Tresadern G. (2010) Progress in the development of positive allosteric modulators of the metabotropic glutamate receptor 2. Curr. Med. Chem., submitted for publication. [DOI] [PubMed] [Google Scholar]
- Brnardic E. J.; Fraley M. E.; Garbaccio R. M.; Layton M. E.; Sanders J. M.; Culberson C.; Jacobson M. A.; Magliaro B. C.; Hutson P. H.; O’Brien J. A.; Huszar S. L.; Uslaner J. M.; Fillgrove K. L.; Tang C.; Kuo Y.; Sylvie M. S.; Hartman G. D. (2010) 3-Aryl-5-phenoxymethyl-1,3-oxazolidin-2-ones as positive allosteric modulators of mGluR2 for the treatment of schizophrenia: hit-to-lead efforts. Bioorg. Med. Chem. Lett. 20, 3129–3133. [DOI] [PubMed] [Google Scholar]
- Tresadern G; Cid J. M.; Macdonald G. J.; Vega J. A.; de Lucas A. I.; García A.; Matesanz E.; Linares M. L.; Oehlrich D.; Lavreysen H.; Biesmans I.; Trabanco A. A. (2010) Scaffold hopping from pyridones to imidazo[1,2-a]pyridines. Bioorg. Med. Chem. Lett. 20, 175–179. [DOI] [PubMed] [Google Scholar]
- The effect of these compounds on glutamate-induced [35S]-GTPγS binding induced by 4 μM glutamate (∼EC20) was characterized using a CHO cell line expressing the human mGluR2 receptor. For this, membranes prepared from human mGluR2 CHO cells were preincubated with compound alone (for determination of agonist effects) or together with an EC20 concentration (f.c. 4 μM) of glutamate (for determination of PAM effects) for 30 min at 30 °C. [35S]GTPγS was added at a final concentration of 0.1 nM and incubated for another 30 min at 30 °C. Final assay mixtures contained 7 μg of membrane protein, and the total reaction volume was 200 μL. Reactions were terminated by rapid filtration through Unifilter-96 GF/B filter plates (Packard, Meriden, CT) using a 96-well Packard filtermate harvester, and filter-bound radioactivity was counted on a Microplate Scintillation and Luminescence Counter from Packard. Z-Primes were ∼0.7. Data were calculated as % of the control agonist challenge. Sigmoid concentration−response curves plotting these percentages versus the log concentration of the test compound were analyzed using nonlinear regression analysis.
- Pinkerton A. B.; Cube R. V.; Hutchinson J. H.; Rowe B. A.; Schaffhauser H.; Zhao X.; Daggett L. P.; Vernier J.-M. (2004) Allosteric potentiators of the metabotropic glutamate receptor 2 (mGlu2). Part 1: identification and synthesis of phenyl-tetrazolyl acetophenones. Bioorg. Med. Chem. Lett. 14, 5329–5332. [DOI] [PubMed] [Google Scholar]
- Pinkerton A. B.; Cube R. V.; Hutchinson J. H.; James J. K.; Gardner M. F.; Schaffhauser H.; Rowe B. A.; Daggett L. P.; Vernier J.-M. (2004) Allosteric potentiators of the metabotropic glutamate receptor 2 (mGlu2). Part 2:4-thiopyridyl acetophenones as non-tetrazole containing mGlu2 receptor potentiators. Bioorg. Med. Chem. Lett. 14, 5867–5872. [DOI] [PubMed] [Google Scholar]
- Compound 8 was tested for agonist or antagonist activity on mGlu receptors in fluorescent Ca2+ assays using HEK293 cells expressing human mGluR3, mGluR5, mGluR7, or mGluR8.
- The CEREP selectivity screen was performed on the following targets: 5HT1A, 5HT2A, 5HT3, 5HT5A, 5HT6, 5HT7, A1, A2A, A3, AT1, Beta1, BK2, CCKA, CCR1, D1, D2, DAT, ETA, GAL2, H1, H2, IL8B, CXCR2, M1, M2, M3, MC4, NET, NK2, NK3, NPY1, NPY2, NT1, OP1, OP3, ORL1, V1A, VIP, SST, 5HT1B, Alpha1, Alpha2, BZD, CaCH, CICH, GABA, KCH, NaCH, and SKCaCH.
- Gleason S. D.; Shannon H. E. (1997) Blockade of phencyclidine-induced hyperlocomotion by olanzapine, clozapine, and serotonin receptor subtype selective antagonists in mice. Psychopharmacology 129, 79–84. [DOI] [PubMed] [Google Scholar]
- Details of the protocol for the in vivo assay: Phencyclidine (PCP)-induced hyperlocomotion in mice Male C57BL6/j mice (Charles River, France) were housed 5 per cage and kept under a 12-h light/dark cycle under constant temperature and humidity conditions. All experimental procedures were performed in full compliance with the French and European legislation governing the protection of vertebrate animals used in scientific research. Tested mGluR2 PAM compounds, (cpd 8, LY487379) were synthesized in house, and phencyclidine (PCP) was purchased from Sigma-Aldrich (France). Nonhabituated animals were treated with the vehicle, or 200 mg/kg ip of either cpd 8 or LY487379, and immediately challenged with either PCP (5.0 mg/kg, ip) or vehicle and individually placed into open-fields for a 30-min period. The distance traveled by animals was measured using video tracking and computerized analysis systems (Viewpoint, France) and the data analyzed using GraphPad Prism (v. 4.01, GraphPad, San Diego, CA, USA).