

Abstract
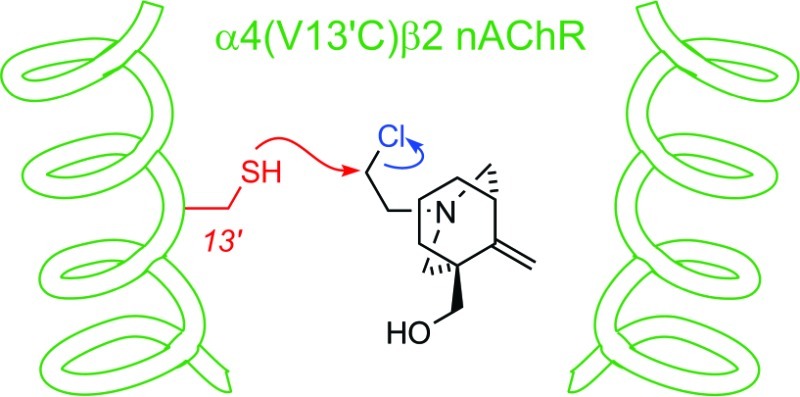
Neuronal nicotinic acetylcholine receptors (nAChR) are ligand gated ion channels that mediate fast synaptic transmission. Methyllycaconitine (MLA) is a selective and potent antagonist of the α7 nAChR, and its anthranilate ester side-chain is important for its activity. Here we report the influence of structure on nAChR inhibition for a series of novel MLA analogs, incorporating either an alcohol or anthranilate ester side-chain to an azabicyclic or azatricyclic core against rat α7, α4β2, and α3β4 nAChRs expressed in Xenopus oocytes. The analogs inhibited ACh (EC50) within an IC50 range of 2.3−26.6 μM. Most displayed noncompetitive antagonism, but the anthranilate ester analogs exerted competitive behavior at the α7 nAChR. At α4β2 nAChRs, inhibition by the azabicyclic alcohol was voltage-dependent suggesting channel block. The channel-lining residues of α4 subunits were mutated to cysteine and the effect of azabicyclic alcohol was evaluated by competition with methanethiosulfonate ethylammonium (MTSEA) and a thiol-reactive probe in the open, closed, and desensitized states of α4β2 nAChRs. The azabicyclic alcohol was found to compete with MTSEA between residues 6′ and 13′ in a state-dependent manner, but the reactive probe only bonded with 13′ in the open state. The data suggest that the 13′ position is the dominant binding site. Ligand docking of the azabicyclic alcohol into a (α4)3(β2)2 homology model of the closed channel showed that the ligand can be accommodated at this location. Thus our data reveal distinct pharmacological differences between different nAChR subtypes and also identify a specific binding site for a noncompetitive channel blocker.
Keywords: Ligand-gated ion channel (LGIC), nicotinic acetylcholine receptor (nAChR), noncompetitive antagonist (NCA), substituted cysteine accessibility method (SCAM), methyllycaconitine (MLA), homology model, reactive probe
Neuronal nicotinic acetylcholine receptors (nAChRs) are members of the ligand-gated ion channel (LGIC) superfamily that mediate fast synaptic transmission between neural cells. nAChRs insert into the cell membrane as a pentameric complex that is arranged around a central ion-conducting pore (1). Each subunit is comprised of a large extracellular N-terminal region, four transmembrane domains (M1−M4), two short loops (M1−M2 and M2−M3), a large intracellular loop (M3−M4), and a short extracellular carboxy terminal. The cationic transmembrane channel is predominately lined by the α-helical M2 domain, where cations passively pass through the receptor.
The nAChRs modulate neuronal function associated with cognition, learning and memory, arousal, cerebral blood flow and metabolism and have been implicated in a variety of pathological conditions, such as Alzheimer’s disease, Parkinson’s disease, nicotine addiction, pain, epilepsy, and schizophrenia (2). For this reason, there is great interest in the development of selective neuronal nAChR ligands as therapies (3). Eleven different nAChR subunits have been identified and cloned thus far from mammalian brain, and these include α2−7,9,10 and β2−4 subunits (4). The α subunits contain a disulfide bridge formed by two adjacent cysteine residues, while the β subunits lack this motif. Both heteromeric and homomeric combinations of these subtypes exist. Heteromeric nAChRs are made up of α and β subunits in mainly a 2:3 and or 3:2 stoichiometric ratios (5,6). Thus, in the pentamer, there are two possible ACh binding sites (1,7), while homomeric receptors contain five (8). In the brain, heteromeric α4β2, α3β4 and homomeric α7-subtypes dominate, but the extent of receptor variability is yet to be determined, largely because of an absence of selective ligands to aid their characterization and location.
Methyllycaconitine (MLA, 1, Figure 1) is a selective and potent antagonist of the α7 nAChR (9), but it is not useful clinically because of its unfavorable molecular properties (10,11). In an attempt to identify more “drug-like” nAChR antagonists, we previously developed a series of simpler azatricyclic MLA analogs incorporating a side-chain. Of particular interest was azatricyclic anthranilate ester 2(12,13), which was based on the ring system of MLA and a competitive antagonist at the α7 nAChR, displaying noncompetitive inhibition at α4β2 and mixed effects on α3β4 nAChRs (Figure 1) (14). The sites of interaction of compound 2 at heteromeric receptors are unknown. Many noncompetitive antagonists (NCAs) are known to inhibit nAChRs by sterically blocking the open ion channel. Here, agonists would be modulating the accessibility of channel blockers to the pore. Other NCAs have been found to block both the open and closed channels (15). Thus the M2 domain incorporates the binding site for many NCAs, including tetracaine, carbamazepine and barbiturates, as well as anesthetics such as phencylidine and ketamine, and antidepressants (See (16) for a comprehensive review of known NCA binding sites).
Figure 1.

Methyllycaconitine 1 and simple analogs. Azatricyclic anthranilate ester 2, azatricyclic alcohol 3, azabicyclic anthranilate ester 4, azabicyclic alcohol 5, and azabicyclic mustard 6.
In this study, a range of simple MLA analogs were investigated at rat α7, α3β4, and α4β2 nAChRs. We specifically sought to more closely define the structural features that give rise to the competitive or noncompetitive inhibition of the previously reported azatricyclic compound 2 at α7, α4β2, and α3β4 nAChRs by evaluating the azatricyclic analog lacking the anthranilate ester side-chain 3(12,13) and their corresponding azabicyclic analogs, 4 and 5(17,18), respectively. In addition, the NCA bicyclic alcohol 5 was used to identify the likely sites of ligand interactions within the channel of α4β2 receptors. Using SCAM in combination with a reactive probe, we show that bicyclic alcohol 5 competes with MTSEA between 6′-13′ of the M2 domain of the α4β2 nAChRs, while the reactive probe, bicyclic mustard 6, bonds solely at the 13′ position.
Results and Discussion
Effects of MLA Analogs on nAChRs
Oocytes expressing α4β2, α3β4, and α7 nAChRs responded to ACh in a concentration-dependent manner with current amplitudes ranging from 0.05−5, 0.05−0.2, and 0.05−1.2 μA, respectively. As shown in Figure 2, the amplitudes of ACh induced currents decreased at higher concentrations, due to receptor desensitization (14).The EC50 values obtained were 136.2 μM (95% CI = 99.54−186.4) for α4β2, 222.3 μM (95% CI = 181.0−273.0) for α3β4, and 277.7 μM (95% CI = 262.1−294.2) for α7 nAChRs.
Figure 2.

Concentration−response curves for ACh alone (◻) and ACh in the presence of compounds (30 μM) 2 (▲), 3 (●), 4 (◼), and 5 (▼) at (A) α4β2 normalized to 1 mM ACh (B) α3β4 normalized to 1 mM ACh and (C) α7 normalized to 10 mM ACh with a 3 min preincubation period. Data are presented as the mean ± SEM (n = 3 − 15 oocytes). All compounds studied were NCAs (represented by a significant drop in Imax and no significant rightward shift of the ACh EC50) at α4β2 and α3β4 nAChRs albeit compound 2 was previously reported (14) to have mixed competitive and noncompetitive properties as there was both a drop in Imax and shift in EC50 (Table 1). At α7, only compounds 3 and 5 were noncompetitive, while compounds 2 and 4 exhibited competitive rather than noncompetitive effects.
The effects of compound 2 have previously been reported (14) and showed competitive antagonism at α7, noncompetitive antagonism at α4β2 and mixed competitive and noncompetitive antagonism at α3β4 nAChRs. This data is summarized in Table 1 for comparison to the activities determined for the MLA analogs 3−6 (Figure 1).
Table 1. Effect of MLA Analogs 2−6 at α4β2, α3β4, and α7 nAChRs.
| α4β2 |
α3β4 |
α7 |
||||
|---|---|---|---|---|---|---|
| analog | IC50 (μM)a (95% CI)b | Imaxc | IC50 (μM)a (95% CI)b | Imaxc | IC50 (μM)a (95% CI)b | Imaxc |
| 2d | 7.15 (4.1−12.3) | 0.39 (0.24−0.54) | 16.0 (6.0−42.4) | 0.89 (0.77−1.00) | 11.2 (5.5−22.8) | 0.92 (0.34−1.5) |
| 3 | 21.1 (13.7−32.3) | 0.24 (0.24−0.27) | 7.9(5.5−11.4) | 0.80 (0.69−0.92) | 26.6 (18−37.6) | 0.58 (0.55−0.59) |
| 4 | 4.66 (2.3−9.2) | 0.52 (0.28−0.71) | 2.3 (0.9−6.2) | 0.26 (0.16−0.36) | 20.7 (7.0−61.4) | 1.06 (0.79−1.32) |
| 5e | 11.6 (5.2−25.9) | 0.58 (0.43−0.72) | 14.3 (2.5−24.8) | 0.53 (0.46−0.61) | 23.5 (12.2−45.1) | 0.53 (0.39−0.68) |
| 5f | 53.2 (18.8−150.6) | 1.06 (0.88−1.24) | ||||
| 6 | 10.9 (2.4−49.8) | 0.70 (0.58−0.82) | ||||
The concentration of the antagonist that inhibit 50% of ACh response (EC50; the concentration that activates 50% of maximum response) at the nAChR specified. The EC50 concentrations for ACh used were 100 (α4β2), 300, (α3β4), and 300 μM (α7).
95% Confidence Intervals (CI); data obtained from 3 to 12 oocytes.
Imax is the maximum current produced by ACh alone or ACh in the presence of antagonist (30 μM) at a specified nAChR.
Data taken from ref (14).
Data for bicyclic alcohol 5 with a 3 min preincubation.
Data for bicyclic alcohol 5 with no preincubation.
Like compound 2, compounds 3−6 did not activate the nAChRs on their own showing that they were not agonists (data not shown). When coapplied with ACh (EC50), they reduced the response in a concentration-dependent manner. Using bicyclic alcohol 5 as a model molecule, we constructed a time course for the inhibition of ACh (100 μM) at α4β2 nAChRs and found that stable and reproducible levels of inhibition were reached only after a 3 min preincubation (Figure 3A). With a 3 min incubation, the IC50 for bicyclic alcohol 5 dropped from 53.2 μM (95% CI = 18.8−50.6 μM) to 11.6 μM (95% CI = 5.2−25.8 μM; Figure 3B and Table 1), which is a 5-fold change. Moreover, bicyclic alcohol 5 (30 μM) inhibited the Imax of ACh from 106% (95% CI = 88−126%) to 58% (95% CI = 43−72%; p < 0.05; Figure 3C; Table 1) in the presence of a 3 min incubation. For all experiments, a 3 min preincubation was therefore used.
Figure 3.

(A) Normalized currents (ACh, 100 μM) versus preincubation times with bicyclic alcohol 5 (30 μM) at α4β2 nAChRs. The maximum inhibition of ACh (100 μM) exhibited by bicyclic alcohol 5 was achieved within 3 min, with no further increase thereafter. (B) Concentration-inhibition curves of bicyclic alcohol 5 in the presence of ACh (100 μM) without (●) and with (○) a 3 min preincubation. The IC50 values for bicyclic alcohol 5 with (11.6 μM; 95% CI = 5.2−25.9) and without (53.2 μM; 95% CI = 18.8−150.6) preincubation were statistically different (Student’s t test; p < 0.05). Data are normalized to ACh (EC50; IACh(100 μM)) and are presented as the mean ± SEM (n = 3−15 oocytes). (C) Concentration−response curves for ACh alone (◻) and ACh in the presence of bicyclic alcohol 5 (30 μM) without preincubation (●) and with a 3 min preincubation (○). Data are normalized to ACh (1 mM; Imax) in the absence of compound and are presented as mean ± SEM (n = 3−5 oocytes).
The IC50 values for compounds 3−5 were determined against an EC50 concentration of ACh (100 μM for α4β2, 300 μM for α3β4 and 300 μM for α7) for each nAChR and were similar to that of compound 2, ranging from 2.3−26.6 μM (Table 1).
At α7 nAChRs, compounds 3 and 5 showed a significant reduction in Imax (58% (95% CI: 55−59%) and 53% (95% CI: 39−68%) respectively; Table 1) for ACh, but compounds 2 and 4 displayed a significant rightward shift in the EC50 of ACh (p < 0.05) with no reduction in the Imax, which is consistent with competitive block. This data highlights the importance of the anthranilate ester moiety for the competitive effects of compounds 2 and 4 at homomeric α7 nAChR.
Recently, Hansen and colleagues presented a cocrystal of the MLA-A-AChBP complex (19). The rigid hexacyclic core of MLA was positioned between two AChBP interfaces, a region homologous to the N-terminal region of α7 subunits. The optimal conformation had the E ring (N-ethylpiperidine) of MLA in a chair conformation and the major interaction was the edge-to-face stacking of the E ring with the amino acid Trp147 of the A-AChBP. Due to the flexibility of the anthranilate side chain of MLA, it was extended to occupy a second pocket within the AChBP and thus had its own distinct interactions with the receptor (19). As compounds 2 and 4 contain the anthranilate side chain, the binding mode may be similar to MLA at this receptor. Interestingly when the anthranilate side chain was removed, the resulting alcohols 3 and 5 were NCAs at α7 nAChR. As there was no significant difference in the activity of the azatricyclic compared to the azabicyclic alcohols or esters, we suggest that the anthranilate side chain directs the compounds to the orthosteric site. In the case of the natural product MLA, removal of the side-chain methyl group, succinimide ring or anthranilate ester side chain results in a 20-, 210- or 1300-fold decrease, respectively, as observed in competitive binding assays at rat brain α7 nAChR (20−22).
Unlike compound 2, the actions of compounds 3−5 at the α3β4 nAChR were insurmountable indicating noncompetitive antagonism (Table 1; Figure 2). As compound 2 had mixed competitive and noncompetitive effects on this receptor, it indicates there may be two binding sites, with the competitive component corresponding to the agonist-binding site (23,24). In contrast the location of the noncompetitive binding site is unknown although a study using computational analysis and blind docking of a series of monocyclic MLA analogs indicated that the noncompetitive binding site may lie at the interface of the α and β subunits and in close proximity to the agonist-binding site in the α3β4 nAChR (25). One reason for why compound 2 has a different pharmacological profile to compound 4 may lie in the size of the molecule; compound 2 contains an azatricyclic core while compound 4 is bicyclic and the ring size difference may account for the observed pharmacological differences between the two compounds at this receptor subtype.
Like compound 2, compounds 3 − 5 were NCAs at α4β2 nAChRs (p < 0.05; Table 1; Figure 2). Furthermore bicyclic alcohol 5 inhibited ACh currents at membrane potentials ranging from −90 mV to 0 mV, but exhibited stronger block at more negative potentials (Figure 4). This indicates that the inhibitory effects of bicyclic alcohol 5 are voltage dependent and that it binds close to or within the channel lumen. In contrast, computational analysis and blind docking of a series of NCA based on monocyclic MLA analogs were found to bind to the N-terminal region of α4β2 nAChRs (26) a site overlapping the site proposed on the α3β4 nAChR (25).
Figure 4.

Voltage-dependence of bicyclic alcohol 5 inhibition at α4β2 wildtype nAChRs. (A) Current−voltage curves obtained by clamping cells (n = 4) between −90 mV and 0 mV in 10 mV steps, with ACh (100 μM) alone (◻) or ACh (100 μM) in the presence of bicyclic alcohol (30 μM) (●). Data is normalized to current generated by ACh (100 μM) alone held at −90 mV. (B) Plot of ACh (100 μM) current block expressed as the ratio of blocked current (30 μM bicyclic alcohol 5) over ACh control current (IDrug/IACh) versus membrane potentials (n = 4). Bicyclic alcohol 5 (30 μM) exerts a stronger block at more negative potentials.
Identifying the Binding Site within the Channel Pore
As all compounds evaluated were NCAs at the α4β2 receptor, we sought to locate the noncompetitive binding site using bicyclic alcohol 5 as the model compound. To this end, bicyclic alcohol 5 was used in competition with MTSEA in substituted cysteine accessibility method (SCAM) studies.
SCAM was used to probe positions within the M2 region of the α4-subunit that were previously known to be accessible (27); T2′ (corresponding to the mutant T278C), S6′ (S282C), L9′ (L285C), V13′ (V289C), and L16′ (L292C). The mutant receptors were coexpressed in oocytes with wildtype β2-subunits, and exposure to 1 mM ACh resulted in rapid inward currents ranging in amplitude from 0.2 μA − 2.0 μA. To assess if cysteine substitutions grossly altered channel structure, ACh concentration−response curves were performed on all cysteine mutants and compared to wildtype responses. EC50 values for ACh ranged from 60.1 − 198.5 μM (Table 2) and were not different from wildtype (EC50 = 136.2 μM; whole curve F-test, p > 0.05), indicating that the mutations did not affect receptor function.
Table 2. Effect of ACh on Wild-Type and Mutant α4β2 nAChRs.
| α4 Subunita | EC50 (μM)b | 95% CI (μM)c | nHd | EC50Mut/EC50WT |
|---|---|---|---|---|
| WT | 136.2 | 99.5−186.4 | 1.0 ± 0.2 | N/A |
| T2′C | 198.5 | 94.1−419.0 | 0.9 ± 0.2 | 1.46 |
| S6′C | 140.3 | 63.6−309.7 | 0.9 ± 0.3 | 1.03 |
| L9′C | 103.0 | 27.7−383.3 | 0.5 ± 0.1 | 0.76 |
| V13′C | 60.1 | 13.0−183.4 | 0.4 ± 0.1 | 0.44 |
| L16′C | 71.7 | 12.2−420.5 | 0.4 ± 0.1 | 0.53 |
ACh-induced currents were recorded in oocytes expressing either wildtype α4 or cysteine-substituted α4 mutants coexpressed with wildtype β2 subunits. Data obtained from 3−12 oocytes over at least 2 batches.
Concentration of agonist that activates 50% of maximal response at the nAChR specified. Values were obtained from 3−8 oocytes over at least two batches.
95% Confidence Intervals (CI).
Hill Coefficients and are mean ± SEM (n = 3−12).
Previous reports have shown that cysteine mutants facing the channel pore of the nAChR react with MTSEA leading to irreversible inhibition of ACh invoked current (27−29). Indeed, mutant α4β2 receptors reacted with 2.5 mM MTSEA leading to irreversible inhibition on all three channel states open, closed, and desensitized (calculated as a reduction of Imax of ACh, using the equation ([1 − (IACh,after/IACh,before)] × 100%)), with the exception of S6′C that showed no inhibition and thus no reaction with MTSEA in the closed or desensitized channel states (Figure 5C). A study performed by Akabas and colleagues showed that in the closed state the 6′ residue of mouse muscle nAChRs was accessible to MTSEA but not methanethiosulfonate ethyltrimethylammonium (MTSET) (27), while the 6′ position GABAA receptors, but not the glycine receptor, is accessible to MTSEA and MTSET (28). This indicates that the accessibility of the 6′ position in the closed state may differ between the various ligand-gated ion channels. At wildtype α4β2 receptors, 2.5 mM MTSEA had no effect on subsequent ACh currents in either the open, closed, or desensitized states, indicating that the reagent does not react with any endogenous cysteine in the protein or the reaction with them had no effect on receptor function (Figure 5).
Figure 5.

Effect of MTSEA on ACh-induced currents. Currents recorded from oocytes expressing either wildtype α4, α4T2′C, α4S6′C, α4L9′C, α4 V13′C, or α4L16′C with wildtype β2. (A) Current trace from an oocyte expressing α4 V13′Cβ2. Two pulses of ACh (100 μM) were applied before and after a 5 min incubation with 2.5 mM MTSEA + and 1 mM ACh. (B) Trace from an oocyte expressing the α4 V13′Cβ2 mutant. Two pulses of ACh (100 μM) were applied before and after a 3 min preincubation with bicyclic alcohol 5 (1 mM), followed by a 5 min incubation of 2.5 mM MTSEA + 1 mM ACh in the presence of bicyclic alcohol 5. (C) Percentage irreversible change in ACh (1 mM) induced currents in three different conformational states of the channel. Open Channel: 2.5 mM MTSEA + 1 mM ACh incubated for 5 min. Closed Channel: 2.5 mM MTSEA incubated in the absence of ACh for 5 min. Desensitized Channel: 1 mM ACh incubated for 1 min, then 1 mM ACh + 2.5 mM MTSEA incubated for 5 min. All mutants were accessible to sulfyhydryl modification in the open and desensitized channel states. Only α4S6′C was not accessible to MTSEA in the closed channel state and possibly the desensitized state. Data are mean ± SEM (n = 3 − 8 oocytes from at least two batches). Statistical significance is indicated as *p < 0.05; **p < 0.01; ***p < 0.001 and compares the effects of MTSEA on the cysteine mutant compared to wildtype. (D) Effects of competing bicyclic alcohol 5 before and after MTSEA on the same mutant. Open Channel: 1 mM bicyclic alcohol 5 was preincubated for 3 min and then treated with 1 mM ACh + 2.5 mM MTSEA for 5 min. Closed Channel: 1 mM bicyclic alcohol 5 incubated for 3 min then competed with 2.5 mM MTSEA in the absence of ACh. Desensitized Channel: 1 mM ACh + 1 mM bicyclic alcohol 5 preincubated for 3 min and then treated with 2.5 mM MTSEA. Data are mean ± SEM of N > 3 oocytes from at least two different batches. Statistical significance is indicated as *p < 0.05; **p < 0.01; ***p < 0.001 and compares differences between the effects of MTSEA in the presence and absence of bicyclic alcohol 5 on cysteine mutants.
Protection from MTSEA modification by bicyclic alcohol 5 was measured at each position within the M2 domain for the open, closed and desensitized states. In all channel states, the addition of bicyclic alcohol 5 protected the V13′C mutant from inhibition by MTSEA as the response to ACh was significantly reduced (Figure 5D). This data suggests that bicyclic alcohol 5 may be binding at the 13′ cysteine and protecting the site from MTSEA reactivity. However, bicyclic alcohol 5 also showed state-dependent protection of the L9′C and S6′C mutants from MTSEA reactivity. The L9′C position was protected in only the closed state, while the V6′C position was protected only in the open state. The effect of bicyclic alcohol 5 at inhibiting MTSEA reactivity in the channel could arise because of bicyclic alcohol 5 binding at multiple points depending on the state of the channel. Alternatively, the protection could arise by binding of the bicyclic alcohol at an allosteric site leading to conformational changes to the channel. The development of a reactive probe was pursued to address these possibilities.
Effects of Bicyclic Mustard 6 at Mutant Receptors
Alternative techniques have been used to identify binding sites for a number of NCAs, including photoaffinity labeling (15), molecular docking (29), and the development of reactive mustards to support traditional SCAM approaches (30,31). These approaches verified that many NCAs bind between the 2′ and 20′ positions of the nAChR channel. To determine whether bicyclic alcohol 5 is directly interacting with the identified amino acids or causing conformational changes to the channel from an allosteric site, bicyclic mustard 6 (Figure 1) was developed and applied to wildtype α4β2 and α4 mutant nAChR receptors.
The design of bicyclic mustard 6 was dictated by a number of considerations. First, the structural changes associated with introducing reactive functionality should be minimal, in order to preserve the location and mode of activity of the reactive ligand on both wildtype and cysteine mutant receptors. Second, the nature of the reactive functionality should provide a traceless ligation to the mutant receptor. Finally, the reactivity of the probe should be balanced to provide reactivity with cysteine but be relatively inert to other residues or system components. Conversion of the ethylamine 5 to its chloroethyl derivative 6 supplied a suitable electrophilic modification that promised to fulfill these requirements.
The synthesis of azabicyclic mustard 6 started with the double Mannich annulation of β-keto ester 7(32), followed by Wittig olefination to give benzyl protected azabicycle 8 (Figure 6). Chemoselective deprotection of the benzyl amine with α-chloroethyl chloroformate, followed by reductive amination with chloroacetic acid gave ethyl ester 9 in moderate yield (33). Reduction of the ester side chain with diisobutylaluminium hydride proceeded smoothly in the presence of the chloroethylamine functionality to afford the azabicyclic mustard compound 6 (synthesis of mustard 6 is reported in the Supporting Information).
Figure 6.

Synthesis of azabicyclic mustard 6: (a) N,N-bis(ethoxymethyl)benzylamine, CH3SiCl3, CH3CN, 87%; (b) CH3PPh3Br, KOtBu, THF, 58%; (c) α-chloroethyl chloroformate, (CH2Cl)2, then CH3OH, 73%; (d) NaBH4, chloroacetic acid, PhCH3, 32%; (e) diisobutylaluminium hydride, CH2Cl2, 78%.
Like bicyclic alcohol 5 (30 μM), the corresponding mustard 6 (30 μM) reduced ACh (100 μM) evoked response by 65% at wildtype α4β2 nAChRs (Figure 7). Both bicyclic mustard 6 (10.9 μM; 95% CI = 2.4−49.8; Figure 7B) and bicyclic alcohol 5 (11.6 μM; 95% CI = 5.2−25.9) had similar IC50 values (p < 0.05; Table 1). In addition, the action of bicyclic mustard 6 in the presence of ACh was insurmountable (Table 1; Figure 7C) indicating noncompetitive antagonism of α4β2 nAChRs. As bicyclic mustard 6 was noncompetitive and had a similar potency to bicyclic alcohol 5, it was a deemed a suitable reactive probe for this study.
Figure 7.

(A) Trace showing the effect of ACh (100 μM; duration indicated by black bar) and ACh in the presence of bicyclic alcohol 5 (30 μM; duration indicated by hatched bar) and bicyclic mustard 6 (30 μM; duration indicated by white bar). Both bicyclic alcohol 5 and bicyclic mustard 6 were preincubated for 3 min before coapplying with ACh. (B) Concentration-inhibition curve for bicyclic mustard 6 (○) in the presence of ACh (100 μM) at α4β2 nAChR. The IC50 value was 10.9 μM (95% CI: 2.40−49.8) and was not statistically different (Student’s t test; p > 0.05) to bicyclic alcohol 5 at α4β2 nAChR under the same conditions (Figure 3). (C) Concentration response curves for ACh alone (◻) and ACh in the presence of bicyclic mustard 6 (30 μM) with a 3 min preincubation (Δ) at α4β2 nAChRs. Bicyclic mustard 6 was a noncompetitive antagonist at α4β2 nAChRs (represented by a significant drop in Imax and no significant rightward shift of the ACh EC50 (Table 1).
Bicyclic mustard 6 was applied to wildtype α4β2 and α4 mutant nAChR receptors (T2′C, S6′C, L9′C, V13′C, and L16′C) following a similar protocol used for MTSEA (Figure 8). In brief, bicyclic mustard 6 (100 μM) was incubated for 5 min in the presence of 1 mM ACh (open state) and in the absence of ACh (closed state) to react directly with the cysteine residues through a nucleophilic substitution reaction to form a covalent adduct.
Figure 8.

Effect of bicyclic mustard 6 on ACh-induced currents evoked from oocytes expressing either wildtype α4, α4T2′C, α4S6′C, α4L9′C, α4 V13′C, or α4L16′C mutants with wildtype β2. (A) Example trace showing bicyclic mustard 6 (100 μM) incubated in the presence of ACh (EC50; 100 μM) for 5 min. Two pulses of ACh (100 μM) were applied before and after incubation. (B) Irreversible change in ACh (100 μM) induced currents in presence of bicyclic mustard 6 (100 μM) for the open and the closed states of α4β2 nAChRs. (C) Irreversible change in ACh (100 μM) induced currents in presence of various bicyclic mustard 6 concentrations ranging from 10 to 100 μM at the α4 V13′Cβ2 mutant. Bicyclic mustard 6 shows a concentration-dependent reactivity at the α4 V13′Cβ2 mutant for a given time. Asterisks indicate statistical significance compared to wildtype and are assigned as follows: *p < 0.05; **p < 0.01; ***p < 0.001. All data presented are mean ± SEM (n = 3−8 oocytes).
Like MTSEA, bicyclic mustard 6 did not show irreversible inhibition of wildtype ACh responses in either the open or closed states (p > 0.05). Furthermore, bicyclic mustard 6 did not react with the T2′C, S6′C, L9′C and L16′C mutant nAChRs. In contrast, bicyclic mustard 6 showed irreversible inhibition of ACh response at the V13′C in the open state compared to wildtype nAChR (73.4% ± 5.5, p < 0.001; Figure 8B). This effect was further investigated for concentration-dependence. Incubation of 30 μM bicyclic mustard 6 with 1 mM ACh gave rise to similar levels of inhibition (62.4% ± 6.0) to that observed at 100 μM, but 10 μM bicyclic mustard 6 was less inhibitory (5.86% ± 14.9) (Figure 8C). These data indicate that bicyclic mustard 6 inhibits ACh-induced currents by bonding to the V13′C in a concentration-dependent manner. No reaction of the bicyclic mustard 6 was observed at the V13′C mutant in the closed state, which may reflect the lower inhibition by MTSEA of the 13′ mutant in the closed state (−62.3% ± 4.4) compared to the open (−80.1% ± 2.6, p < 0.05; Figure 5C).
The S6′ and L9′ cysteine mutants were unaffected by bicyclic mustard 6 in both the open and closed states, respectively. This is despite the protection of these residues from MTSEA reaction afforded by bicyclic alcohol 5 (Figure 5). The apparent protection of the S6′C mutant from reaction with MTSEA may arise through binding of the bicyclic alcohol 5 at the 13′ site maintaining the channel in a closed-like conformation. The apparent protection of the L9′C mutant by bicyclic alcohol 5 in the closed state may reflect the proximity to the 13′ binding site or alternatively, the lower inhibition by MTSEA of the 13′, 9′ and 2′ mutants observed in competition experiments with bicyclic alcohol 5 in the closed relative to open state (p < 0.05; Figure 5D). Taken together, the data obtained with bicyclic alcohol 5 and mustard 6 provides strong evidence that the compounds bind within the channel lumen, specifically at the 13′ position.
Homology Modeling and Docking
To visualize the binding of bicyclic alcohol 5 at the 13′ position we docked this compound into a homology model of the closed (α4)3(β2)2 receptor. A homology model of the pore region of the α4β2 nAChR was generated based on the low resolution structure (4 Å; 2BG9) of the nAChR (Figure 9) (34). Both (α4)3(β2)2 and (α4)2(β2)3 receptors exist, but based on the higher ACh EC50 values of our studies the model was generated with a stoichiometry of (α4)3(β2)26,35. Amino acid sequence alignment of α4 and β2 with the α, β, γ and δ subunits from the template structure showed a strong conservation within the M2 pore region (Figure 9). Overall target-template amino acid sequence identity was 54% (α4−α), 47% (β2−β), 39% (α4−δ), and 46% (β2−γ) but identity within M2 was higher (>50%, Figure 9A). Structural comparisons of M2-helices from the homology model and template structure gave an rmsd of 1.8 Å and z-score of 10.6, which would yield confidence levels of >95% and thus compares favorably with other published results (36). The average pore diameter for the (α4)3(β2)2 homology model and the original template were also very similar, suggesting that in this region the homology model accurately represents the native receptor. As the template we used for homology modeling is proposed to be in the closed conformation, we assume that docking was also performed in this closed conformation.
Figure 9.

Comparison of the channel from the template 2BG9 and the homology model used in this study. (A) An amino acid sequence alignment of the α4 and β2 subunits with those in the 2BG9 structure (T. marmorata α, β, δ, and γ). The position of M2 and the pore-lining residues are highlighted. A pairwise sequence comparison (% identity) of α4 with α or δ subunits, and β2 with β or γ subunits is shown to the right. (B) A surface representation of a single α4 subunit from the homology model, with V13′ highlighted in green. The dark line shows the subunit profile that faces the pore. (C) The same profile is plotted as the average pore diameter for the whole pentamer (solid line, (α4)3(β2)2 homology model; dotted line, 2BG9) and was calculated using HOLE (18).
Flexible docking of bicyclic alcohol 5 into the channel of the (α4)3(β2)2 homology model yielded a series of docking solutions that fell into two main clusters, both of which located bicyclic alcohol 5 close to the 13′ residue. In the first cluster, bicyclic alcohol 5 was in the same plane as the 13′ residue (Figure 10A). In the second, the ligand was slightly lower, lying equidistant between 9′ and 13′ (Figure 10B). This second pose is consistent with some protection of the L9′C mutant from MTSEA reactivity because of the binding of the bicyclic alcohol 5 in the closed state. However, no reaction was seen at this position with bicyclic mustard 6. For all docked poses, bicyclic alcohol 5 was stabilized by van der Waals contacts and no hydrogen bonds were predicted.
Figure 10.

Examples of the two main docked poses generated using flexible ligand docking of bicyclic alcohol 5 into a (α4)3(β2)2 homology model of the closed channel. The backbones of the M2 helices are shown as ribbons. The side-chains of residues 6′ and 13′ are highlighted, but all others have been removed for clarity. The left-hand panels show the docked pose as seen looking from the extracellular domain, down through the receptor pore. The right-hand panels are the same poses from the side, but with the closest β2 subunit removed so that the ligand can be more clearly viewed. Bicyclic alcohol 5 is shown in CPK.
Rigid docking of bicyclic alcohol 5 into the channel of the (α4)3(β2)2 homology model identified three main groups. The first two clusters were similar to those described for flexible ligand docking (Figure 11A and B). In the third, bicyclic alcohol 5 extended lower into the channel, between residues 6′ and 13′ (Figure 11C)). As the 6′ mutant was unaltered in the closed state by MTSEA (even in the absence of the bicyclic alcohol 5), the competition experiments cannot provide evidence to verify this pose. However, no reaction was seen at either the L9′C or S6′C positions with bicyclic mustard 6. van der Waals forces could account for all protein−ligand contacts with no predicted hydrogen bonds. Data obtained from both flexible and rigid docking runs is shown in the Supporting Information.
Figure 11.

Examples of the three main docked poses generated using rigid ligand docking of bicyclic alcohol 5 into a (α4)3(β2)2 homology model of the closed channel. M2 helices are shown as ribbons, and apart from the side chains of residues 6′ and 13′, all others have been removed for clarity. The left-hand panels show the docked pose as seen looking from the extracellular domain, down through the receptor pore. The right-hand panels are the same poses viewed from the side. Bicyclic alcohol 5 is shown in CPK.
Conclusions
The nAChRs modulate neuronal function and have been implicated in a variety of pathological conditions such nicotine addiction, pain, epilepsy, and schizophrenia (2). For this reason, there is great interest in the development of selective neuronal nAChR ligands as therapies (3).
In an attempt to identify more “drug-like” nAChR antagonists based on the MLA template, we evaluated a series of simpler azatricyclic and azabicyclic ligands, compounds 2−5 that incorporate the anthranilate or alcohol side-chains. From this study, we identified the MLA anthranilate side-chain of to be an important structural determinant that directs the binding of these compounds to the orthosteric site of the α7 nAChR. In contrast, this side chain had no influence on the pharmacological effects of the compounds at other nAChRs such as the α4β2 subtype.
As all compounds were NCAs at the α4β2 nAChR, we used bicyclic alcohol 5 as a model ligand to identify the binding site within the channel lumen. In all channel states, the addition of bicyclic alcohol 5 protected the V13′C mutant from inhibition by MTSEA. The reactive probe, bicyclic mustard 6, reacted with the V13′C mutant in the open state providing further evidence for drug binding in the 13′ region. Docking of bicyclic alcohol 5 into a homology model of the (α4)3(β2)2 closed channel shows that this ligand can bind close to the 13′ residue which is located in the narrowest region of the pore (9′−17′). Thus our data reveal distinct pharmacological differences between different nAChR, and also identify a specific binding site for a noncompetitive channel blocker.
Methods
Materials
ACh, MLA, tricaine, sodium pyruvate, theophylline, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), kanamycin and gentamycin were obtained from Sigma (Australia).
The cDNA encoding wildtype rat nAChR α3, α4 subunit subcloned in pSP64, β2 subunit subcloned in the pSP65 vector and α7, β4 subunit subcloned in vector pBS SK (+) were generous gifts from Professor Jim Boulter (University of California, Los Angeles, CA).
Synthesis of Bicyclic Mustard
Bicyclic mustard 6 was synthesized (Figure 6) according to the experimental procedures outlined in the Supporting Information. Spectroscopic data and reproductions of 1H and 13C NMR spectra associated with the synthesis of mustard 6 are also reported in the Supporting Information.
Site-Directed Mutagenesis
Cysteine mutations were generated within the pore lumen of the α4 nAChR subunit by using sense and antisense oligonucleotide primers and the QuickChange II Site-directed Mutagenesis kit protocol (Stratagene, La Jolla, CA). The design of the primers incorporated a series of silent mutations that allowed for quick screening of colonies. The plasmids were then isolated with the Wizard Plus Minipreps DNA Purification System (Promega, New South Wales, Australia). Mutants were screened by restriction enzyme analysis and then confirmed by full DNA sequencing at the Sydney University Prince Alfred Molecular Analysis Centre, Australia. The sense oligonucleotides primers are shown in Table 1 of the Supporting Information.
Preparation of nAChR subunit mRNAs− Plasmids containing α7 insert was linearized with SmaI, while plasmids containing wildtype α3, α4 and mutant α4 inserts were linearized with EcoRI. Wildtype β2 and β4 inserts were linearized with HindIII and XhoI, respectively. α3, α4, and β2 mRNAs were synthesized using the SP6 mMESSAGE mMACHINE kit (Ambion, Austin, TX), α7 with the T7 mMESSAGE mMACHINE kit (Ambion, Austin, TX) and β4 with the T3 mMESSAGE mMACHINE kit (Ambion, Austin, TX).
Expression of Wildtype and Mutant nAChRs
Oocytes from Xenopus laevis (South Africa clawed frogs) were surgically removed while under general anesthetic (tricaine, 850 mg/500 mL) in accordance with the National Health and Medical Research Council of Australia’s ethical guidelines and approved by the University of Sydney Animal Ethics Committee. Harvested lobes were treated with collagenase A (2 mg mL−1; Roche Diagnostics, Australia) in oocyte releasing buffer 2 (OR-2; 82.5 mM NaCl, 2 mM KCl, 1 mM MgCl2, 5 mM HEPES, pH 7.5) for 1.5 h to release and defolliculate the oocytes. 1−10 ng cRNA was mixed in a ratio of 1:1 for α3/β4 and α4/β2 in a 50.6 nL volume and injected into selected healthy stage V−VI oocytes. Injected oocytes were kept in frog Ringer storage solution (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2 and 5 mM HEPES) containing 2.5 mM sodium pyruvate, 0.5 μM theophylline, and 4 μg/mL of kanamycin. Oocytes were stored for 2−5 days at 18 °C in an orbital shaker before recording.
Electrophysiological Recordings
Two-electrode voltage-clamp recordings were undertaken as previously described (14). Briefly, oocytes expressing α4β2, α3β4, and α7 nACh receptors were clamped at −60 mV and continually perfused with Ca2+-free solution (115 mM NaCl, 2.5 mM KCl, 1.8 mM BaCl2, 10 mM HEPES) where Ca2+ was replaced with Ba2+ to maintain osmolarity. The solution was supplemented with 1 μM atropine to block any muscarinic receptors that may be endogenously expressed on the oocyte. Glass electrodes were used for recording and had a resistance ranging from 0.2−2 MΩ. The internal electrode solution contained 3 M KCl. Compounds were stored at −20 °C and made up to the required concentrations in Ca2+-free solution before applying to the oocyte by gravity flow. Whole-cell currents were measured using a GeneClamp 500 amplifier (Axon Instruments, Foster City, CA, USA), a MacLab 2e recorder (AD Instruments, Sydney, NSW, Australia) and Chart Version 4.0.1 program.
Concentration Response Curves for ACh
Concentration−response curves for ACh at wildtype α7, α3β4, α4β2, and mutant α4*β2 receptors (Figures 2 and 7, Tables 1 and 2) were constructed from the peak current response of a range of concentrations (0.1−10000 μM) and normalized to maximal ACh currents (Imax; 1 mM). Recording oocytes were washed for 10 min between doses of ACh for all nAChRs except for the 6′ α4S262Cβ2 mutant, which required a 20 min washout time and the α7 nAChR which required 15 min washout time. This allowed the receptor to recover from the desensitized state. To determine whether the compounds were competitive or noncompetitive, concentration response curves for ACh were constructed in the presence of a fixed concentration of 3−6 (30 μM).
Time Course Experiments
The time required for maximal inhibition of the compounds (Figure 3) was determined by preincubating the α4β2 nAChR with bicyclic alcohol 5 at 0, 1, 3, and 10 min. Since there was no difference in activity between 3 and 10 min, all compounds were preincubated for 3 min before the addition of ACh in all the experiments described.
Inhibitory Concentration Response Curves
To determine the activities of 2−5 and mustard 6, inhibition concentration response curves (Figures 3 and 7, Table 1) of the compounds were constructed using increasing antagonist concentrations (0.1−1000 μM) in the presence of 100 μM ACh for α4β2, 300 μM for α3β4, and 300 μM for α7 nAChRs. Inhibited responses were normalized to the responses to respective concentrations of ACh.
Voltage Sensitivity of Bicyclic Alcohol Block
Voltage dependence block of bicyclic alcohol 5 (Figure 4) was investigated by measuring currents induced by ACh in 10 mV steps at a membrane potential range of 0 to −90 mV. Currents induced by (i) 100 μM ACh alone and (ii) 100 μM ACh in the presence of bicyclic alcohol (30 μM) were measured. To compare between different oocytes, all ACh induced currents were normalized by the current induced by 100 μM ACh at −90 mV.
Data Analysis
The normalized current (I/Imax unless otherwise stated) recorded in response to each drug was fitted using the Gaussian fit equation from GraphPad Prism 4 for Macintosh (GraphPad Software, San Diego, California), while activity was determined using sigmoidal fit (variable slope) equation from GraphPad Prism 4.0:
where [A] is the ligand concentration and nH is the Hillslope. From this equation, the concentration of the agonist that activates 50% of expressed receptors (EC50) or in the case of inhibitory concentration response curves the concentration of the antagonist that inhibits 50% of the evoked ACh current (IC50) were calculated. Data are presented as mean ± SEM or as mean (95% confidence intervals) from a minimum of 3 oocytes over a minimum of 2 batches. Unless otherwise stated statistical differences between groups were calculated using Student’s t test.
Substituted Cysteine Accessibility Studies
Stock solutions of methanethiosulfonate ethylammonium (1 M; MTSEA; Toronto Research Chemicals Inc., Ontario, Canada) were made up in distilled water, aliquoted and stored at −20 °C. For each application, the solution was thawed, diluted to the working concentration in Ca2+ free solution, and used immediately.
The irreversible effects of 2.5 mM MTSEA and bicyclic mustard 6 (10, 30, and 100 μM) were assayed by measuring the baseline average of two peak currents evoked by two applications of 100 μM ACh before incubating with either MTSEA or bicyclic mustard in Ca2+ free solution for 5 min (Figures 5 and 8). This was followed by measuring the average of two peak currents evoked by two applications of 100 μM ACh. Three different channel states were evaluated: (i) the open state with the presence of 1 mM ACh, (ii) the closed state in the absence of ACh, and (iii) the slow onset desensitized state where the receptor was first desensitized with 1 mM ACh for 1 min before MTSEA incubation.
To investigate the ability of bicyclic alcohol 5 to protect the cysteine mutant from sulfydryl modification, a similar assay was used. This time, 1 mM bicyclic alcohol 5 was incubated for 3 min to allow it to occupy its binding site before adding MTSEA to the perfusion system for a further 5 min in the described channel states. For the desensitized channel state, 1 mM ACh was added with 1 mM bicyclic alcohol 5 for the 3 min.
The effect of the reagents was taken as I − (IACh(After) − IACh(Before)) and statistical significance performed using the Student’s t test. For the effects of MTSEA or bicyclic mustard on cysteine mutants, statistical significance was evaluated compared to wildtype. For investigating the ability of bicyclic alcohol 5 in protecting the cysteine mutant from sulfydryl modification, statistical significance was taken compared to the corresponding effects of MTSEA in the same mutant and channel state.
Homology Modeling
Using FUGUE, the protein sequences nAChR α4 (accession number P43681) and β2 (P17787) were aligned with α, β, γ, and δ subunits taken from the ACh receptor at 4 Å (PDB ID 2BG9) (34). The α4 peptide sequence was substituted onto the α and δ chains of 2BG9, and the β2 peptide was substituted onto β and γ chains. On the basis of the concentration−response parameters described herein, a (α4)3(β2)2 stoichiometry was used, and a three-dimensional homology model was generated using MODELER 6v2 (35,37,38). The intracellular domain was removed from the final models and the best model selected by Ramachandran plot using RAMPAGE (39).
The three-dimensional structure of bicyclic alcohol 5 was drawn in ChemDraw Ultra 7.0.1 (CambridgeSoft, Cambridge, MA) and energy minimized using the MM2 force field in Chem3D v 7.0.0. To ensure a reliable bicyclic alcohol 5 structure, the energy minimized version was imported into PyMOL (Schrödinger, NY) and compared to an active conformation of methyllycaconitine bound to AChBP (PDB ID 2BYR). To improve the fit of the molecules, dihedral angles of bicyclic alcohol 5 were manually changed and energy minimized in Chem3D.
Model Evaluation
Model quality was measured by comparisons with the template structure using ALADYN (40). Structural comparisons of M2-helices from the homology model and template structure gave an rmsd of 1.8 Å and z-score of 10.6, which would yield confidence levels of >95%.
Docking
Rigid and flexible ligand docking of the protonated form of bicyclic alcohol 5 into the (α4)3(β2)2 receptor homology model was carried out using GOLD 3.0 (The Cambridge Crystallographic Data Centre, Cambridge, U.K.). The binding site was constrained to be within 10 Å of the V13′ residue (a region that encompassed the positions between 6′ and 20′) of all three α4 subunits (chains A, C, and D). These amino acids were chosen based on the data presented in this manuscript. Ten genetic algorithm runs were performed for both rigid and flexible ligand docking, giving a total of 20 solutions. Automatic GA settings were used, and docked clusters were identified using the rms analysis implemented in GOLD (Supporting Information). A cutoff of <3.0 Å rmsd was used for the purposes of illustrating the main poses shown in Figures 10 and 11.
Acknowledgments
We would like to thank Professor Jim Boulter (University of California, Los Angeles, CA) for the generous gifts of rat nAChRs.
Abbreviations
A-AChBP, Aplysia acetylcholine binding protein; ACh, acetylcholine; EC50, effective concentration that activates 50% of the receptors; GABA, gamma-amino butyric acid; HEPES, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; IC50, the effective concentration that inhibits 50% of the receptors; LGIC, ligand-gated ion channel; MLA, methyllycaconitine; MTSEA, methanethiosulfonate ethylammonium; MTSET, methanethiosulfonate ethyltrimethylammonium; nAChR, nicotinic acetylcholine receptor; NCA, noncompetitive antagonist; nH, Hill slope or hill coefficient; SCAM, substituted cysteine accessibility method.
Supporting Information Available
Oligonucleotide primers, experimental procedures, docking information spectroscopic data, and reproductions of 1H and 13C NMR spectra associated with the synthesis of mustard 6. This information is available free of charge via the Internet at http://pubs.acs.org/.
Experiments were designed by A.J.T., M.L., S.C.R.L., M.D.M., and M.C. Experiments were performed by G.X.J.Q., D.L., J.I.H., J.I.A., and A.J.T. Data analyses were performed by G.X.J.Q., D.L., J.I.H., N.A., J.I.A., A.J.T., S.C.R.L., M.D.M., and M.C. Manuscript was written by G.X.J.Q., N.A., A.J.T., S.C.R.L., M.D.M., and M.C.
This research was supported by a Discovery Project (DP0986469) from the Australian Research Council (M.D.M., M.C.), Australian Postgraduate Award (G.Q., J.I.H., D.L.), John A. Lamberton (G.Q., J.I.H.) scholarships, and the Wellcome Trust (S.C.R.L., A.J.T.). S.C.R.L. is a Wellcome Trust Senior Research Fellow in Basic Biomedical Studies.
Supplementary Material
References
- Unwin N. (1993) Nicotinic acetylcholine receptor at 9 A resolution. J. Mol. Biol. 229, 1101–1124. [DOI] [PubMed] [Google Scholar]
- Paterson D.; Nordberg A. (2000) Neuronal nicotinic receptors in the human brain. Prog. Neurobiol. 61, 75–111. [DOI] [PubMed] [Google Scholar]
- Romanelli M. N.; Gratteri P.; Guandalini L.; Martini E.; Bonaccini C.; Gualtieri F. (2007) Central nicotinic receptors: Structure, function, ligands, and therapeutic potential. ChemMedChem 2, 746–767. [DOI] [PubMed] [Google Scholar]
- Lukas R. J.; Changeux J. P.; Le Novere N.; Albuquerque E. X.; Balfour D. J.; Berg D. K.; Bertrand D.; Chiappinelli V. A.; Clarke P. B.; Collins A. C.; Dani J. A.; Grady S. R.; Kellar K. J.; Lindstrom J. M.; Marks M. J.; Quik M.; Taylor P. W.; Wonnacott S. (1999) International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol. Rev. 51, 397–401. [PubMed] [Google Scholar]
- Anand R.; Conroy W. G.; Schoepfer R.; Whiting P.; Lindstrom J. (1991) Neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes have a pentameric quaternary structure. J. Biol. Chem. 266, 11192–11198. [PubMed] [Google Scholar]
- Zwart R.; Vijverberg H. P. (1998) Four pharmacologically distinct subtypes of alpha4beta2 nicotinic acetylcholine receptor expressed in Xenopus laevis oocytes. Mol. Pharmacol. 54, 1124–1131. [PubMed] [Google Scholar]
- Luetje C. W.; Patrick J. (1991) Both alpha- and beta-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J. Neurosci. 11, 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma E.; Bertrand S.; Binzoni T.; Bertrand D. (1996) Neuronal nicotinic alpha 7 receptor expressed in Xenopus oocytes presents five putative binding sites for methyllycaconitine. J. Physiol. 491, 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J. M.; Cockcroft V. B.; Lunt G. G.; Smillie F. S.; Wonnacott S. (1990) Methyllycaconitine: A selective probe for neuronal alpha-bungarotoxin binding sites. FEBS Lett. 270, 45–48. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Delivery Rev. 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H. Y.; Smith B. R.; Ward K. W.; Kopple K. D. (2002) Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- Barker D.; Brimble M. A.; McLeod M. D.; Savage G. P. (2004) Synthesis of tricyclic analogues of methyllycaconitine using ring closing metathesis to append a B ring to an AE azabicyclic fragment. Org. Biomol. Chem. 2, 1659–1669. [DOI] [PubMed] [Google Scholar]
- Barker D.; McLeod M. D.; Brimble M. A.; Savage G. P. (2002) Application of olefin metathesis to the synthesis of ABE ring analogues of methyllycaconitine. Tetrahedron Lett. 43, 6019–6022. [Google Scholar]
- Barker D.; Lin D. H.; Carland J. E.; Chu C. P.; Chebib M.; Brimble M. A.; Savage G. P.; McLeod M. D. (2005) Methyllycaconitine analogues have mixed antagonist effects at nicotinic acetylcholine receptors. Bioorg. Med. Chem. 13, 4565–4575. [DOI] [PubMed] [Google Scholar]
- Chiara D. C.; Hamouda A. K.; Ziebell M. R.; Mejia L. A.; Garcia G. 3rd; Cohen J. B. (2009) [(3)H]chlorpromazine photolabeling of the torpedo nicotinic acetylcholine receptor identifies two state-dependent binding sites in the ion channel. Biochemistry 48, 10066–10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias H. R.; Bhumireddy P.; Bouzat C. (2006) Molecular mechanisms and binding site locations for noncompetitive antagonists of nicotinic acetylcholine receptors. Int. J. Biochem. Cell Biol. 38, 1254–1276. [DOI] [PubMed] [Google Scholar]
- Barker D.; Brimble M. A.; McLeod M.; Savage G. P. (2001) A High yielding synthesis of anthranilate esters from sterically hindered alcohols. Tetrahedron Lett. 42, 1785–1788. [Google Scholar]
- Barker D.; Brimble M. A.; McLeod M.; Savage G. P. (2004) Synthesis of Simple Analogues of Methyllycaconitine − An Efficient Method for the Preparation of the N-Substituted Anthranilate Pharmacophore. Tetrahedron 60, 5953–5963. [Google Scholar]
- Hansen S. B.; Sulzenbacher G.; Huxford T.; Marchot P.; Taylor P.; Bourne Y. (2005) Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. Embo. J. 24, 3635–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardick D. J.; Blagbrough I. S.; Cooper G.; Potter B. V.; Critchley T.; Wonnacott S. (1996) Nudicauline and elatine as potent norditerpenoid ligands at rat neuronal alpha-bungarotoxin binding sites: Importance of the 2-(methylsuccinimido)benzoyl moiety for neuronal nicotinic acetylcholine receptor binding. J. Med. Chem. 39, 4860–4866. [DOI] [PubMed] [Google Scholar]
- Hardick D. J.; Cooper G.; Scott-Ward T.; Blagbrough I. S.; Potter B. V.; Wonnacott S. (1995) Conversion of the sodium channel activator aconitine into a potent alpha 7-selective nicotinic ligand. FEBS Lett. 365, 79–82. [DOI] [PubMed] [Google Scholar]
- Jacyno J. M.; Harwood J. S.; Lin N. H.; Campbell J. E.; Sullivan J. P.; Holladay M. W. (1996) Lycaconitine revisited: partial synthesis and neuronal nicotinic acetylcholine receptor affinities. J. Nat. Prod. 59, 707–709. [DOI] [PubMed] [Google Scholar]
- Free R. B.; Bryant D. L.; McKay S. B.; Kaser D. J.; McKay D. B. (2002) [3H]Epibatidine binding to bovine adrenal medulla: Evidence for alpha3beta4* nicotinic receptors. Neurosci. Lett. 318, 98–102. [DOI] [PubMed] [Google Scholar]
- Free R. B.; von Fischer N. D.; Boyd R. T.; McKay D. B. (2003) Pharmacological characterization of recombinant bovine alpha3beta4 neuronal nicotinic receptors stably expressed in HEK 293 cells. Neurosci. Lett. 343, 180–184. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Cestari T. F.; Henderson B. J.; Pavlovicz R. E.; McKay S. B.; El-Hajj R. A.; Pulipaka A. B.; Orac C. M.; Reed D. D.; Boyd R. T.; Zhu M. X.; Li C.; Bergmeier S. C.; McKay D. B. (2009) Effect of novel negative allosteric modulators of neuronal nicotinic receptors on cells expressing native and recombinant nicotinic receptors: implications for drug discovery. J. Pharmacol. Exp. Ther. 328, 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson B. J.; Pavlovicz R. E.; Allen J. D.; Gonzalez-Cestari T. F.; Orac C. M.; Bonnell A. B.; Zhu M. X.; Boyd R. T.; Li C.; Bergmeier S. C.; McKay D. B. (2010) Negative allosteric modulators that target human α4β2 neuronal nicotinic receptors. J. Pharmacol. Exp. Ther. 334, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akabas M. H.; Kaufmann C.; Archdeacon P.; Karlin A. (1994) Identification of acetylcholine receptor channel-lining residues in the entire M2 segment of the alpha subunit. Neuron 13, 919–927. [DOI] [PubMed] [Google Scholar]
- Shan Q.; Haddrill J. L.; Lynch J. W. (2002) Comparative surface accessibility of a pore-lining threonine residue (T6′) in the glycine and GABA(A) receptors. J. Biol. Chem. 277, 44845–44853. [DOI] [PubMed] [Google Scholar]
- Arias H. R.; Rosenberg A.; Targowska-Duda K. M.; Feuerbach D.; Jozwiak K.; Moaddel R.; Wainer I. W. (2010) Tricyclic antidepressants and mecamylamine bind to different sites in the human alpha4beta2 nicotinic receptor ion channel. Int. J. Biochem. Cell Biol. 42, 1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Shi L.; Karlin A. (2003) Structural effects of quinacrine binding in the open channel of the acetylcholine receptor. Proc. Natl. Acad. Sci. U.S.A. 100, 3907–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen S. E.; Sharp S. D.; Liu W. S.; Cohen J. B. (1992) Structure of the noncompetitive antagonist-binding site of the Torpedo nicotinic acetylcholine receptor. J. Biol. Chem. 267, 10489–10499. [PubMed] [Google Scholar]
- Halliday J. I.; Chebib M.; Turner P.; McLeod M. D. (2006) Double-Mannich annulation of cyclic ketones using N,N-bis(ethoxymethyl)alkylamine reagents. Org. Lett. 8, 3399–3401. [DOI] [PubMed] [Google Scholar]
- Yang B. V.; O’Rourke D.; Li J. (1993) Mild and selective debenzylation of tertiary amines using alpha-chloroethyl chloroformate. Synlett 3, 195–196. [Google Scholar]
- Shi J.; Blundell T. L.; Mizuguchi K. (2001) FUGUE: Sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310, 243–257. [DOI] [PubMed] [Google Scholar]
- Moroni M.; Zwart R.; Sher E.; Cassels B. K.; Bermudez I. (2006) Alpha4beta2 nicotinic receptors with high and low acetylcholine sensitivity: pharmacology, stoichiometry, and sensitivity to long-term exposure to nicotine. Mol. Pharmacol. 70, 755–768. [DOI] [PubMed] [Google Scholar]
- Dalton J. A.; Jackson R. M. (2007) An evaluation of automated homology modelling methods at low target template sequence similarity. Bioinformatics 23, 1901–1908. [DOI] [PubMed] [Google Scholar]
- Sali A.; Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815. [DOI] [PubMed] [Google Scholar]
- Moroni M.; Bermudez I. (2006) Stoichiometry and pharmacology of two human alpha4beta2 nicotinic receptor types. J. Mol. Neurosci. 30, 95–96. [DOI] [PubMed] [Google Scholar]
- Lovell S. C.; Davis I. W.; Arendall W. B. 3rd; de Bakker P. I.; Word J. M.; Prisant M. G.; Richardson J. S.; Richardson D. C. (2003) Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50, 437–450. [DOI] [PubMed] [Google Scholar]
- Potestio R., Aleksiev T., Pontiggia F., Cozzini S., Micheletti C. (2010) ALADYN: a web server for aligning proteins by matching their large-scale motion, Nucleic Acids Res 38 Suppl, published online May 05, 2010. DOI: 2010.1093/nar/gkq2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.