

Abstract
Dopamine (DA) is a neurotransmitter implicated in multiple functions, including movement, cognition, motivation, and reward. The DA transporter (DAT) is responsible for clearing extracellular DA, thereby terminating DA neurotransmission. Previously, it has been shown that insulin signaling through protein kinase B/Akt regulates DAT function by fine-tuning DAT cell surface expression. Importantly, specific Akt isoforms (e.g., Akt1, Akt2) serve distinct physiological functions. Here, we demonstrate using isoform-specific Akt inhibitors that basal activity of Akt2, rather than Akt1, regulates DAT cell surface expression. Since Akt2 activation is mediated by insulin, these data further implicate insulin signaling as an important modulator of DAT function and dopaminergic tone.
Keywords: Dopamine, transporter, Akt, insulin
Dopamine (DA) is a neurotransmitter that plays an important role in movement, motivation, and cognition. DA is also a key regulator of reward (1). An essential element in fine-tuning DA neurotransmission is the DA transporter (DAT) (2,3). DAT function is required to clear released DA by active reuptake into the presynaptic bouton (4,5), thereby terminating DA signaling. Therefore, changes in DAT function have profound implications in DA homeostasis and signaling (6,7).
Both function and trafficking of the DAT are tightly regulated by several signaling pathways, including protein kinase C (PKC), mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K), and importantly, protein kinase B (Akt) (8,9). It is well documented that inhibition of PI3K decreases surface levels of DAT and reduces DAT function, including DA clearance measured in vivo(10−13). Similarly, inhibition of Akt results in a decrease in DAT surface expression and function (14).
While it is known that Akt regulates DAT cell surface expression, it is unclear which Akt isoform is involved in this regulation. Importantly, different Akt isoforms appear to serve distinct functions in brain (15,16). Akt exists as three isoforms; Akt1 and Akt2 are ubiquitously expressed, whereas Akt3 is found only in the brain and testes (16). Knockout studies reveal that Akt1 is primarily associated with cell survival and growth (17) and Akt3 appears to have similar functions in the brain (15). In contrast, Akt2 is associated with insulin modulation of glucose homeostasis, including regulation of glucose transporter (GLUT4) trafficking (18,19).
Many studies have linked dysfunctions in Akt signaling to the underlying mechanisms of disorders such as schizophrenia, which also involves dysregulation of DA signaling (20−22). Therefore, defining the isoform of Akt that is responsible for altering DAT cell surface expression will further advance our knowledge of how aberrant Akt signaling leads to abnormal DA neurotransmission and may be clinically relevant to central dopaminergic disorders.
Results
Pharmacological inhibition of Akt has been shown to reduce DAT cell surface expression (14). However, the isoform of Akt involved is unknown. Therefore, we utilized allosteric, isoform-specific inhibitors of Akt1 (I-Akt1) and Akt2 (I-Akt2), as well as a dual Akt1 and Akt2 inhibitor (I-Akt1/2), to identify the Akt isoform that regulates DAT trafficking (Figure 1). These selective inhibitors were developed and characterized in cell lines and primary tissue (23−25). HEK-293 cells stably expressing the human DAT (hDAT cells) were plated at the same density and treated for 1 h with either I-Akt1 (12 μM), I-Akt2 (12 μM), or I-Akt1/2 (5 μM). These concentrations have previously been shown to be isoform-specific (23,24). Using biotinylation assays (14,26), we examined surface expression of hDAT after both vehicle treatment and drug treatment. Figure 2A (inset) shows representative immunoblots for hDAT obtained from hDAT cells treated with vehicle (control), I-Akt1, I-Akt2, or I-Akt1/2. Densitometric analysis of the immunoblots was performed, and the level of surface hDAT was normalized to total hDAT. These data demonstrated that only inhibition of Akt2 significantly reduced hDAT cell surface expression (Figure 2A, surface hDAT was normalized to total hDAT and expressed as percent of control; ∗ indicates p < 0.01 by one-way ANOVA followed by Bonferroni post hoc test). Consistently, dual inhibition of both isoforms significantly decreased surface hDAT as well (Figure 2A). Importantly, inhibition of Akt1 had no significant effect on hDAT surface expression.
Figure 1.

Structures of isoform-selective, allosteric Akt inhibitors. The structures of the potent dual Akt1/Akt2-selective (1), Akt1-selective (2), and Akt2-selective (3) inhibitors are shown. IC50 for each Akt isoform is shown for each compound.
Figure 2.

Inhibition of Akt2 reduces hDAT cell surface expression in hDAT cells: (A) Representative immunoblots obtained from HEK cells expressing hDAT after treatment for 1 h with isoform-specific inhibitors of Akt1 (I-Akt1, 12 μM) or Akt2 (I-Akt2, 12 μM) or a dual inhibitor of Akt1 and Akt2 (I-Akt1/2, 5 μM). For quantification, the density of each biotinylated hDAT band was normalized to the density of its corresponding total hDAT band and expressed as a percentage of control (∗ = p < 0.01; one-way ANOVA, followed by Bonferroni posthoc test; n = 4). All data are represented as mean ± SEM. (B) Immunoprecipitation of Akt1 and Akt2 was performed from hDAT cells after treatment with each inhibitor as described in panel A. Immunoblots were probed for p-Akt (Thr308) to assess the phosphorylation state of each isoform after drug treatment.
Active Akt is phosphorylated at Thr308 and Ser473, and therefore application of isoform-specific inhibitors should reveal reduction of the phosphorylation of the appropriate isoform. Therefore, to confirm the isoform specificity of these inhibitors in our assay, hDAT cells were treated as described above and specific Akt isoforms were immunoprecipitated and analyzed by immunoblot analysis using phospho-specific antibodies. We probed for phosphorylated Akt (Thr308) to gauge the active, phosphorylated state of each Akt isoform after drug treatment. Figure 2B demonstrates that the each inhibitor dramatically decreased basal phosphorylation of the relevant isoform(s), confirming that these inhibitors are isoform specific at the concentrations and treatment time used in our assay. Thus, these data suggest that Akt2, not Akt1, is responsible for regulating hDAT cell surface expression in this heterologous expression system.
Next we confirmed our findings ex vivo by biotinylation of striatal slices (300 μm), which are enriched in DAT-positive nerve termini (27). Slices were treated with either I-Akt1, I-Akt2, or I-Akt1/2 as shown in Figure 3. The samples from each tissue punch were normalized to total level of proteins. Similar to the results obtained from hDAT cells, treatment of striatal slices with I-Akt2 reduced rat DAT (rDAT) cell surface expression, as did treatment with the dual inhibitor I-Akt1/2 (Figure 3A, inset). The cytosolic protein CaMKII was found only in the cytosolic fraction, confirming the plasma membrane nature of the biotinylated fraction of the striatal preparation. Importantly, CaMKII levels (loading control) were not affected by Akt inhibitor treatment (Figure 3A, inset). Densitometric analysis of the immunoblots showed that inhibition of Akt2 significantly decreased DAT cell surface expression with respect to vehicle-treated control (Figure 3A). Similarly, dual inhibition of both Akt1 and Akt2 significantly decreased DAT cell surface expression (Figure 3A). Importantly, inhibition of Akt1 alone had no effect on DAT trafficking in our striatal preparation (Figure 3A).
Figure 3.

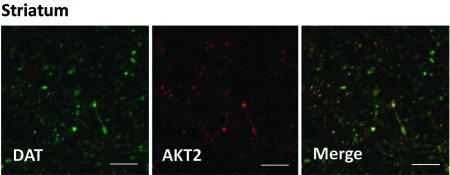
Inhibition of Akt2 reduces rDAT cell surface expression in rat striatal tissue: (A) Representative immunoblots obtained from striatal slices of biotinylated and total rat DAT (rDAT) after treatment for 1 h with isoform-specific inhibitors of Akt1 (I-Akt1; 12 μM) or Akt2 (I-Akt2; 12 μM), or dual inhibitor of Akt1 and Akt2 (I-Akt1/2; 5 μM). Immunoblots of CaMKII were used to determine the plasma membrane identity of the biotinylated fraction and control for loading. For quantification, the density of each biotinylated rDAT band was normalized to that of its corresponding total rDAT band and expressed as a percentage of control (∗ = p < 0.05; one-way ANOVA followed by Bonferroni posthoc test; n = 11). All data are represented as mean ± SEM. (B) Confocal imaging of rat striatal slices, where green indicates DAT-positive regions and red indicates Akt2-positive regions (scale bar, 12 μm). The merged image depicts yellow regions indicating high levels of expression of both Akt2 and DAT in dopaminergic projections (n = 3). Similar results were obtained for Akt1.
To demonstrate further the role of Akt2 in the regulation of DAT, we determined whether Akt2 is expressed in striatum in DAT-positive projections. Figure 3B shows that in striatal slices, Akt2 is heavily enriched in dopaminergic terminals marked by DAT immunoreactivity. These data further support our finding that Akt2 is involved in regulating DAT trafficking, demonstrating for the first time isoform specificity of Akt regulation of DAT trafficking in native tissue.
Discussion
DA clearance is regulated both by DAT turnover rate and by the number of active transporters at the plasma membrane (28). As a consequence, DAT membrane expression is thought to fine-tune DA homeostasis and dopaminergic signaling (6,29,30). Previously, we have shown that insulin signaling through Akt regulates DAT cell surface expression, DA clearance, and the ability of psychostimulants such as amphetamine to target the DAT and thereby increase extracellular DA levels (11,12,14,31). Here, we demonstrate by using selective inhibitors of Akt1 and Akt2 that, in striatum, Akt2 activity regulates DAT trafficking, whereas Akt1 does not.
Akt activation has diverse functions ranging from cell survival and growth to glucose homeostasis (17−19,32−34). In order to contribute to such diverse physiological processes, it is thought that each Akt isoform serves a distinct role. This hypothesis is postulated from the phenotypes observed in the Akt isoform-specific knockout mouse models (17,18). In particular, Akt2 knockout mice are hyperglycemic and insulin resistant (18). These observations obtained from knockout models are consistent with data supporting a pivotal role of Akt2 in the increase of glucose uptake (19). This insulin-mediated increase in glucose uptake is supported by trafficking of the glucose transporter GLUT4 to the plasma membrane, an effect shown to be mediated through Akt2, not Akt1 (19). Although insulin crosses the blood−brain barrier (35), neurons use insulin-independent mechanisms to transport glucose. Insulin receptors (IRs) are found throughout the brain, including DA-rich areas such as the striatum (36). Importantly, abnormal insulin status has been shown to alter DAT cell surface expression and function (11−13,37). Therefore, it is conceivable that insulin signaling regulates DAT surface expression by modulating Akt2 activity. Furthermore, disease states with dysfunctional insulin signaling, such as type II diabetes, could alter Akt2 activity in brain and affect DAT function and DA homeostasis. Further work is needed to determine the extent to which improper insulin tone affects DAT function and DA homeostasis.
Notably, these isoform-specific, allosteric Akt inhibitors exert their effects by blocking the phosphorylation of Akt itself (23), making Akt unable to activate downstream targets. Importantly, they do not have inhibitory activity on other cellular kinases, such as protein kinase A (PKA) or protein kinase C (PKC) (24). Previously, these inhibitors have been used to examine the isoform specificity of increased Akt activity in tumor cell lines and tissues (24,25). While each drug stimulates apoptosis, inhibition of both isoforms by the dual inhibitor I-Akt1/2 was most effective, providing evidence that tumor cell growth is somehow not isoform specific (24). Conversely, in striatal preparations, we found isoform specificity to Akt regulation of DAT trafficking, despite the fact that Akt1 is also expressed in dopaminergic terminals (data not shown).
While the data shown here firmly point to Akt2 as an important regulator of DAT, only Akt1 and Akt2 were examined. A third isoform, Akt3, is expressed in the brain and testes (38). Previous work demonstrates that Akt3 plays a role similar to Akt1 in the brain in regulating cell growth and survival, whereas Akt2 contributes to mediating insulin receptor signaling (15,16). The possibility that Akt3 regulates monoamine transporter trafficking is intriguing and warrants further evaluation in future studies when a specific inhibitor becomes available.
In summary, our data indicate that basal Akt2 activity is responsible for maintaining DAT cell surface expression, implicating Akt2 as a key regulator of DAT function and DA homeostasis. Akt2 is known to be coupled to insulin receptor activation, further confirming insulin signaling as an important modulator of DAT function and dopaminergic tone.
Methods
Cell Surface Protein Biotinylation
Biotinylation experiments were performed on intact cells as described previously (14,39,41). Briefly, HEK-293 stably transfected with hDAT (hDAT cells) were plated at a density of 1 × 106 per well in a six-well poly-(d-lysine) coated plate. Cells were washed with cold phosphate-buffered saline (PBS) containing Ca2+/Mg2+ and treated for the indicated times. Then, cells were incubated with 1.0 mg/mL sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithiopropionate [NHS-SS-biotin] (Pierce/Thermo Scientific, Rockford, IL) for 30 min, washed, quenched with 100 mM glycine, and extracted in lysis buffer (PBS Ca2+/Mg2+, 1% Triton 100-X, and 0.5 mM phenylmethylsulfonyl fluoride (PMSF) at 4 °C). Lysates were centrifuged, total fractions reserved, and supernatants incubated with immobilized streptavidin beads (Pierce/ThermoScientific) for 1 h at room temperature. Beads were washed three times in lysis buffer, and bound proteins were eluted with 2× sample buffer containing 2-mercaptoethanol. Proteins were separated by sodium dodecyl sulfate−polyacrylamide gel electrophoresis (SDS−PAGE) and immunoblotted. For estimation of relative amounts of proteins, the exposed films of the immunoblots were scanned, and band intensities were measured with Scion Image (Scion Corporation, Frederick, MD).
Brain Slice Preparation
Methods are as described previously (42). All procedures were conducted in accordance with the Vanderbilt Institutional Animal Care and Use Committee. Briefly, Sprague−Dawley rats (approximately 300 g) were decapitated. The brains were quickly removed and placed in an ice-cold, low-sodium/high-sucrose dissecting solution. Hemisected (300 μm) coronal brain slices containing the striatum were prepared on a vibratome. Slices were allowed to recover in a submerged holding chamber (37 °C) containing oxygenated (95% O2, 5% CO2) artificial cerebrospinal fluid (aCSF) that contained 124 mM NaCl, 4.4 mM KCl, 2.5 mM CaCl2, 1.3 mM MgSO4, 1 mM NaH2PO4, 10 mM glucose, and 26 mM NaHCO3 for a recovery period of 60 min before beginning experiments. Slices were then incubated with Akt inhibitors, 12 μM (I-Akt1 and I-Akt2) or 5 μM (I-Akt 1/2), in aCSF or aCSF containing the vehicle DMSO for 1 h at 37 °C. Biotinylation assays were then performed.
Biotinylation Assays
For slice assays, hemisected coronal slices (300 μm) were transferred to multiwell submerged chambers containing oxygenated aCSF with NHS-SS-biotin (1 mg/mL) on ice at 4 °C and incubated for 45 min, then washed twice for 10 min each in aCSF, and finally incubated in aCSF containing glycine (100 mM) for two 20 min periods. Slices were then placed onto dishes on dry ice, and the striatum was removed and placed into eppendorf tubes. Tissue punches of the striatum were homogenized in ice-cold homogenization buffer (1% Triton, 2 mM sodium orthovanadate, 2 mM sodium fluoride, 25 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 150 mM NaCl, 10 μg/mL aprotinin, 10 μg/mL leupeptin, and 100 μM phenylmethylsulfonyl fluoride) and centrifuged for 30 min at 1000g at 4 °C. Protein levels were obtained, and equal amounts were added to strepavidin beads with pulldown buffer (0.1% Triton, 25 mM HEPES, 150 mM NaCl, 2 mM sodium orthovanadate, 2 mM sodium fluoride, 10 μg/mL aprotinin, 10 μg/mL leupeptin, and 100 μM phenylmethylsulfonyl fluoride) and incubated overnight at 4 °C. Samples were washed and eluted, and immunoblot analysis was carried out. Total slice lysates and the biotinylated (slice surface) fraction underwent immunodetection for rDAT.
Immunoprecipitation
After treatment with either vehicle or each inhibitor as described, hDAT expressing HEK cells were washed with ice-cold PBS/Ca2+/Mg2+ and incubated in 400 μL/well of lysis buffer containing 50 mM NaH2PO4, 10 mM Tris, 100 mM NaCl, 0.5 mM PMSF, pH 8.0, and 1% Triton X-100 for 1 h at 4 °C. Cell lysates recovered by centrifugation at 20 000g for 30 min were incubated overnight at 4 °C either with Akt1 (1:250; Cell Signaling Technology; Danvers, MA) or Akt2 (1:800; Santa Cruz Biotechnology, Sana Cruz, CA) antibodies. Complexes were retrieved by the addition of 20 μL of protein G-Sepharose (GE Healthcare, Little Chalfont, Buckinghamshire, U.K.), washed three times with lysis buffer. Bound proteins were then eluted and processed for immunoblot analysis as described.
Immunoblotting
Determination of immunoreactivity was conducted according to previously described methods (13,14). Briefly, tissue samples were separated by SDS−PAGE, and resolved proteins were transferred to poly(vinylidene difluoride) (PVDF) membranes (BioRad), which were incubated for 1 h in blocking buffer (5% BSA and 0.1% Tween20 in Tris-buffered saline). The blots were incubated with primary antibody overnight at 4 °C. Primary antibodies used for immunostaining were CaMKII (1:2000; Affinity BioReagents, Rockford, IL), p-Akt (Thr308; 1:1000; Millipore, Billerica, MA), and hDAT (1:1000, Cell Signaling Technology, Danvers, MA). For rat DAT (rDAT) immunostaining, mouse monoclonal primary antibodies were used (antibody 16, 1:1000; (43)). All proteins were detected using HRP-conjugated secondary antibodies (1:5000; Santa Cruz Biotechnology, Santa Cruz, CA). After chemiluminescent visualization (ECL-Plus; Amersham; Piscataway, NJ) on Hyper-film ECL film (Amersham), protein band densities were quantified (Scion Image; Frederick, MD) and normalized to control.
Immunohistochemistry
For tissue staining, slices were prepared as described above and subsequently fixed with PBS Ca2+/Mg2+ and 4% paraformaldehyde, washed three times with PBS, permeabilized, blocked with PBS with 4% bovine serum albumin (BSA)/0.15% Tween-20, and immunostained with the appropriate antibody dissolved in PBS with 4% BSA/0.05% Tween-20. Primary antibodies used for immunostaining were Akt2 (1:200; Santa Cruz Technology; Santa Cruz, CA) and rDAT (1:400; antibody 16; (43)) overnight at 4 °C. After incubation with secondary fluorophores, immunofluorescence was imaged using a Perkin-Elmer UltraView confocal with a Nikon Eclipse 2000-U microscope equipped with a 60× lens with an N.A. of 1.49. Image processing was performed using Image J and Adobe Photoshop.
Work supported by NIH Grant DA14684 (A.G.).
Funding Statement
National Institutes of Health, United States
References
- Wise R. A. (1998) Drug Alcohol Depend. 51, 13–22. [DOI] [PubMed] [Google Scholar]
- Borowsky B.; Adham N.; Jones K. A.; Raddatz R.; Artymyshyn R.; Ogozalek K. L.; Durkin M. M.; Lakhlani P. P.; Bonini J. A.; Pathirana S.; Boyle N.; Pu X.; Kouranova E.; Lichtblau H.; Ochoa F. Y.; Branchek T. A.; Gerald C. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 8966–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giros B.; el Mestikawy S.; Godinot N.; Zheng K.; Han H.; Yang-Feng T.; Caron M. G. (1992) Mol. Pharmacol. 42, 383–90. [PubMed] [Google Scholar]
- Giros B.; Jaber M.; Jones S. R.; Wightman R. M.; Caron M. G. (1996) Nature 379, 606–12. [DOI] [PubMed] [Google Scholar]
- Jones S. R.; Gainetdinov R. R.; Jaber M.; Giros B.; Wightman R. M.; Caron M. G. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 4029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer T. J.; Biederman J.; Madras B. K.; Faraone S. V.; Dougherty D. D.; Bonab A. A.; Fischman A. J. (2005) Biol. Psychiatry 57, 1293–300. [DOI] [PubMed] [Google Scholar]
- Gelernter J.; Kranzler H. R.; Satel S. L.; Rao P. A. (1994) Neuropsychopharmacology 11, 195–200. [DOI] [PubMed] [Google Scholar]
- Gonzalez M. I.; Robinson M. B. (2004) Curr. Opin. Pharmacol. 4, 30–5. [DOI] [PubMed] [Google Scholar]
- Torres G. E. (2006) J. Neurochem. 97, 3–10. [DOI] [PubMed] [Google Scholar]
- Carvelli L.; Moron J. A.; Kahlig K. M.; Ferrer J. V.; Sen N.; Lechleiter J. D.; Leeb-Lundberg L. M.; Merrill G.; Lafer E. M.; Ballou L. M.; Shippenberg T. S.; Javitch J. A.; Lin R. Z.; Galli A. (2002) J. Neurochem. 81, 859–69. [DOI] [PubMed] [Google Scholar]
- Owens W. A.; Sevak R. J.; Galici R.; Chang X.; Javors M. A.; Galli A.; France C. P.; Daws L. C. (2005) J. Neurochem. 94, 1402–10. [DOI] [PubMed] [Google Scholar]
- Lute B. J.; Khoshbouei H.; Saunders C.; Sen N.; Lin R. Z.; Javitch J. A.; Galli A. Biochem. Biophys. Res. Commun. 2008, 372, 656–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. M.; Owens W. A.; Turner G. H.; Saunders C.; Dipace C.; Blakely R. D.; France C. P.; Gore J. C.; Daws L. C.; Avison M. J.; Galli A. (2007) PLoS Biol. 5, 2369–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia B.; Wei Y.; Moron J. A.; Lin R. Z.; Javitch J. A.; Galli A. (2005) Mol. Pharmacol. 68, 102–9. [DOI] [PubMed] [Google Scholar]
- Tschopp O.; Yang Z. Z.; Brodbeck D.; Dummler B. A.; Hemmings-Mieszczak M.; Watanabe T.; Michaelis T.; Frahm J.; Hemmings B. A. (2005) Development 132, 2943–54. [DOI] [PubMed] [Google Scholar]
- Dummler B.; Tschopp O.; Hynx D.; Yang Z. Z.; Dirnhofer S.; Hemmings B. A. (2006) Mol. Cell. Biol. 26, 8042–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H.; Thorvaldsen J. L.; Chu Q.; Feng F.; Birnbaum M. J. (2001) J. Biol. Chem. 276, 38349–52. [DOI] [PubMed] [Google Scholar]
- Cho H.; Mu J.; Kim J. K.; Thorvaldsen J. L.; Chu Q.; Crenshaw E. B. 3rd; Kaestner K. H.; Bartolomei M. S.; Shulman G. I.; Birnbaum M. J. (2001) Science 292, 1728–31. [DOI] [PubMed] [Google Scholar]
- Bae S. S.; Cho H.; Mu J.; Birnbaum M. J. (2003) J. Biol. Chem. 278, 49530–6. [DOI] [PubMed] [Google Scholar]
- Emamian E. S.; Hall D.; Birnbaum M. J.; Karayiorgou M.; Gogos J. A. (2004) Nat. Genet. 36, 131–7. [DOI] [PubMed] [Google Scholar]
- Bajestan S. N.; Sabouri A. H.; Nakamura M.; Takashima H.; Keikhaee M. R.; Behdani F.; Fayyazi M. R.; Sargolzaee M. R.; Bajestan M. N.; Sabouri Z.; Khayami E.; Haghighi S.; Hashemi S. B.; Eiraku N.; Tufani H.; Najmabadi H.; Arimura K.; Sano A.; Osame M. (2006) Am. J. Med. Genet., Part B 141B, 383–6. [DOI] [PubMed] [Google Scholar]
- Schwab S. G.; Hoefgen B.; Hanses C.; Hassenbach M. B.; Albus M.; Lerer B.; Trixler M.; Maier W.; Wildenauer D. B. (2005) Biol. Psychiatry 58, 446–50. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Zhao Z.; Leister W. H.; Robinson R. G.; Barnett S. F.; Defeo-Jones D.; Jones R. E.; Hartman G. D.; Huff J. R.; Huber H. E.; Duggan M. E. (2005) Bioorg. Med. Chem. Lett. 15, 761–4. [DOI] [PubMed] [Google Scholar]
- DeFeo-Jones D.; Barnett S. F.; Fu S.; Hancock P. J.; Haskell K. M.; Leander K. R.; McAvoy E.; Robinson R. G.; Duggan M. E.; Lindsley C. W.; Zhao Z.; Huber H. E.; Jones R. E. (2005) Mol. Cancer Ther. 4, 271–9. [PubMed] [Google Scholar]
- Zhao Z.; Robinson R. G.; Barnett S. F.; Defeo-Jones D.; Jones R. E.; Hartman G. D.; Huber H. E.; Duggan M. E.; Lindsley C. W. (2008) Bioorg. Med. Chem. Lett. 18, 49–53. [DOI] [PubMed] [Google Scholar]
- Saunders C.; Ferrer J. V.; Shi L.; Chen J.; Merrill G.; Lamb M. E.; Leeb-Lundberg L. M.; Carvelli L.; Javitch J. A.; Galli A. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 6850–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giros B.; Caron M. G. (1993) Trends Pharmacol. Sci. 14, 43–9. [DOI] [PubMed] [Google Scholar]
- Giros B.; el Mestikawy S.; Bertrand L.; Caron M. G. (1991) FEBS Lett. 295, 149–54. [DOI] [PubMed] [Google Scholar]
- Blakely R. D.; Defelice L. J.; Galli A. (2005) Physiology (Bethesda) 20, 225–31. [DOI] [PubMed] [Google Scholar]
- Amara S. G. (1996) Rev. Bras. Biol. 56(Suppl 1 Part 1), 5–19. [PubMed] [Google Scholar]
- Williams J. M.; Galli A. (2006) Handb. Exp. Pharmacol. 215–32. [DOI] [PubMed] [Google Scholar]
- Krizman-Genda E.; González M. I.; Zelenaia O.; Robinson M. B. (2005) Neuropharmacology 49, 872–82. [DOI] [PubMed] [Google Scholar]
- Somwar R.; Kim D. Y.; Sweeney G.; Huang C.; Niu W.; Lador C.; Ramlal T.; Klip A. (2001) Biochem. J. 359, 639–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Z.; Tschopp O.; Baudry A.; Dummler B.; Hynx D.; Hemmings B. A. (2004) Biochem. Soc. Trans. 32, 350–4. [DOI] [PubMed] [Google Scholar]
- Banks W. A.; Jaspan J. B.; Kastin A. J. (1997) Peptides 18, 1257–62. [DOI] [PubMed] [Google Scholar]
- Schulingkamp R. J.; Pagano T. C.; Hung D.; Raffa R. B. (2000) Neurosci. Biobehav. Rev. 24, 855–72. [DOI] [PubMed] [Google Scholar]
- Patterson T. A.; Brot M. D.; Zavosh A.; Schenk J. O.; Szot P.; Figlewicz D. P. (1998) Neuroendocrinology 68, 11–20. [DOI] [PubMed] [Google Scholar]
- Gonzalez E.; McGraw T. E. (2009) Cell Cycle 8, 2502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung U.; Apparsundaram S.; Galli A.; Kahlig K. M.; Savchenko V.; Schroeter S.; Quick M. W.; Blakely R. D. (2003) J. Neurosci. 23, 1697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dipace C.; Sung U.; Binda F.; Blakely R. D.; Galli A. (2007) Mol. Pharmacol. 71, 230–9. [DOI] [PubMed] [Google Scholar]
- Grueter B. A.; Winder D. G. (2005) Neuropsychopharmacology 30, 1302–11. [DOI] [PubMed] [Google Scholar]
- Gaffaney J. D.; Vaughan R. A. (2004) Mol. Pharmacol. 65, 692–701. [DOI] [PubMed] [Google Scholar]