

Abstract
The difficulty in developing successful treatments to facilitate nerve regeneration has prompted a number of new in vitro experimental methods. We have recently shown that functional presynaptic boutons can be formed when neuronal cells are cocultured with surface-modified artificial substrates including poly(d-lysine)-coated beads and supported lipid bilayer-coated beads (Lucidoet al. (2009) J. Neurosci. 1, 12449−12466; Gopalakrishnanet al. (2010) ACS Chem. Neurosci. 1, 86−94). We demonstrate here, using confocal microscopy combined with immunocytochemistry, that it is possible to isolate such in vitro presynaptic endings in an exclusive fashion onto glass substrates through a simple “sandwich/lift-off” technique (Perezet al. (2006) Adv. Funct. Mater. 1, 306−312). Isolated presynaptic complexes are capable of releasing and recycling neurotransmitter in response to an external chemical trigger. These bead−presynaptic complexes are facile to prepare and are readily dispersible in solution. They are thus compatible with many experimental methods whose focus is the study of the neuronal presynaptic compartment.
Keywords: Neurons, synapse, cell-free system, synaptic vesicle exocytosis, neurotransmitter release, neurodegenerative diseases, presynaptic complex
The understanding of fundamental aspects of the central nervous system has substantially advanced in recent years, particularly at the molecular and cellular level. Much has been learned about the molecular composition, assembly, and function of synapses, the cellular junctions through which neurons communicate (4−6). However, translating these advances in fundamental understanding into studies that can address and potentially correct nervous system damage remains a great challenge. While various experimental approaches to restore the function of damaged nerve cells and their outgrowing axons have been met with some success (for review, see ref (7 and 8), it remains necessary to engineer complementary tools and methods that facilitate the reformation of functional synapses (8−10).
We have recently developed three-dimensional artificial substrates (coated latex and silica beads) that induce certain specific cellular responses (1,2). For example, formation of functional synapses in vitro was achieved by coculturing neurons with beads coated with cationic synthetic molecules, including poly(d-lysine) (PDL) and specific lipid bilayers (1,2). These findings support the possibility that artificial substrates, when combined with other “smart” engineering devices (7,8), could become a successful strategy to ameliorate damaged CNS neurons and their associated synapses caused by disease. However, much remains to be learned about the mechanism of induction and molecular nature of these “artificial synapses”. This is in part due to the experimental challenges of culturing hippocampal neurons and the difficulty in isolating single synapses from a mixed culture comprising several thousand synapses in close spatial proximity to one another.
In this context, we have adapted a protocol established by Vogel and co-workers, involving HEK-293 cells (3). In our work, we have made cell-free presynaptic−bead complexes from in vitro neuronal cultures. These three-dimensional complexes offer considerable freedom to work with a range of experimental techniques under ambient rather than 37 °C/5% CO2 conditions. These complexes are very robust and structurally stable (as observed by confocal microscopy) for weeks when stored at 4 °C without fixation.
In a typical procedure, hippocampal neurons were dissected from embryonic (E17/18) rats and grown in culture for 7 or more days in vitro (DIV) prior to the addition of PDL-coated 7 μm-diameter polystyrene−sulfonate (PSS) beads. These beads were introduced via dropwise addition to the culture medium. Within hours, the coated beads were capable of inducing presynaptic assembly at bead−axon contacts (1,2). The neurons used were cocultured with PDL-coated or lipid-coated beads for 24 h prior to preparation of the isolated complexes.
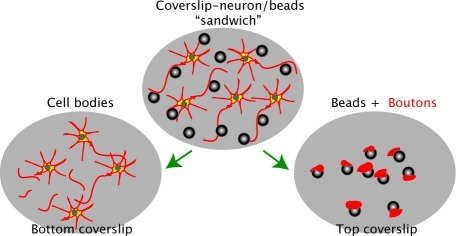
Scheme 1 depicts the experimental steps involved in the preparation of the bead−presynaptic complexes using a “sandwich/lift-off” method (3). After 24 h of coculture, the coverslip containing the neuron−bead culture was removed from the incubator and a second coverslip, coated with PDL, was laid on top. Pressure was briefly applied to the second coverslip (60 s or less). Lateral movement was carefully avoided during the procedure. This process was followed by separation of the top and bottom coverslips.
Scheme 1. Scheme Illustrating the Method of Preparation of Isolated Bead-Presynaptic Complexes (Not to Scale).

(i) Hippocampal neurons are first cultured on a coverslip for 7-15 DIV and then incubated with PDL-coated beads for 24 h. (ii) Following incubation, a second coverslip (pre-coated with PDL) is placed on top and brief pressure is applied. (iii) Careful removal of the top coverslip reveals beads to which isolated presynaptic compartments are attached.
To monitor the isolation of the bead−presynaptic complexes, the neuron−bead cultures were incubated with a fluorescent carbocyanine dye (DiI) 1 h prior to preparation of the bead−presynaptic complexes. Confocal microscopy revealed several distinct DiI-labeled puncta along the surface of the isolated bead. The fluorescent aggregates observed around the beads (arrowheads, Figure 1B) suggested that the membrane fragments were indeed being isolated during this process, and their cluster-like appearance is suggestive of presynaptic endings (Figure 1B,C).
Figure 1.

Isolated bead−axonal membrane complexes detach and readily adhere to the applied PDL-coated coverslip. (A−C) Representative image panel of a bead complex isolated from DIV14 neurons incubated with PDL beads for 24 h. The neuronal cultures were labeled with the membrane dye DiI for 1 h prior to patch preparation. Note the puncta-like shape of the DiI clusters along the bead surface (B, white arrowheads), suggestive of presynaptic-like endings. Scale bar, 5 μm.
To confirm that these isolated puncta contained proteins characteristic of native presynaptic boutons, the isolated bead−presynaptic complexes (prepared following 24 h of neuron−bead culture) were fixed and immunostained for a variety of presynaptic markers. Figure 2 is a representative image panel of two such complexes. Figure 2A establishes the location of the PDL-coated beads, while Figure 2B,C are the corresponding fluorescence images. Figure 2B reveals that both beads were positively labeled for the synaptic vesicle protein synaptophysin (red), the presynaptic scaffolding molecule bassoon (blue), and the cytoskeletal protein F-actin (green), all of which are typical constituents of native presynaptic endings (4,5). Figure 2C is a highlighted view of one of the two beads in a zoomed-in image panel and reveals a significant accumulation of all three proteins, some having highly overlapping distributions. These isolated presynaptic complexes closely resemble those found at native presynaptic endings in typical neuronal cultures (Figure 3), both at axon−bead contact sites (Figure 3B,C) and between native axon−dendrite contact sites (Figure 3B). By visual inspection, it appears that the fluorescence intensity is lower for the isolated bead−presynaptic complexes compared with native complexes. This may be due to the lift-off procedure, which may not remove all of the presynaptic complexes attached to the bead in situ. These results nonetheless show that the “sandwich/lift-off” method indeed allows one to isolate presynaptic boutons that contain synaptic vesicles and other presynaptic proteins as a “single package”. Furthermore, given our previous observations that PDL-/lipid bilayer-coated beads do not form presynaptic specializations when attached to cell bodies (data not shown) and that dendrites do not form synaptic specializations when directly associated with PDL-/lipid bilayer-coated beads (1, 2), we conclude that our isolated boutons contain minimal neuronal cell body and/or any postsynaptic factors physically associated.
Figure 2.

Isolated bead−presynaptic complexes resemble native complexes observed along axons. (A−C) Image panel of two isolated PDL bead−presynaptic complexes (A) fixed and stained for synaptophysin (red), bassoon (blue) and F-actin (green) (B). Similar to the native complexes in Figure 3, these isolated complexes contain the presynaptic proteins clustered at hippocampal axon−bead sites. Box in the merged fluorescence image (B) represents the bead−presynaptic complex shown in panel C as a zoomed image panel. Arrowheads in panel C highlight puncta that are colabeled for all three presynaptic proteins. Scale bar, 5 μm.
Figure 3.

Presynaptic bouton formation triggered along hippocampal axons by PDL-coated beads in culture. (A−C) Image panel of a typical DIV13 hippocampal neuron incubated with PDL beads for 24 h (A) and stained for the presynaptic proteins synaptophysin (red), bassoon (blue), and F-actin (green) (B). The white box in panel B) represents the zoomed image panels shown in panel C). Note the dense and large presynaptic clusters observed along the bead-axon contact site. Scale bar, 20 μm.
These presynaptic complexes were subsequently assessed for their ability to associate with other active zone proteins when isolated from the whole cell environment. In addition to synaptophysin (red) and F-actin (green) (Figure 4), we observed that accumulation of Rab3a-interacting molecule occurs at these sites (RIM, blue) (Figure 4B,C). RIM is present in the presynaptic active zones and is responsible for regulating multiple steps of synaptic vesicle cycling, including the docking of presynaptic vesicles and priming of synaptic vesicles for activity-dependent neurotransmitter release (4,11). Our ability to detect RIM further supports the assertion that we are able to isolate critical components of the presynaptic complex, including the active zone containing “ready-to-fire” synaptic vesicles.
Figure 4.

Isolated bead−presynaptic complexes contain active zones. (A−C) Image panel of a PDL bead−presynaptic complex (A) fixed and stained for synaptophysin (red), RIM (blue), and actin (green) (B). Bead-presynaptic complex shown in panel C is a zoomed image panel of B. Scale bar, 5 μm
If the isolated bead−presynaptic complexes retain active zones with docked synaptic vesicles (SVs), then one should be able to effect the functional fusion and recycling of these SVs in the complexes attached to the bead. To test this, we employed a stimulation assay to induce the release of neurotransmitter by fusion of SVs with the plasma membrane of isolated bead−presynaptic complexes. Ca2+-mediated SV fusion and its associated exocytosis/endocytosis cycles during neurotransmission have been well documented in the literature (12). To monitor SV fusion, the bead−presynaptic complexes were stimulated using CaCl2 in the presence of the fluorescent vital dye FM4-64, which binds to the outer leaflet of neuronal membranes and thus becomes integrated into the SV membranes following SV endocytosis (12). In this assay, the complexes were exposed to a stimulation buffer containing 20 mM Ca2+and FM4-64 for 90 s. Time lapse imaging of the bead−presynaptic complexes (Figure 5) shows that high Ca2+ stimulation indeed causes a marked accumulation of the FM4-64 dye (Figure 5B). To remove nonspecifically bound dye, we then washed the coverslip 3−4 times in buffer without FM dye or supplemented Ca2+. Although we did observe a decrease in fluorescence in images acquired during the first wash compared with those acquired during stimulation (compare Figure 5, panels B and C), we observed no further decrease in fluorescence during the washout period, suggesting that little photobleaching occurred during these experiments (see Supporting Information). Figure 5D shows an image of the same bead cluster acquired during a second stimulation in the presence of 20 mM Ca2+ but in the absence of FM4-64, so as to induce release of the dye from SV membranes during neurotransmitter release. As expected, the FM4-64 fluorescence intensity was markedly decreased compared with both panels B and C in Figure 5, consistent with Ca2+-stimulated SV fusion (Figure 5D). Furthermore, little difference in fluorescence in frames acquired during or following the second unloading stimulation was observed (Figure 5D,E, respectively), suggesting that the FM dye was rapidly released (see movies showing the uptake with associated washing steps and the release with the associated washing steps). Finally, quantification of these images reveals a statistically significant decrease in fluorescence at isolated bead−presynaptic complexes (Stim 1, 1.0 ± 0.0001 vs Stim 2, 0.1480 ± 0.1304, p < 0.0001) (Figure 5F). Taken together, these results clearly indicate that the presynaptic complexes attached to the bead are functional in the absence of any other cellular compartments. It must be emphasized that these experiments were performed under ambient conditions and do not require special temperature adjustments, thus offering added ease in carrying out similar functional imaging experiments.
Figure 5.

Isolated bead−presynaptic complexes are capable of recycling synaptic vesicles. The membrane dye FM 4-64 was used to visualize synaptic vesicle recycling. (A−E) Representative image panel of a cluster of three bead−presynaptic complexes (A) that have been stimulated using HBSS containing FM4-64 dye and 10 mM Ca2+ (Stim 1; B,C). This stimulation triggers SV fusion with the plasma membrane and internalization of the dye via incorporation into SV membranes. The second stimulation involves the use of HBSS with Ca2+ alone (Stim 2; D,E). In panels B and D, the images were acquired during stimulation, while in panels C and E, the images were captured during the wash (rest) phase of the protocol. (F) Quantification of the fluorescence changes in response to uptake (Stim 1) and unload (Stim 2) of the dye. The ∗∗∗ indicate p < 0.0001, n = 14 beads pooled from at least four independent coverslips. Scale bar, 10 μm.
Collectively, these results show that it is possible to readily isolate functional presynaptic endings using three-dimensional artificial substrates. This method is simple, versatile, and can be used to better understand the constitution and function of presynaptic boutons. The mechanism by which the formation of these presynaptic endings are triggered remains the subject of ongoing study. The evidence reveals that they strongly resemble those found at native axodendritic synapses (1,2). In the present study, we have further manipulated these presynaptic complexes to create a cell-free system of isolated presynaptic endings that could be used in a variety of studies in which a whole cell configuration would be difficult to pursue. Studies addressing the molecular composition of presynaptic endings, the kinetics of SV cycling, and the molecular triggers of presynapse formation will be facilitated by this three-dimensional cell-free system. Another approach could be a complete mass spectrometry-based analysis of the synaptic components (proteins, lipids etc.) being recruited to bead−presynaptic complexes. Furthermore, these types of studies are not limited to normal neuronal cultures as used here but may also be undertaken in cultures exposed to various experimental paradigms including knockdown or overexpression of proteins of interest, exposure to growth or inhibitory soluble factors, or following induced neuronal damage. It is also interesting to mention that in contrast to biochemical purification of synaptic complexes, this method better retains their native conformation and functionality.
In sum, we have exploited a versatile method to isolate presynaptic-like structures that are fully functional in a “cell-free” format. Previous studies have reported that biologically relevant molecules can be readily bound on spherical supports such as polystyrene and silica beads, either by direct adsorption onto the bead surface or incorporation into lipid bilayers (13), and these artificial substrates can induce the localized formation of synaptic-like endings (14−18). Combining such triggered responses with the isolation method described here provides an enhanced entry into the study of synaptogenesis mechanisms and may be potentially useful in studies aimed at developing regenerative therapeutic approaches.
Methods
Primary Neuronal Culture
All animal experimentation was approved by the MNI institutional animal care committee and conformed to the guidelines of the Canadian Council of Animal Care. Hippocampal neurons were dissected from embryonic day 17−18 Sprague−Dawley rat embryos (Charles River, Quebec) as described previously (2). Dissociated hippocampal neurons were resuspended in serum-free neurobasal medium supplemented with l-glutamine and B27 (all Invitrogen) and plated onto 18 mm (Fisher Scientific) or 42 mm (Hemogenix) glass coverslips coated with poly(d-lysine) (50 μg/mL, Sigma) at a density of 50 000−80 000 or 400 000−500 000 neurons per coverslip, respectively. Cells were then maintained in a humidified atmosphere (37 °C, 5% CO2) for a minimum of 7 days in vitro (DIV) and up to 15 DIV prior to experimentation. During this time, one-third of the medium was changed every 2−3 days and replaced with fresh medium supplemented with l-glutamine and B27.
Preparation of Poly(d-lysine) (PDL)-Coated Beads
Seven micrometer polystyrene latex beads (Bangs Laboratories) were coated with PDL by incubating the beads in a solution of 50 μg/mL PDL in sterile phosphate-buffered saline (PBS, pH 7.4; usually about 10 million beads per 10 mL of PDL solution) overnight at 4 °C with end-to-end mixing. The beads were then washed 3× in PBS by centrifugation, resuspended in PBS, and added dropwise to the cultures at a concentration of 100 000 or 500 000 beads per 18 mm or 42 mm coverslip, respectively. The neuron−bead cultures were subsequently incubated for 24 h.
Isolation of Presynaptic−Bead Complexes
For preparation of the presynaptic−bead complexes, the neuron−bead coverslip was removed from the humidified incubator and washed twice in warmed Hank’s buffered salt solution (HBSS, Invitrogen). Following this, another PDL-coated coverslip was laid on top of the culture (matched to the diameter of the coverslip beneath). Pressure was applied by placing the wide end of a pipet tip onto the second coverslip and maintaining the pressure for approximately 60 s. The second coverslip was then carefully removed by inserting fine forceps [e.g., Dumont #5 (Fine Science Tools) or similar] between the two coverslips and gently pulling them apart. Great care was taken to avoid any lateral motion during the application of pressure as well as during separation of the two coverslips. The second coverslip was then either placed into another Petri dish containing HBSS for time-lapse imaging or fixed in 4% PFA for immunocytochemistry.
Synaptic Vesicle Recycling Experiments Using FM 4-64 Dye
The presynaptic−bead complexes were obtained using the protocol described above. The coverslips containing presynaptic−bead complexes were mounted on a sample chamber for glass coverslips (Bioscience tools, San Diego, CA) in regular warmed HBSS (without Ca2+ or Mg2+). The first stimulation, to load the FM 4-64 dye into the SV membrane, was performed using HBSS containing 20 mM CaCl2 and 15 μM FM 4-64 for 90 s. The sample was then washed (3×) with HBSS (without Ca2+ or Mg2+). The second stimulation, to unload the FM 4-64 dye, was performed with HBSS containing 20 mM CaCl2 for another 90 s. The coverslip was then washed again (3×) with regular HBSS.
Experiments were recorded by time-lapse confocal microscopy, with the time-lapse series initiated just prior to the stimulation steps. Individual image frames were acquired using a Zeiss LSM510 META laser scanning confocal microscope with a 63× PlanNeofluar oil-immersion objective (1.4 NA) on a Zeiss Axiovert 100 M inverted microscope. Laser intensity was set to approximately 25% of the maximum to minimize photobleaching and phototoxicity. We captured images every 4 s during both the stimulation and washout periods, with great care being taken not to disrupt the bead−presynaptic complexes held in place by the coverslip. Both fluorescence and brightfield (DIC) images were taken for each frame.
Quantification and Statistics of the FM 4-64 SV Recycling Experiments
Quantification of the individual fluorescence images was performed using NIH ImageJ software. For each coverslip, the brightfield (DIC) image was used to locate and highlight the bead−presynaptic complexes, and the corresponding fluorescence intensity at a bead site was measured for each frame in the time-lapse series. For each bead site, we then grouped the corresponding fluorescence values from the first stimulation (including those obtained during the wash periods), calculated the average fluorescence intensity, and normalized all values in the time-lapse series (Stim 1 + Stim 2) to this average. In this way, we were able to measure changes in fluorescence according to the maximum fluorescence intensity observed for each bead (normalized value = 1). For these experiments, we quantified a total of 14 beads obtained from at least four separate coverslips. These results were then analyzed and graphed using GraphPad Prism software, whereby the normalized values for each stimulation were averaged for each bead, and statistically compared using the paired t test (Stim 1 vs Stim 2; ∗∗∗, p < 0.0001).
Confocal Imaging of DiI-Labeled Bead−Presynaptic Complexes
One hour prior to preparation of the isolated bead−presynaptic complexes, cells were incubated with the fluorescent membrane dye DiI (Invitrogen; 1:500 from stock prepared according to the manufacturer’s directions) in their neuronal medium. The bead−presynaptic complexes were prepared as described above and mounted for imaging as described for the FM dye studies. DiI-labeled coverslips were maintained in a solution of warmed HBSS during the imaging session. Images were acquired with 1× zoom and 2−4× Kalman averaging along with the corresponding DIC image.
Immunocytochemistry
Coverslips were fixed and stained with antibodies against synaptic proteins as previously described (1). We used three primary antibodies for our studies: rabbit polyclonal anti-synaptophysin (Invitrogen), mouse monoclonal anti-bassoon (Assay Designs), and mouse monoclonal anti-Rab3a-interacting molecule (RIM) (BD Biosciences). All secondary fluorochrome-coupled antibodies (species-specific, highly cross-adsorbed IgG) were purchased from either Jackson ImmunoResearch or Invitrogen and used at a dilution of 1:200 to 1:500. Alexa-488-conjugated phalloidin (Invitrogen), to stain F-actin filaments, was added to the secondary antibody solution at a dilution of 1:50 (stock prepared according to the manufacturer’s directions). For every experiment, we performed staining controls by incubating a subset of cultures with dilution buffer in the absence of primary antibody.
Confocal Microscopy of Antibody-Labeled Cultures
Fixed bead−presynaptic complexes were imaged using an Olympus Fluoview FV1000 laser scanning confocal microscope with a 60× PlanApo oil-immersion objective [1.4 numerical aperture (NA)] on an IX81 inverted microscope. For each coverslip, scanning parameters were adjusted manually to avoid image saturation. Images were acquired via sequential scanning of each individual fluorescence channel to avoid bleed-through, along with the corresponding brightfield (differential interference contrast, or DIC) image. Images were acquired as stacks of 0.3−0.5 μm thickness with 2× Kalman averaging and either 2× or 3× digital zoom. At least four or five individual stacks (10−15 frames per stack) were taken for each combination of antibodies. Bead−presynaptic complexes from at least three separate cultures were imaged.
All images were processed for print using Photoshop (Adobe) software.
Supporting Information Available
Detailed experimental details, as well as additional data including extended figures. This material is available free of charge via the Internet at http://pubs.acs.org.
A.L.L. and G.G. contributed equally to this work. G.G. conceived the idea. A.L.L. and G.G. designed and performed the experiments. P.T.Y. supported the experiments. A.L.L., G.G., D.R.C., and R.B.L. analyzed the experimental data. G.G. and A.L.L. wrote the manuscript, and all the authors contributed to the editing of the manuscript.
This work is supported by a grant from the Regenerative Medicine and Nanomedicine Initiative of the Canadian Institutes of Health Research (CIHR) to D.R.C. and R.B.L. The NeuroEngineering Program of McGill University is also supported by grants from Rio Tinto Alcan and the Molson Foundation.
Supplementary Material
References
- Zhai R. G.; Bellen H. J. (2004) The architecture of the active zone in the presynaptic nerve terminal. Physiology 19, 262–270. [DOI] [PubMed] [Google Scholar]
- Waites C. L.; Craig A. M.; Garner C. C. (2005) Mechanisms of vertebrate synaptogenesis. Annu. Rev. Neurosci. 28, 251–274. [DOI] [PubMed] [Google Scholar]
- Dittman J.; Ryan T. A. (2009) Molecular circuitry of endocytosis at nerve terminals. Annu. Rev. Cell Dev. Biol. 25, 133–160. [DOI] [PubMed] [Google Scholar]
- Schmidt C. E.; Leach J. B. (2003) Neural tissue engineering: Strategies for repair and regeneration. Annu. Rev. Biomed. Eng. 5, 293–347. [DOI] [PubMed] [Google Scholar]
- Orive G.; Anitua E.; Pedraz J. L.; Emerich D. F. (2009) Biomaterials for promoting brain protection, repair and regeneration. Nat. Rev. Neurosci. 10, 682–692. [DOI] [PubMed] [Google Scholar]
- Filbin M. T. (2003) Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat. Rev. Neurosci. 4, 703–713. [DOI] [PubMed] [Google Scholar]
- Giovanni S. D. (2009) Molecular targets for axon regeneration: Focus on the intrinsic pathways. Expert Opin. Ther. Targets 13, 1387–1398. [DOI] [PubMed] [Google Scholar]
- Lucido A. L.; Suarez Sanchez F.; Thostrup P.; Kwiatkowski A. V.; Leal-Ortiz S.; Gopalakrishnan G.; Liazoghli D.; Belkaid W.; Lennox R. B.; Grutter P.; Garner C. C.; Colman D. R. (2009) Rapid assembly of functional presynaptic boutons triggered by adhesive contacts. J. Neurosci. 29, 12449–12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan G.; Thostrup P.; Rouiller I.; Lucido A. L.; Belkaid W.; Colman D. R.; Lennox R. B. (2010) Lipid bilayer membrane-triggered presynaptic vesicle assembly. ACS Chem. Neurosci. 1, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez J.-B.; Martinez K. L.; Segura J.-M.; Vogel H. (2006) Supported cell-membrane sheets for functional fluorescence imaging of membrane proteins. Adv. Funct. Mater. 16, 306–312. [Google Scholar]
- Kaeser P. S.; Sudhof T. C. (2005) RIM function in short- and long-term synaptic plasticity. Biochem. Soc. Trans. 33, 1345–1349. [DOI] [PubMed] [Google Scholar]
- Ryan T. (2001) Presynaptic imaging techniques. Curr. Opin. Neurobiol. 11, 544–549. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan G.; Rouiller I.; Colman D. R.; Lennox R. B. (2009) Supported bilayers formed from different phospholipids on spherical silica substrates. Langmuir 25, 5455–5458. [DOI] [PubMed] [Google Scholar]
- Kim S.; Burette A.; Chung H. S.; Kwon S.-K.; Woo J.; Lee H. W.; Kim K.; Kim H.; Weinberg R. J.; Kim E. (2006) NGL family PSD-95-interacting adhesion molecules regulate excitatory synapse formation. Nat. Neurosci. 9, 1294–1301. [DOI] [PubMed] [Google Scholar]
- Graf E. R.; Zhang X. Z.; Jin S.-X.; Linhoff M. W.; Craig A. M. (2004) Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119, 1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baksh M. M.; Dean C.; Pautot S.; DeMaria S.; Isacoff E.; Groves J. T. (2005) Neuronal activation by GPI-linked neuroligin-1 displayed in synthetic lipid bilayer membranes. Langmuir 21, 10693–10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledda F.; Paratcha G.; Sandoval-Guzman T.; Ibanez C. (2007) GDNF and GFRα1 promote formation of neuronal synapses by ligand-induced cell adhesion. Nat. Neurosci. 10, 293–300. [DOI] [PubMed] [Google Scholar]
- Ko J.; Kim S.; Chung H.; Kim K.; Han K.; Kim H.; Jun H.; Kaang B.; Kim E. (2006) SALM Synaptic cell adhesion-like molecules regulate the differentiation of excitatory synapses. Neuron 50, 233–245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.