

Abstract
To evaluate in vivo brain penetration of α2C-adrenoceptor (α2C-AR) antagonists as a therapeutic agent, we synthesized two new 11C-labeled selective α2C-AR antagonists 4-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)methyl-2-aryl-7-methoxybenzofuran ([11C]MBF) and acridin-9-yl-[4-(4-methylpiperazin-1-yl)phenyl]amine ([11C]JP-1302) as α2C-AR-selective positron emission tomography (PET) probes. The radiochemical yield, specific activity, and radiochemical purity of these probes was appropriate for injection. To evaluate whether the brain penetration of these probes is related to the function of two major drug efflux transporters, P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), we performed PET studies using wild-type and P-gp/Bcrp knockout mice. In wild-type mice, the radioactivity level after injection with [11C]MBF initially increased and effluxed immediately from the brain, whereas that with [11C]JP-1302 was distributed throughout the brain. However, the regional distribution of radioactivity after injection with [11C]JP-1302 in the brain was different from that of α2C-ARs. In P-gp/Bcrp knockout mice, uptake of [11C]MBF was approximately 3.7-fold higher and that of [11C]JP-1302 was approximately 1.6-fold higher than those in wild-type mice. These results indicate that brain penetration of the two PET probes was affected by modulation of P-gp and Bcrp functions.
Keywords: α2C-Adrenoceptor; 11C; 4-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)methyl-2-aryl-7-methoxybenzofuran (MBF); acridin-9-yl-[4-(4-methylpiperazin-1-yl)phenyl]amine (JP-1302); P-glycoprotein (P-gp); breast cancer resistance protein (Bcrp)
Drug efflux transporters are expressed at various protective organ and tissue surfaces and play a pivotal role in the absorption (small intestine), distribution (placenta and blood−brain barrier), and elimination (liver and small intestine) of xenobiotics. The most extensively characterized drug efflux transporters are the ATP-binding cassette (ABC) transporters, which include P-glycoprotein (P-gp), multidrug resistance-associated protein (MRP), and breast cancer resistance protein (BCRP) (1,2). Some positron emission tomography (PET) probes for imaging the central nervous system (CNS) functions have revealed behavior as P-gp substrates (3−9). These PET probes showed a relatively rapid clearance of radioactivity from the brain; however, their uptake was noticeably enhanced by treatment with a P-gp inhibitor or modulator (8). To discern whether P-gp substrates are modulated by drug efflux transporters is important for development of new PET probes.
α2C-Adrenoceptors (α2C-ARs) are a subtype of α2-ARs (10) and are feedback regulators of catecholamine release from adrenal chromaffin cells (11,12). In the CNS, activation of α2C-ARs influences several complex memory and behavioral functions (13,14). Involvement of α2C-ARs has been reported in hypothermic (15), locomotor (15,16), cognitive (17), and analgesic effects (18,19). In the mouse brain, α2C-ARs are present in the accumbens nuclei, caudate putamen, olfactory tubercles, lateral septum, hippocampus, amygdala, and frontal cortex (20,21). To achieve a better understanding of the effects of α2C-ARs in CNS, selective probes with a high specificity that could detect low concentration sites would be valuable.
Although several PET probes for α2-ARs have been synthesized (22−26), a selective PET probe for α2C-ARs has not been synthesized until now. Recently, 4-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)methylbenzofuran derivatives were synthesized as selective α2C-ARs antagonists (27). Of these, 4-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)methyl-2-aryl-7-methoxybenzofuran (MBF) exhibited high and selective affinity for α2C-ARs (Ki for α2C, 20 nM; Ki for α2A, 1700 nM; Ki for α2B, 750 nM) (27). Furthermore, acridin-9-yl-[4-(4-methylpiperazin-1-yl)phenyl]amine (JP-1302) was recently synthesized and characterized as a selective α2C-AR antagonist (Ki for α2C, 28 nM; Ki for α2A, 3500 nM; Ki for α2B, 1500 nM; Ki for αD, 1700 nM) (28). Previous work with α2-AR ligands for CNS imaging agents showed that low nanomolar affinity is required (29), and general criteria for PET probes are proposed for lipophilicity (log D < 3.5) to predict brain penetration of PET probes (30). Calculated log D values of MBF and JP-1302 were 5.72 and 5.13, respectively (Pallas 3.4, CompuDrug, Sedona, AZ). MBF and JP-1302 may be too lipophilic for imaging agents of central α2C-adrenoceptors due to few dozens of nanomolar affinity and relatively high log D value. MBF and JP-1302 were indicated as possible therapeutic agents for psychiatric and neurological disorders (27,31). Here, we synthesized 11C-labeled MBF and JP-1302 as a PET probes to evaluate in vivo brain penetration of therapeutic agents and evaluated the P-gp and BCRP functions that mediated the brain penetration of these probes using small animal PET.
Results and Discussion
Radiosynthesis of [11C]MBF and [11C]JP-1302
We successfully synthesized [11C]MBF by methylation of the O-desmethyl precursor with [11C]methyl iodide (Figure 1). The decay-corrected radiochemical yield of [11C]MBF from [11C]carbon dioxide was 27% ± 4.7% (n = 3), and the specific activity was 62 ± 15 TBq/mmol (n = 3) at 30 min after the end of radionuclide production. The radiochemical yield and specific activity were applicable for injection as a PET probe. We achieved appropriate radiochemical purity (>97%) and stability of [11C]MBF injection over 1 h after the end of synthesis (EOS).
Figure 1.

Synthesis of precursor 6 (A) and [11C]MBF (B). Reagents and conditions: (i) boron tribromide, CH2Cl2, 0 °C, 2 h; (ii) chloromethyl methyl ether, N,N′-diisopropylethylamine, CH2Cl2, room temperature (rt), 4 h; (iii) normal butyl lithium, THF then DMF, −78 °C− to rt, 1 h; (iv) 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride, sodium tri(acetoxy)borohydride, CH2Cl2, rt, 16.5 h; (v) p-toluenesulfonic acid, methanol, rt, 7 h, 55.3% yield from 1, 98.7% chemical purity; (vi) [11C]CH3I, tetrabutylammonium hydroxide, DMF, 80 °C, 5 min, 27% radiochemical yield (RCY) from [11C]CO2.
We also successfully synthesized [11C]JP-1302 by methylation of the N-desmethyl precursor with [11C]methyl triflate (Figure 2). The decay-corrected radiochemical yield of [11C]JP-1302 from [11C]carbon dioxide was 26% ± 5.8% (n = 7). As a result of the relatively low dose of precursor (0.2 mg), the radiochemical yield of [11C]JP-1302 was the similar to that of [11C]MBF, as achieved by methylation with [11C]methyl iodide, although the methylation with [11C]methyl triflate possibly improves the radiochemical yield (32). The specific activity was 95 ± 24 TBq/mmol (n = 7) at 30 min after the end of radionuclide production. The radiochemical yield and specific activity were suitable for injection as a PET probe. We achieved appropriate radiochemical purity (>96%) and stability of [11C]JP-1302 injection over 1 h after EOS.
Figure 2.

Synthesis of precursor 11 (A) and [11C]JP-1302 (B). Reagents and conditions: (i) K2CO3, methyl chloroformate, CH2Cl2, rt, 0.5 h; (ii) 10% Pd/C, H2, ethanol, rt, 2 h; (iii) 9-chloroacridine, ethylene glycol, 150 °C, 1 h; (iv) KOH, water, ethylene glycol, rt, 150 °C, 4 h, 18.8% yield from 7, 95.2% chemical purity; (v) [11C]methyl triflate, acetone, rt, 26% RCY from [11C]CO2.
In Vivo Biodistribution in Mice
To evaluate in vivo brain penetration of [11C]MBF and [11C]JP-1302, we investigated the time courses of radioactivity in the brain and blood after the injection of [11C]MBF and [11C]JP-1302 in normal mice (Figure 3). The radioactivity level of [11C]MBF in the brain was lower than that in the blood (Figure 3A), whereas the radioactivity level of [11C]JP-1302 in the brain was higher than that in the blood (Figure 3B). Next, we investigated specific uptake of radioactivity by α2C-ARs in the brain by conducting a blocking study using cold MBF, cold JP-1302, and the high-affinity α2C-AR antagonist MK-912 ((2S,12bS)1′,3′-dimethylspiro(1,3,4,5′,6,6′,7,12b-octahydro-2H-benzo[b]furo[2,3-a]quinolizine)-2,4′-pyrimidin-2′-one, Ki for α2C = 0.03 nM) (22) (Figure 4). Treatment with any investigated α2C-AR ligands did not affect the brain-to-blood ratio of [11C]JP-1302 (Figure 4B). Therefore, the uptake of [11C]JP-1302 in the brain may be nonspecific. On the other hand, the treatment with cold MBF induced a significant increase in the brain-to-blood ratio of [11C]MBF, although the brain-to-blood ratio of [11C]MBF was not affected by the treatment with cold JP-1302 and MK-912 (Figure 4A). Furthermore, treatment with the P-gp/BCRP inhibitor GF120918 induced a significant increase in the brain-to-blood ratio of [11C]MBF, and treatment with GF120918 plus cold MBF induced a significant increase in the brain-to-blood ratio compared with treatment with GF120918 alone (Figure 5A). These results suggest that the penetration of [11C]MBF and cold MBF into the brain is related to the function of drug efflux transporters as a substrate. Unexpectedly, the treatment with GF120918 induced a significant increase in the brain-to-blood ratio of [11C]JP-1302 (Figure 5B). These results suggested that the penetration of [11C]JP-1302 into the brain is also related to the function of drug efflux transporters as a substrate.
Figure 3.

In vivo distribution of radioactivity in the brain and blood after injection of [11C]MBF (A) and [11C]JP-1302 (B) into mice (n = 3 per group). Values are means ± standard deviation (SD). Injected dose of 11C ligands was 7.4−12 MBq/0.07−0.12 nmol. SUV, standardized uptake value.
Figure 4.

Effects of treatment with α2C-adrenoceptor (α2C-AR) ligands on the brain-to-blood ratio at 15 min after the injection of [11C]MBF or at 30 min after the injection of [11C]JP-1302 into mice (n = 3−7 per group). Values are means ± SD. Injected dose of 11C-labeled ligands was 5.9−11 MBq/0.13−0.25 nmol. One of the α2C-AR ligands (MBF, JP-1302, MK912; 1.0 mg/kg) was intravenously co-injected with [11C]MBF or injected 15 min before administration of [11C]JP-1302. The ∗ indicates P < 0.05 compared with control (one-way ANOVA and Dunnett’s post hoc tests).
Figure 5.

Effects of the inhibition of P-gp and Bcrp functions on the brain-to-blood ratio at 30 min after the injection of [11C]MBF (A) and [11C]JP-1302 (B). The 11C-labeled ligands (11−12 MBq/0.12−0.17 nmol), P-gp/BCRP inhibitor GF120918 (5.0 mg/kg), or cold ligand (MBF or JP-1302; 1.0 mg/kg) were intravenously co-injected into mice (n = 4 per group). Values are means ± SD; ∗ indicates P < 0.05 (one-way ANOVA and Bonferroni’s multiple comparison tests).
Metabolites Analysis in the Brain and Plasma of Mice
The percentages of the unchanged of [11C]MBF in the brain and plasma at 30 min after the injection were 88.4% ± 6.3% and 84.0% ± 6.8%, respectively (n = 3). [11C]MBF demonstrated a high metabolic stability at 30 min after injection. A radiolabeled metabolite with high polarity was observed on HPLC charts (retention time, 2.5 min).
The percentages of unchanged [11C]JP-1302 in the brain and plasma at 30 min after the injection were 88.4% ± 1.8% and 17.4% ± 5.9%, respectively (n = 4). Therefore, [11C]JP-1302 seemed to readily metabolize in peripheral organs. A radiolabeled metabolite with high polarity was observed on HPLC charts (retention time, 2.5 min).
PET Studies in Wild-Type and P-gp/Bcrp Knockout Mice
To evaluate whether the brain penetration by [11C]MBF and [11C]JP-1302 is related to P-gp and Bcrp functions, we further performed PET studies using [11C]MBF and [11C]JP-1302 in P-gp/Bcrp knockout and wild-type mice (Figures 6 and 7). In wild-type mice after injection with [11C]MBF, the radioactivity level was low in the brain (Figure 6A). In P-gp/Bcrp knockout mice after injection with [11C]MBF, the radioactivity level was distributed throughout the brain (Figure 6C); however, the regional distribution of radioactivity in the brain was different from that of α2C-ARs (20,21). The radioactivity level in the wild-type and P-gp/Bcrp knockout mice after injection with [11C]JP-1302 was also distributed throughout the brain (Figure 6B,D), and it initially increased and then remained constant (Figure 7B). This result indicates that [11C]JP-1302 may bind nonspecifically and irreversibly in the brain and suggests that [11C]MBF and [11C]JP1302 are inadequate for evaluation of α2C-ARs in the brain. In P-gp/Bcrp knockout mice after injection with [11C]MBF, the radioactivity level in the brain increased gradually after initial uptake for 20 min after injection and then decreased (Figure 7A). The area under the time−activity curve of the region of interest in the brain from 0 to 60 min after the injection (AUCbrain[0−60min]) in P-gp/Bcrp knockout mice was approximately 3.7-fold higher than that in wild-type mice (74.4 ± 6.0 in P-gp/Bcrp knockout mice and 19.8 ± 0.8 in wild-type mice; n = 3 each group). These results suggested effective exclusion of [11C]MBF from the brain by P-gp and Bcrp function-mediated drug efflux. In P-gp/Bcrp knockout mice after injection with [11C]JP-1302, the kinetics of radioactivity in the brain followed a time course similar to that in wild-type mice after injection with [11C]JP-1302, but the radioactivity level was higher than that in wild-type mouse (Figure 7B). The AUCbrain[0−60min] in P-gp/Bcrp knockout mice was approximately 1.6-fold higher than that in wild-type mice (82.1 ± 4.9 in P-gp/Bcrp knockout mice; 51.0 ± 0.7 in wild-type mice; n = 3 each group). These results suggest that brain penetration of [11C]JP-1302 is mediated by P-gp and Bcrp functions.
Figure 6.
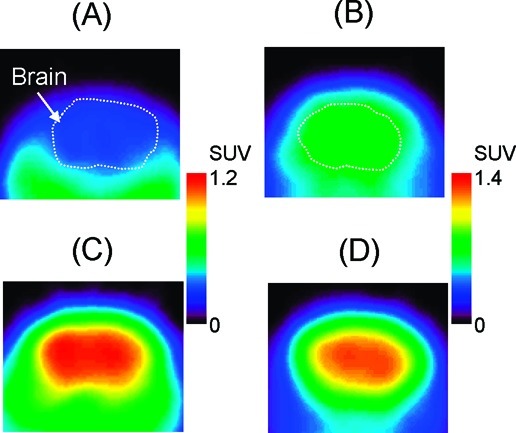
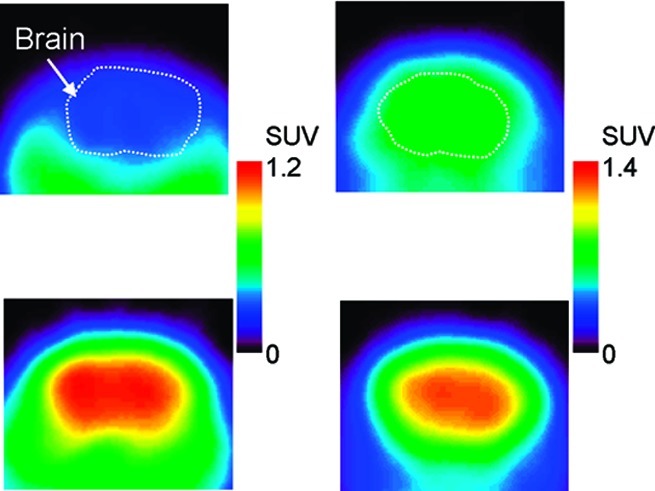
Typical transaxial positron emission tomography (PET) images showing [11C]MBF (A, C) and [11C]JP-1302 (B, D) in the brain of a wild-type mouse (A, B) and a P-gp/Bcrp knockout mouse (C, D). Injected dose was 3.7−4.7 MBq/0.071−0.56 nmol. PET images were acquired from 5 to 60 min after the injection. Mice were anesthetized with isoflurane and fixed in a prone position on the bed of the scanner.
Figure 7.

Time−activity curves of the brain after injection of [11C]MBF (A) and [11C]JP-1302 (B) in wild-type mice and P-gp/Bcrp knockout mice (n = 3 per group). Values are means ± SD. The injected dose was 3.6−8.1 MBq/0.033−1.22 nmol. SUV, standardized uptake value.
Consequently, [11C]MBF and [11C]JP-1302 are inadequate for evaluation of the function of α2C-AR in the brain as PET probes due to the effect of drug efflux transporters. As recent evidence from P-gp and Bcrp knockout mice suggests (33), PET study using P-gp/Bcrp knockout mice is more effective than that using P-gp knockout or Bcrp knockout mice alone to study brain penetration of probes related to P-gp and Bcrp functions (34−36). Therefore, PET study using P-gp/Bcrp knockout mice is more useful for evaluating the P-gp and Bcrp function-mediated brain penetration of PET probes.
Conclusions
We reliably synthesized [11C]MBF and [11C]JP-1302 as a PET probes to evaluate in vivo brain penetration of α2C-adrenoceptor antagonists as therapeutic agents. Brain penetration of these two PET probes was affected by modulation of drug efflux transporter functions. We also proposed that PET study using P-gp/Bcrp knockout mice is more useful for evaluating the P-gp and Bcrp function-mediated brain penetration of PET probes.
Methods
General
Reagents and organic solvents were commercially available and used without further purification. JP-1302 dihydrochloride was purchased from Tocris Bioscience (Bristol, U.K.). MBF was prepared in our laboratory as described previously (27) with some modifications. GF120918 was prepared in our laboratory as described previously (37,38) with some modifications.
Proton nuclear magnetic resonance (1H NMR) and carbon-13 nuclear magnetic resonance (13C NMR) spectra were recorded on the JNM-AL300 spectrometer (Jeol, Tokyo, Japan) or the JNM-GX-270 spectrometer (Jeol). High-resolution mass spectra (MS) were obtained on the JEOL NMS-SX 102-SX spectrometer (Jeol). Column chromatography was carried out using Kieselgel gel 60 F254 (70−230 mesh; Merck, Darmstadt, Germany) or Wakogel C-200 (100−200 mesh; Wako Pure Chemical Industries, Osaka, Japan).
Male FVB mice were purchased from CLEA Japan (Tokyo, Japan). Male P-gp/Bcrp knockout [Mdr1a/1b(−/−)-Abcg2(−/−)] mice (39) were purchased from Taconic Farm (Hudson, NY). The animals were maintained and handled in accordance with the recommendations by the US National Institutes of Health and in-house guidelines (National Institute of Radiological Sciences, Chiba, Japan). The animal experiments were approved by the Animal Ethics Committee of the National Institute of Radiological Sciences.
Synthesis of 4-(6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline-2-ylmethyl)-2-phenylbenzofuran-7-ol (6)
Boron tribromide in dichloromethane (1.0 M, 20 mL, 20 mmol) was added to a solution of 4-bromo-7-methoxy-2-phenyl-2,3-dihydrobenzofuran (1, 0.86 g, 2.8 mmol) (27) in dichloromethane (71 mL) at 0 °C and stirred for 2 h. The mixture was poured into a saturated solution of monopotassium phosphate and extracted with diethyl ether. The organic layer was washed with brine, dried over sodium sulfate, and concentrated. The residue was purified by column chromatography on silica gel using a mixture of n-hexane and ethyl acetate (5:1, v/v) as the mobile phase, yielding 4-bromo-2-phenylbenzofuran-7-ol (2; 0.73 g, 2.5 mmol) as a white-colored solid.
N,N′-Diisopropylethylamine (0.05 mL, 0.27 mmol) and chloromethyl methyl ether (0.02 mL, 0.27 mmol) were added to a solution of compound 2 (0.053 g, 0.18 mmol) in dichloromethane (3.6 mL). The solution was warmed to ambient temperature and stirred for 4 h. The reaction was quenched with a saturated solution of ammonium chloride and extracted with dichloromethane. The organic layer was washed with brine, dried over sodium sulfate, and concentrated. The residue was purified by column chromatography on silica gel using a mixture of n-hexane ethyl acetate (10:1, v/v) as the mobile phase, yielding 4-bromo-7-(methoxymethoxy)-2-phenylbenzofuran (3; 0.059 g, 0.18 mmol) as a yellow-colored solid.
Normal butyl lithium (1.6 M in hexane 1.3 mL, 2.07 mmol) and N,N-dimethylformamide (DMF) (0.21 mL, 2.76 mmol) were added to a solution of compound 3 (0.46 g, 1.38 mmol) in tetrahydrofuran (14 mL) at −78 °C. The solution was warmed to ambient temperature and stirred for 1 h. The reaction mixture was quenched with water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, and concentrated. The residue was purified by column chromatography on silica gel using a mixture of n-hexane and ethyl acetate (from 10:1 [v/v] to 5:1 [v/v], gradient elution) as the mobile phase, yielding 7-(methoxymethoxy)-2-phenylbenzofuran-4-carbaldehyde (4; 0.34 g, 1.22 mmol) as a white-colored solid.
6,7-Dimethoxy-1,2,3,4-tetrahydroisoquinoline hydrochloride (0.25 g, 1.06 mmol) and sodium tri(acetoxy)borohydride (0.40 g, 1.77 mmol) were added to a solution of compound 4 (0.25 g, 0.89 mmol) in 1,2-dichloroethane (7.4 mL) at 0 °C. The solution was warmed to ambient temperature and stirred for 16.5 h. The reaction mixture was quenched with 1 M sodium hydroxide and extracted with dichloromethane. The organic layer was washed with brine, dried over potassium carbonate, and concentrated. The residue was purified by column chromatography on silica gel using a mixture of n-hexane and ethyl acetate (1:1, v/v) as the mobile phase, yielding 4-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-2-ylmethyl)-7-methoxymethoxy-2-phenylbenzofuran (5; 0.324 g, 0.71 mmol) as a white-colored solid.
p-Toluenesulfonic acid (0.067 g, 0.35 mmol) was added to a solution of compound 5 (0.027 g, 0.059 mmol) in methanol (3.0 mL). After the mixture was stirred for 7 h, the solvent was evaporated. The residue was suspended in dichloromethane, made basic using 28% ammonia−water, and extracted with dichloromethane. The organic layer was washed with brine, dried over sodium sulfate, and concentrated. The residue was purified by column chromatography on silica gel using a mixture of n-hexane and ethyl acetate (1:2, v/v) as the mobile phase, yielding compound 6 (0.022 g, 0.054 mmol, 55.3% from 1) as a yellow-colored solid. 1H NMR (300 MHz,CDCl3): δ (ppm) 2.87 (4H, s), 3.68 (2H, s), 3.79 (3H, s), 3.83 (3H, s), 3.85 (2H, s), 6.48 (1H, s), 6.59 (1H, s), 6.64 (1H, d, J = 7.80 Hz), 7.02 (1H, d, J = 7.80 Hz), 7.14 (1H, s), 7.28 (1H, brt), 7.37 (2H, brt), 7.78 (2H, d, J = 7.20 Hz). 13C NMR (75.5 MHz, CDCl3): δ (ppm) 27.70, 50.62, 55.12, 55.82, 55.84, 59.04, 100.87, 109.45, 111.17, 111.24, 120.71, 124.89, 125.11, 125.58, 125.70, 128.30, 128.51, 130.23, 130.90, 141.27, 143.78, 147.24, 147.57, 155.51. High-resolution MS m/z: 416.1862 (calculated for C26H25NO4, 416.4967). Chemical purity: 98.7% (Supporting Information).
Synthesis of Acridin-9-yl-[4-(piperazin-1-yl)phenyl]amine (11)
Potassium carbonate (0.47 g, 3.4 mmol) and methyl chloroformate (0.2 mL, 2.6 mmol) were added a solution of 1-(4-nitrophenyl)piperazine (7; 0.35 g, 1.7 mmol) in dichloromethane (7 mL) at room temperature (rt). The resulting solution was stirred for 30 min, and 1 M sodium hydroxide (5 mL) was added at rt The reaction mixture was extracted with dichloromethane. The organic layer was dried with sodium sulfate and evaporated in vacuo. The residue was purified by column chromatography on silica gel using a mixture of dichloromethane and methanol (9:1, v/v) as the mobile phase, yielding 4-(4-nitrophenyl)piperazine-1-carboxylate (8; 0.45 g, 1.7 mmol) as a yellow-colored solid.
Compound 8 (0.15 g, 0.57 mmol) was dissolved in ethanol (5 mL) and tetrahydrofuran (3.5 mL). The solution was purged with hydrogen gas, and 10% palladium carbon (70 mg) was added. The resulting solution was stirred for 2 h under hydrogen gas and then filtered. The filtrate was condensed and purified by column chromatography on silica gel using a mixture of dichloromethane and methanol (9:1, v/v) as the mobile phase, yielding methyl 4-(4-aminophenyl)piperazine-1-carboxylate (9; 67 mg, 0.29 mmol) as a white-colored solid.
Compound 9 (0.80 g, 3.4 mmol) was dissolved in ethylene glycol (20 mL). The solution was heated to 150 °C and 9-chloroacridine (0.87 g, 4.1 mmol) was added. The resulting solution was stirred for 1 h. The solution was condensed and added to a mixture of ethyl acetate and water. The extraction was carried out with ethyl acetate. The organic layer was dried with sodium sulfate, and concentrated. The residue was purified by column chromatography on silica gel using a mixture of n-hexane and ethyl acetate (1:1, v/v) as the mobile phase, yielding 4-(4-(acridin-9-ylamino)phenyl)piperazine-1-carboxylate (10; 0.90 g, 2.2 mmol) as an orange-colored solid.
Compound 10 (0.10 g, 0.25 mmol) was dissolved in ethylene glycol (5 mL). The resulting solution was heated to 150 °C; potassium hydroxide solution (0.18 mg/15 mL, 3.2 mmol) was dropped into the solution and stirred for 4 h. The solution was condensed and was added to a mixture of dichloromethane and water. The mixture was extracted with dichloromethane. The organic layer was dried with sodium sulfate and concentrated. The residue was purified by column chromatography on silica gel using a mixture of dichloromethane and methanol (9:1, v/v) as the mobile phase, yielding compound 11 (41 mg, 0.12 mmol, 18.8% from 7) as an orange-colored solid. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.97 (4H, m, Ar−H), 7.62 (2H, m, Ar−H), 7.26 (2H, m, Ar−H), 6.89 (4H, m), 3.11 (4H, m), 3.08 (4H, m). High-resolution MS m/z: 355.1912 (calculated for C23H22N4, 355.4624). Chemical purity: 95.2% (Supporting Information).
Radiosynthesis of [11C]MBF
11C was produced by 14N(p, α)11C nuclear reaction using a CYPRIS HM-18 cyclotron (Sumitomo Heavy Industries, Tokyo, Japan). [11C]Methyl iodide was prepared from [11C]carbon dioxide via [11C]methanol using an automated system as described previously (40). [11C]Methyl iodide was trapped in the solution of DMF (0.35 mL) containing desmethyl MBF (1.0 mg) and 1.0 M tetrabutylammonium hydroxide in methanol (3 μL) on cooling, and the solution was then heated to 80 °C for 5 min. After cooling again, 0.5 mL of eluent for the preparative high-performance liquid chromatography (HPLC) was added, and the reaction mixture was applied to the preparative HPLC. Preparative HPLC was carried out using a Capcell Pak C18 UG 80 column (10 mm internal diameter × 250 mm length; Shiseido, Tokyo, Japan) with an ultraviolet detector at 254 nm and a radioactivity detector. Elution was carried out using a mixture of acetonitrile and 0.1 M ammonium formate (80:20, v/v/v) as the mobile phase at a flow rate of 5 mL/min. The retention times of desmethyl MBF and [11C]MBF were 4.5 and 7.5 min, respectively. The HPLC fraction of [11C]MBF was collected into a flask to which 25% ascorbic acid (100 μL) and Tween 80 (75 μL) in ethanol (0.3 mL) had been added before radiosynthesis, and then evaporated to dryness. The residue was dissolved in physiological saline.
The products were analyzed by HPLC using a Capcell Pak C18 UG 80 column (4.6 mm internal diameter × 250 mm length; Shiseido). Elution was carried out using the same mobile phase as that used for preparative HPLC at a flow rate of 2.0 mL/min. The retention time of [11C]MBF was 3.4 min.
Radiosynthesis of [11C]JP-1302
[11C]Methyl iodide was prepared as described above. [11C]Methyl iodide was passed through a glass column containing silver triflate at 180 °C with a nitrogen gas flow to convert it into [11C]methyl triflate. This [11C]methyl triflate was trapped in the solution of acetone (0.3 mL) containing desmethyl JP-1302 (0.3 mg) at rt. After cooling, 0.5 mL of eluent for the preparative HPLC was added, and the reaction mixture was applied to the preparative HPLC. Preparative HPLC was carried out using a Capcell Pak C18 UG 80 column (10 mm internal diameter × 250 mm length; Shiseido) with an ultraviolet detector at 254 nm and a radioactivity detector. Elution was carried out using a mixture of acetonitrile and 0.1% triethylamine in water (50:50, v/v/v) as the mobile phase at a flow rate of 5 mL/min. The retention time of desmethyl JP-1302 and [11C]JP-1302 were 5.5 and 8.5 min, respectively. The HPLC fraction of [11C]JP-1302 was collected into a flask to which 25% ascorbic acid (100 μL) and Tween 80 (75 μL) in ethanol (0.3 mL) had been added before radiosynthesis and then evaporated to dryness. The residue was dissolved in physiological saline.
The products were analyzed by HPLC using an X Bridge C18 column (4.6 mm internal diameter × 150 mm length; Waters, Milford, MA). Elution was carried out using a mixture of acetonitrile and 0.1% triethylamine in water (40:60, v/v/v) as the mobile phase at a flow rate of 1.5 mL/min. The retention time of [11C]JP-1302 was 4.0 min.
In Vivo Biodistribution in Mice
The tissue distribution of radioactivity after the injection of [11C]MBF and [11C]JP-1302 was investigated. The 11C-labeled ligand (7.4−12 MBq/0.07−0.12 nmol) was intravenously injected into normal mice (age, 7 weeks). Mice were sacrificed by cervical dislocation at 5, 15, 30, or 60 min after injection (n = 3 per group).
The effects of treatment with a cold α2C-AR ligand on tissue distribution of [11C]MBF and [11C]JP-1302 were investigated. The 11C-labeled ligand (5.9−11 MBq/0.13−0.25 nmol) was intravenously injected into normal mice (age, 8−10 weeks, n = 3−7 per group). One of the α2C-AR ligands (MBF, JP-1302, MK912; dose, 1.0 mg/kg) was intravenously co-injected with [11C]MBF or injected 15 min before administration of [11C]JP-1302. Mice were sacrificed by cervical dislocation at 15 min ([11C]MBF) or 30 min ([11C]JP-1302) after the injection.
The effects of the inhibition of P-gp and Bcrp functions on tissue distribution of [11C]MBF and [11C]JP-1302 were investigated. The 11C-labeled ligand (11−12 MBq/0.12−0.17 nmol), P-gp/BCRP inhibitor GF120918 (5.0 mg/kg), or cold ligand (MBF or JP-1302; 1.0 mg/kg) were intravenously co-injected into mice (age, 7−8 weeks, n = 4 per group). Mice were sacrificed by cervical dislocation at 30 min after the injection.
Blood samples were collected by heart puncture, and the tissues were dissected out and weighed. Radioactivity in samples was measured using an automatic gamma counter (Wizard 3′′ 1480, PerkinElmer, Waltham, MA). Tissue uptake of 11C was expressed as the standardized uptake value [SUV, (tissue radioactivity/gram or milliliter of tissue)/(injected radioactivity/gram of body weight)].
Metabolite Analysis in the Brain and Plasma of Mice
[11C]MBF (44 MBq/1.5 nmol) or [11C]JP1302 (59 MBq/1.3 nmol) was intravenously injected into male ddY mice (age, 9 weeks; weight, 35−41 g; n = 3−4 per tracer). Mice were sacrificed by cervical dislocation at 30 min after the injection. Blood samples were collected by heart puncture into a heparinized syringe, and the whole brain was dissected out. Deproteinization was carried out using previously described methods (34−36) mentioned below. Plasma and homogenized brain tissues were deproteinized with the same volume of ice-cold acetonitrile. The mixture was then vortexed and centrifuged at 20 000g for 2 min, and the supernatant was collected. Supernatants were analyzed using HPLC with a radioactivity detector (41) and an ultraviolet detector at 254 nm on a Novapak C18 column (100 mm internal diameter × 8 mm length; Waters, Milford, MA) contained within a radial compression module (RCM-100, Waters) for [11C]MBF or a Capcell Pak C18 UG 80 column (4.6 mm internal diameter × 250 mm length; Shiseido) for [11C]JP-1302. Elution was carried out using a mixture of acetonitrile and 0.1% triethylamine in water (50:50, v/v) for [11C]MBF or a mixture of methanol and 50 mM sodium acetate buffer (80:20, v/v) for [11C]JP-1302 at a flow rate of 1.5 mL/min. The retention times of [11C]MBF and [11C]JP-1302 were 4.1 and 4.9 min, respectively. Radioactivity in the supernatants, residual precipitates after centrifugation, and waste solution from HPLC were measured using an automatic γ counter. Percentages of the unchanged form were then determined.
PET Study in Mice
PET scans were obtained using an Inveon dedicated PET scanner (Siemens Medical Solutions, Knoxville, TN). P-gp/Bcrp knockout mice (age, 31−34 weeks; weight, 34−38 g; n = 3 per tracer) and wild-type mice (age, 11−14 weeks; weight, 26−43 g; n = 3 per tracer) were used in the PET study.
Mice were anesthetized with isoflurane (1.0−1.5%, v/v) and placed in a prone position on the bed. [11C]MBF (4.1−7.8 MBq/0.12−1.22 nmol) or [11C]JP-1302 (3.6−8.1 MBq/0.033−0.078 nmol) was intravenously injected into mice. A time sequential scan was carried out for 60 min in the three-dimensional (3D) list mode with an energy window of 350−650 keV. List-mode data were sorted into 3D sinograms (6 × 10 s, 4 × 15 s, 5 × 1 min, 4 × 2 min, 3 × 5 min, and 3 × 10 min), which were then Fourier rebinned into two-dimensional sinograms. Dynamic images were reconstructed with filtered back-projection using a ramp filter. Decay-corrected radioactivity was expressed as SUV. Regions of interests (ROIs) were masked on the brain using ASIPro VM software (Siemens Medical Solutions). The area under the time−activity curve of the ROIs in the brain (AUCbrain, SUV·min) was calculated starting from 0 to 60 min.
Statistical Analysis
Quantitative data are expressed as the mean ± standard deviation (SD). In the in vivo distribution study, differences between control and treatment groups were tested by one-way analysis of variance (ANOVA) and Dunnett’s post hoc tests. In the in vivo distribution study, different groups were assessed using one-way ANOVA and Bonferroni’s multiple comparison tests. The data was analyzed using GraphPad Prism 5 software (GraphPad Software, San Diego, CA). Differences were considered significant if P < 0.05.
Acknowledgments
We are grateful to Ms. Teruno Nakaguma (National Institute of Radiological Sciences, NIRS; WDB) for her technical assistance with the synthesis and Mr. Kenji Furutuka (SHI Accelerator Service, SAS) for his technical assistance with the high-resolution LC-MS experiments. We are also grateful to Mr. Yukio Nakamura (Tokyo Nuclear Services), Mr. Masanao Ogawa (SAS), Mr. Yuichiro Yoshida (SAS) and Mr. Hisashi Suzuki (NIRS) for their technical assistance with the radiosynthesis. We thank the staff of Cyclotron Operation Section NIRS for their technical assistance in radioisotope production and also the staff of the Department of Molecular Probes NIRS for their assistance.
Supporting Information Available
HPLC results for compounds 6 and 11 and MBF. This material is available free of charge via the Internet at http://pubs.acs.org.
Drs. Ming-Rong Zhang and Toshimitsu Fukumura designed compounds and directed the synthetic chemistry experiments, synthetic radiopharmaceutical chemistry experiments, and molecular imaging. Drs. Megumi Akiyama and Katsushi Kumata performed synthetic chemistry experiments and radiopharmaceutical chemistry experiments. Drs. Joji Yui, Tomoteru Yamazaki, Akiko Hatori, Kazuhiko Yanamoto, and Hidekatsu Wakizaka performed the in vivo biology experiments and molecular imaging. Drs. Makoto Takei and Nobuki Nengaki performed synthetic radiopharmaceutical chemistry experiments.
This study was partially supported by consignment expenses from the Molecular Imaging Program from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of the Japanese Government.
Supplementary Material
References
- Gottesman M. M.; Fojo T.; Bates S. E. (2002) Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58. [DOI] [PubMed] [Google Scholar]
- Borst P.; Oude Elferink R. (2002) Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 71, 537–592. [DOI] [PubMed] [Google Scholar]
- Doze P.; Van Waarde A.; Elsinga P. H.; Hendrikse N. H.; Vaalburg W. (2000) Enhanced cerebral uptake of receptor ligands by modulation of P-glycoprotein function in the blood-brain barrier. Synapse 36, 66–74. [DOI] [PubMed] [Google Scholar]
- Passchier J.; vanWaarde A.; Doze P.; Elsinga P. H.; Vaalburg W. (2000) Influence of P-glycoprotein on brain uptake of [18F]MPPF in rats. Eur. J. Pharmacol. 407, 273–280. [DOI] [PubMed] [Google Scholar]
- Elsinga P. H.; Hendrikse N. H.; Bart J.; Vaalburg W.; van Waarde A. (2004) PET studies on P-glycoprotein function in the blood-brain barrier: How it affects uptake and binding of drugs within the CNS. Curr. Pharm. Des. 10, 1493–1503. [DOI] [PubMed] [Google Scholar]
- Elsinga P. H.; Hendrikse N. H.; Bart J.; van Waarde A.; Vaalburg W. (2005) Positron emission tomography studies on binding of central nervous system drugs and P-glycoprotein function in the rodent brain. Mol. Imaging Biol. 7, 37–44. [DOI] [PubMed] [Google Scholar]
- Liow J. S.; Lu S.; McCarron J. A.; Hong J.; Musachio J. L.; Pike V. W.; Innis R. B.; Zoghbi S. S. (2007) Effect of a P-glycoprotein inhibitor, Cyclosporin A, on the disposition in rodent brain and blood of the 5-HT1A receptor radioligand, [11C](R)-(−)-RWAY. Synapse 61, 96–105. [DOI] [PubMed] [Google Scholar]
- Ishiwata K.; Kawamura K.; Yanai K.; Hendrikse N. H. (2007) In vivo evaluation of P-glycoprotein modulation of 8 PET radioligands used clinically. J. Nucl. Med. 48, 81–87. [PubMed] [Google Scholar]
- Zoghbi S. S.; Liow J. S.; Yasuno F.; Hong J.; Tuan E.; Lazarova N.; Gladding R. L.; Pike V. W.; Innis R. B. (2008) 11C-loperamide and its N-desmethyl radiometabolite are avid substrates for brain permeability-glycoprotein efflux. J. Nucl. Med. 49, 649–656. [DOI] [PubMed] [Google Scholar]
- Murphy T. J.; Bylund D. B. (1988) Characterization of alpha-2 adrenergic receptors in the OK cell, an opossum kidney cell line. J. Pharmacol. Exp. Ther. 244, 571–578. [PubMed] [Google Scholar]
- Brede M.; Nagy G.; Philipp M.; Sorensen J. B.; Lohse M. J.; Hein L. (2003) Differential control of adrenal and sympathetic catecholamine release by α2-adrenoceptor subtypes. Mol. Endocrinol. 17, 1640–1646. [DOI] [PubMed] [Google Scholar]
- Moura E.; Afonso J.; Hein L.; Vieira-Coelho M. A. (2006) α2-Adrenoceptor subtypes involved in the regulation of catecholamine release from the adrenal medulla of mice. Br. J. Pharmacol. 149, 1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björklund M.; Sirviö J.; Sallinen J.; Scheinin M.; Kobilka B. K.; Riekkinen P. Jr. (1999) α2C-Adrenoceptor overexpression disrupts execution of spatial and non-spatial search patterns. Neuroscience 88, 1187–1198. [DOI] [PubMed] [Google Scholar]
- Scheinin M; Sallinen J; Haapalinna A. (2001) Evaluation of the α2C-adrenoceptor as a neuropsychiatric drug target studies in transgenic mouse models. Life Sci. 68, 2277–2285. [DOI] [PubMed] [Google Scholar]
- Sallinen J.; Link R. E.; Haapalinna A.; Viitamaa T.; Kulatunga M.; Sjöholm B.; Macdonald E.; Pelto-Huikko M.; Leino T.; Barsh G. S.; Kobilka B. K.; Scheinin M. (1997) Genetic alteration of α2C-adrenoceptor expression in mice: Influence on locomotor, hypothermic, and neurochemical effects of dexmedetomidine, a subtype-nonselective α2-adrenoceptor agonist. Mol. Pharmacol. 51, 36–46. [DOI] [PubMed] [Google Scholar]
- Tanila H.; Mustonen K.; Sallinen J.; Scheinin M.; Riekkinen P Jr. (1999) Role of α2C-adrenoceptor subtype in spatial working memory as revealed by mice with targeted disruption of the α2C-adrenoceptor gene. Eur. J. Neurosci. 11, 599–603. [DOI] [PubMed] [Google Scholar]
- Björklund M.; Sirviö J.; Puoliväli J.; Sallinen J.; Jäkälä P.; Scheinin M.; Kobilka B. K.; Riekkinen P. Jr. (1998) α2C-Adrenoceptor-overexpressing mice are impaired in executing nonspatial and spatial escape strategies. Mol. Pharmacol. 54, 569–576. [DOI] [PubMed] [Google Scholar]
- Khasar S. G.; Green P. G.; Chou B.; Levine J. D. (1995) Peripheral nociceptive effects of α2-adrenergic receptor agonists in the rat. Neuroscience 66, 427–432. [DOI] [PubMed] [Google Scholar]
- Aley K. O.; Levine J. D. (1997) Multiple receptors involved in peripheral α2, μ, and A1 antinociception, tolerance, and withdrawal. J. Neurosci. 17, 735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dossin O.; Moulédous L.; Baudry X.; Tafani J. A.; Mazarguil H.; Zajac J. M. (2000) Characterization of a new radioiodinated probe for the α2C adrenoceptor in the mouse brain. Neurochem. Int. 36, 7–18. [DOI] [PubMed] [Google Scholar]
- Holmberg M.; Fagerholm V.; Scheinin M. (2003) Regional distribution of α2C-adrenoceptors in brain and spinal cord of control mice and transgenic mice overexpressing the α2C-subtype: An autoradiographic study with [3H]RX821002 and [3H]rauwolscine. Neuroscience 117, 875–898. [DOI] [PubMed] [Google Scholar]
- Shiue C.; Pleus R. C.; Shiue G. G.; Rysavy J. A.; Sunderland J. J.; Cornish K. G.; Young S. D.; Bylund D. B. (1998) Synthesis and biological evaluation of [11C]MK-912 as an α2-adrenergic receptor radioligand for PET studies. Nucl. Med. Biol. 25, 127–133. [DOI] [PubMed] [Google Scholar]
- Hume S. P.; Hirani E.; Opacka-Juffry J.; Osman S.; Myers R.; Gunn R. N.; McCarron J. A.; Clark R. D.; Melichar J.; Nutt D. J.; Pike V. W. (2000) Evaluation of [O-methyl-11C]RS-15385-197 as a positron emission tomography radioligand for central α2-adrenoceptors. Eur. J. Nucl. Med. 27, 475–484. [DOI] [PubMed] [Google Scholar]
- Van der Mey M.; Windhorst A. D.; Klok R. P.; Herscheid J. D.; Kennis L. E.; Bischoff F.; Bakker M.; Langlois X.; Heylen L.; Jurzak M.; Leysen J. E. (2006) Synthesis and biodistribution of [11C]R107474, a new radiolabeled α2-adrenoceptor antagonist. Bioorg. Med. Chem. 14, 4526–4534. [DOI] [PubMed] [Google Scholar]
- Jakobsen S.; Pedersen K.; Smith D. F.; Jensen S. B.; Munk O. L.; Cumming P. (2006) Detection of α2-adrenergic receptors in brain of living pig with 11C-yohimbine. J. Nucl. Med. 47, 2008–2015. [PubMed] [Google Scholar]
- Smith D. F.; Dyve S.; Minuzzi L.; Jakobsen S.; Munk O. L.; Marthi K.; Cumming P. (2006) Inhibition of [11C]mirtazapine binding by α2-adrenoceptor antagonists studied by positron emission tomography in living porcine brain. Synapse 59, 463–471. [DOI] [PubMed] [Google Scholar]
- Hagihara K.; Kashima H.; Iida K.; Enokizono J.; Uchida S.; Nonaka H.; Kurokawa M.; Shimada J. (2007) Novel 4-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)methylbenzofuran derivatives as selective α2C-adrenergic receptor antagonists. Bioorg. Med. Chem. Lett. 17, 1616–1621. [DOI] [PubMed] [Google Scholar]
- Sallinen J.; Höglund I.; Engström M.; Lehtimäki J.; Virtanen R.; Sirviö J.; Wurster S.; Savola J. M.; Haapalinna A. (2007) Pharmacological characterization and CNS effects of a novel highly selective α2C-adrenoceptor antagonist JP-1302. Br. J. Pharmacol. 150, 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume S. P.; Lammertsma A. A.; Opacka-Juffry J.; Ahier R. G.; Myers R.; Cremer J. E.; Hudson A. L.; Nutt D. J.; Pike V. W. (1992) Quantification of in vivo binding of [3H]RX 821002 in rat brain: Evaluation as a radioligand for central α2-adrenoceptors. Int. J. Radiat. Appl. Instrum. B 19, 841–849. [DOI] [PubMed] [Google Scholar]
- Waterhouse R. N. (2003) Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 5, 376–389. [DOI] [PubMed] [Google Scholar]
- Fagerholm V.; Rokka J.; Nyman L.; Sallinen J.; Tiihonen J.; Tupala E.; Haaparanta M.; Hietala J. (2008) Autoradiographic characterization of α2C-adrenoceptors in the human striatum. Synapse 62, 508–515. [DOI] [PubMed] [Google Scholar]
- Kawamura K.; Ishiwata K. (2004) Improved synthesis of [11C]SA4503, [11C]MPDX and [11C]TMSX by use of [11C]methyl triflate. Ann. Nucl. Med. 18, 165–168. [DOI] [PubMed] [Google Scholar]
- Zhou L.; Schmidt K.; Nelson F. R.; Zelesky V.; Troutman M. D.; Feng B. (2009) The effect of breast cancer resistance protein and P-glycoprotein on the brain penetration of flavopiridol, imatinib mesylate (Gleevec), prazosin, and 2-methoxy-3-(4-(2-(5-methyl-2-phenyloxazol-4-yl)ethoxy)phenyl)propanoic acid (PF-407288) in mice. Drug Metab. Dispos. 37, 946–955. [DOI] [PubMed] [Google Scholar]
- Kawamura K.; Yamasaki T.; Yui J.; Hatori A.; Konno F.; Kumata K.; Irie T.; Fukumura T.; Suzuki K.; Kanno I.; Zhang M. R. (2009) In vivo evaluation of P-glycoprotein and breast cancer resistance protein modulation in the brain using [11C]gefitinib. Nucl. Med. Biol. 36, 239–246. [DOI] [PubMed] [Google Scholar]
- Kawamura K., Yamasaki T., Konno F., Yui J., Hatori A., Yanamoto K., Wakizaka H., Takei M., Kimura Y., Fukumura T., and Zhang M. R. (2010) Evaluation of limiting brain penetration related to P-glycoprotein and breast cancer resistance protein using [11C]GF120918 by positron emission tomography in mice. Mol. Imaging. Biol. DOI: 10.1007/s1130701003131. [DOI] [PubMed] [Google Scholar]
- Kawamura K., Konno F., Yui J., Yamasaki T., Hatori A., Yanamoto K., Wakizaka H., Takei M., Nengaki N., Fukumura T., and Zhang M. R. (2010) Synthesis and evaluation of [11C]XR9576 to assess the function of drug efflux transporters using PET. Ann. Nucl. Med. DOI: 10.1007/s121490100373y. [DOI] [PubMed] [Google Scholar]
- Dodic N.; Dumaitre B.; Daugan A.; Pianetti P. (1995) Synthesis and activity against multidrug resistance in Chinese hamster ovary cells of new acridone-4-carboxamides. J. Med. Chem. 38, 2418–2426. [DOI] [PubMed] [Google Scholar]
- Gilday J. P., andMoody D. (2004) Process for the preparation of 4-(3′-chlorp-4’-fluoroanilino)-7-methoxy-6-(3-morpholinopropoxy) quinazoline. PCT. Int. Appl. WO/2004/024703 A1.
- Jonker J. W.; Freeman J.; Bolscher E.; Musters S.; Alvi A. J.; Titley I.; Schinkel A. H.; Dale T. C. (2005) Contribution of the ABC transporters Bcrp1 and Mdr1a/1b to the side population phenotype in mammary gland and bone marrow of mice. Stem Cells 23, 1059–1065. [DOI] [PubMed] [Google Scholar]
- Suzuki K.; Inoue O.; Hashimoto K.; Yamasaki T.; Kuchiki M.; Tamate K. (1985) Computer-controlled large scale production of high specific activity [11C]Ro15−1788 for PET studies of benzodiazepine receptors. Int. J. Appl. Radiat. Isot. 36, 971–976. [DOI] [PubMed] [Google Scholar]
- Takei M.; Kida T.; Suzuki K. (2001) Sensitive measurement of positron emitters eluted from HPLC. Appl. Radiat. Isot. 55, 229–234. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.