

Abstract
Background
Huanglongbing (HLB) is one of the most destructive citrus diseases in the world. The disease is associated with the presence of a fastidious, phloem-limited α- proteobacterium, 'Candidatus Liberibacter asiaticus', 'Ca. Liberibacter africanus' or 'Ca. Liberibacter americanus'. HLB-associated Liberibacters have spread to North America and South America in recent years. While the causal agents of HLB have been putatively identified, information regarding the worldwide population structure and epidemiological relationships for 'Ca. L. asiaticus' is limited. The availability of the 'Ca. L. asiaticus' genome sequence has facilitated development of molecular markers from this bacterium. The objectives of this study were to develop microsatellite markers and conduct genetic analyses of 'Ca. L. asiaticus' from a worldwide collection. Two hundred eighty seven isolates from USA (Florida), Brazil, China, India, Cambodia, Vietnam, Taiwan, Thailand, and Japan were analyzed.
Results
A panel of seven polymorphic microsatellite markers was developed for 'Ca. L. asiaticus'. Microsatellite analyses across the samples showed that the genetic diversity of 'Ca. L. asiaticus' is higher in Asia than Americas. UPGMA and STRUCTURE analyses identified three major genetic groups worldwide. Isolates from India were genetically distinct. East-southeast Asian and Brazilian isolates were generally included in the same group; a few members of this group were found in Florida, but the majority of the isolates from Florida were clustered separately. eBURST analysis predicted three founder haplotypes, which may have given rise to three groups worldwide.
Conclusions
Our results identified three major genetic groups of 'Ca. L. asiaticus' worldwide. Isolates from Brazil showed similar genetic makeup with east-southeast Asian dominant group, suggesting the possibility of a common origin. However, most of the isolates recovered from Florida were clustered in a separate group. While the sources of the dominant 'Ca. L. asiaticus' in Florida were not clearly understood, the less-pervasive groups may have been introduced directly from Asia or via Brazil. Notably, the recent outbreak of HLB in Florida probably occurred through multiple introductions. Microsatellite markers developed in this study provide adequate discriminatory power for the identification and differentiation of closely-related isolates, as well as for genetic studies of 'Ca. L. asiaticus'.
Background
Huanglongbing (HLB) is one of the most devastating diseases of citrus, which is characterized by the development of yellow shoots and stunted growth of infected trees combined with a decline in quantity and quality of fruit production [1]. HLB-affected fruit are abnormally-pigmented, developmentally flawed, and have a bitter taste- making them unusable for juice production or as table fruit [2,3]. Typically, trees with HLB succumb to the effects of infection and die within a few years after showing the first signs of the disease [4].
HLB is associated with three 'Candidatus Liberibacter' species worldwide: 'Ca. L. asiaticus', 'Ca. L. africanus' and 'Ca. L. americanus'; the nomenclature is based on the presumptive origin of each bacterium in Asia, Africa and South America, respectively [1]. HLB has been known in Asian countries since the 1870s [1,5,6] and found to be associated with the presence of a fastidious α-proteobacterium named 'Candidatus Liberibacter asiaticus'. In the western hemisphere, it was reported in São Paulo, Brazil in 2004 and in Florida, USA in 2005- two of the largest citrus growing regions in the world [1]. Although 'Ca. L. americanus' initially constituted a major proportion of the total bacterial population in Brazil, this ratio has changed since 2004, and 'Ca. L. asiaticus' is now the most prevalent citrus-destroying species [4]. Both 'Ca. L. americanus' and 'Ca. L. asiaticus' are transmitted by a psyllid vector, Diaphorina citri (also known as the Asian citrus psyllid, or ACP) in Asia, North America, and South America [7,8]. The HLB-associated Liberibacters can also be transmitted by grafting propagative material from infected plants onto nursery stock. The continued economic losses associated with HLB are a serious threat to the U.S. citrus industry [9]. HLB affects all citrus cultivars [10] and to date there are no known HLB-resistant citrus cultivars.
The genetic structure within a given pathogen population can be a valuable resource for determining the source or origin of the pathogen and risk management of the disease. The availability of a genome sequence for 'Ca. L. asiaticus' has provided information on the metabolic features of this bacterium and key insights into HLB pathogenesis [11]. In addition, the genome sequence has facilitated the development of DNA markers for genetic analysis; these molecular genetic markers are critical for understanding the genetic diversity of global populations and the epidemiology of HLB. DNA markers have been used for characterization of microbial populations associated with plant diseases, including RAPDs, SNPs, MLST and SSRs (microsatellites) [12-15]. Molecular genetic markers not only aid in the general characterization of a given population, but can help identify the source of an introduced pathogen [16].
Among the three HLB-associated Liberibacter species, 'Ca. L. asiaticus' is the most widespread and is responsible for increasing economic losses of citrus industries. Much attention has been drawn by researchers in the last few years to the importance of understanding the epidemiology and ecology of the disease associated with 'Ca. L. asiaticus'. 'Ca. L. asiaticus' isolates were characterized in some previous studies; most of these studies focused on the Asian continent and utilized conserved genes as genetic markers. For example, southeast Asian isolates were characterized by sequencing the 16S rDNA and 16S/23S regions, omp, the rpl gene cluster, and the bacteriophage-type DNA polymerase [17]. The 16S rDNA was employed for understanding genetic diversity of 'Ca. L. asiaticus' in India [18] and a prophage gene was used to reveal variations in China [19]. However, genetic variation within conserved genes has limited discriminatory power to differentiate closely-related isolates within populations.
Microsatellite DNA markers associated with hypervariable sequence regions can provide sufficient resolution for differentiating closely-related isolates and for tracking genotypes of interest; additionally, these markers may help identify the source of invasive strains. Recently, similar types of markers have been used for differentiating 'Ca. L. asiaticus' in Japan [20]. Chen et al. [21] studied populations from Guangdong province in China and Florida in the United States. However, the single variable locus used in that study provided limited characterization of 'Ca. L. asiaticus' genetic diversity.
Here, we report a panel of seven polymorphic microsatellite markers for conducting genetic analyses of 'Ca. L. asiaticus' isolates from Asia (India, China, Cambodia, Vietnam, Thailand, Taiwan, and Japan), North America (Florida, USA) and South America (São Paulo, Brazil). The microsatellite profile for each isolate was compared with all members of the sample set to make predictions on the possible origin and dissemination of HLB in Florida.
Results
PCR amplification and characteristic of microsatellite loci
A total of 287 'Ca. L. asiaticus' isolates obtained from HLB-affected plants across Asia and the Americas (Additional file 1 and Table 2) were successfully amplified by all seven microsatellite primer pairs. The number of alleles and haploid genetic diversity per locus ranged from 2 to 30, and 0.204 to 0.881, respectively (Table 1). In the clone-corrected data set, genotypic linkage disequilibrium was not detected by pairwise comparison of loci across the overall isolates (P > 0.01).
Table 2.
Descriptive statistics and genetic diversity of 'Candidatus Liberibacter asiaticus' isolates across seven microsatellite loci in the samples obtained from nine different countries from Asia, North (Florida, USA) and South Americas (São Paulo, Brazil)
| Country | Location ID | Location Information | Total number of individuals | Number of individuals in clone corrected data | Alleles per locus | Haploid genetic diversity |
|---|---|---|---|---|---|---|
| Brazil | BRA | São Paulo | 22 | 14 | 2.7 | 0.313 |
| USA | FL-A | Charlotte County, Florida | 5 | 4 | 1.6 | 0.161 |
| FL-B | Collier County, Florida | 46 | 11 | 2.1 | 0.234 | |
| FL-C | DeSoto County, Florida | 30 | 5 | 1.7 | 0.194 | |
| FL-D | Hardee County, Florida | 8 | 5 | 1.7 | 0.160 | |
| FL-E | Hendry County, Florida | 13 | 5 | 1.6 | 0.171 | |
| FL-F | Highlands County, Florida | 19 | 6 | 1.7 | 0.119 | |
| FL-G | Indian River, County, Florida | 23 | 7 | 1.9 | 0.175 | |
| FL-H | Martin County, Florida | 10 | 7 | 1.9 | 0.175 | |
| FL-I | Okechobee County, Florida | 4 | 2 | 1.3 | 0.143 | |
| FL-J | Polk County, Florida | 6 | 4 | 2.0 | 0.304 | |
| FL-K | St. Lucie County, Florida | 6 | 4 | 1.4 | 0.179 | |
| FL-L | Pasco County, Florida | 2 | 2 | 1.1 | 0.071 | |
| FL-M | Manatee County, Florida | 2 | 2 | 1.3 | 0.143 | |
| FL-N | Hillsborough County, Florida | 2 | 2 | 1.3 | 0.071 | |
| FL-O | Lake County, Florida | 1 | 1 | 1.0 | 0.000 | |
| USA-Florida-overall | 177 | 67 | 3.6 | 0.247 | ||
| CHINA | CHN-A | Baise, Guangxi Province | 3 | 2 | 1.1 | 0.071 |
| CHN-B | Guilin, Guangxi Province | 3 | 3 | 1.4 | 0.190 | |
| CHN-C | Hezhou, Guangxi Province | 3 | 2 | 1.3 | 0.143 | |
| CHN-D | Nanning, Guangxi province | 14 | 14 | 3.0 | 0.276 | |
| CHN-E | Fuzhou, Fujian Province | 7 | 5 | 2.1 | 0.320 | |
| CHN-F | Tangshan, Yunnan Province | 3 | 2 | 1.3 | 0.143 | |
| CHN-G | Gangzhoou, Jiangxi Province | 1 | 1 | 1.0 | 0.000 | |
| CHN-H | Guangzhou, Guangdong Province | 1 | 1 | 1.0 | 0.000 | |
| CHN-I | Hunan Province | 1 | 1 | 1.0 | 0.000 | |
| China-overall | 36 | 31 | 5.7 | 0.342 | ||
| CAMBODIA | CAM-A | Pursat Province | 7 | 6 | 2.4 | 0.341 |
| CAM-B | Battambang Province | 4 | 4 | 1.9 | 0.304 | |
| Cambodia-overall | 11 | 10 | 3.1 | 0.423 | ||
| VIETNAM | VIET | Hung Yen Province, Hoa Binh Province, Hanoi | 3 | 3 | 1.9 | 0.317 |
| THAILAND | THAI | Unknown | 1 | 1 | 1.0 | 0.000 |
| TAIWAN | TIW | Unknown | 1 | 1 | 1.0 | 0.000 |
| JAPAN | JPN | Unknown | 1 | 1 | 1.0 | 0.000 |
| INDIA | IND-A | Anantapur District, Andhra Pradesh | 7 | 7 | 2.4 | 0.297 |
| IND-B | Chittoor District, Andhra Pradesh | 6 | 6 | 2.0 | 0.254 | |
| IND-C | Kadapa District, Andhra Pradesh | 4 | 4 | 1.9 | 0.250 | |
| IND-D | Mahaboobnagar District, Andhra Pradesh | 3 | 3 | 1.4 | 0.159 | |
| IND-E | Nalgonda District, Andhra Pradesh | 4 | 4 | 1.7 | 0.196 | |
| IND-F | Prakasam District, Andhra Pradesh | 4 | 3 | 2.4 | 0.540 | |
| IND-G | Tirupati District, Andhra Pradesh | 5 | 5 | 2.0 | 0.274 | |
| IND-H | Kurnool District, Andhra Pradesh | 1 | 1 | 1.0 | 0.000 | |
| IND-I | Ludhiana District, Punjab | 1 | 1 | 1.0 | 0.000 | |
| India-overall | 35 | 34 | 5.4 | 0.360 | ||
Allele per locus: average number of alleles per locus
Clone corrected data (removed repeated genotypes within a population)
Table 1.
Characteristics of seven microsatellite markers developed from 'Candidatus Liberibacter asiaticus'
| SSR Markers | Primer sequences (5'-----3') | Repeats | Location in genome | ORF | Ta (°C) | Size range (bp) | No. of alleles | H |
|---|---|---|---|---|---|---|---|---|
| LasSSR-A-f LasSSR-A-r |
FAM-CGCCTACAGGAATTTCGTTACG TCTCATCTTGTTGCTTCGTTTATCC |
(TATTCTG)8 | 255477-255753 | adenosine deaminases | 50°C | 241-434 | 30 | 0.881 |
| LasSSR-B-f LasSSR-B-r |
VIC-ATCGCCTATAAATCCCTTTACTGATATGTTTCC TGGTAACGGAAGTGATAATAACTACAGCAATAAG |
(TTTAA)6 | 669257-669458 | hypothetical protein | 60°C | 196-206 | 3 | 0.216 |
| LasSSR-C-f LasSSR-C-r |
VIC-CGATTGTTGATGAATTACC GAATAGAAGAACCCTAAGC |
(CAGT)8 | 666722-666947 | phosphohydrolases | 50°C | 208-254 | 15 | 0.613 |
| LasSSR-D-f LasSSR-D-r |
NED-CGGTGTCGGTATCGGTATCATTC CGAAGAAGAGACGGAGGTTAAGC |
(TTC)5 | 377678-377850 | hypothetical protein | 55°C | 158-174 | 3 | 0.391 |
| LasSSR-E-f LasSSR-E-r |
NED-GATCAGTAGTCTATCACCAC TACTGGAAACAAATGGAATAC |
(CTTGTGT)5 | 354424-354613 | transcriptional regulator | 50°C | 173-290 | 17 | 0.587 |
| LasSSR-F-f LasSSR-F-r |
FAM-TCGTCTTATCGTATATCACTCC TTCACTATTAAAGGATCAAGGC |
(TTTACATC)3 | 520542-520307 | repair ATPase | 52°C | 227-235 | 2 | 0.204 |
| LasSSR-G-f LasSSR-G-r |
FAM-CGGGAGAAATTAAAGATGATGG CGCTGTTAATACATACTTACGC |
(TTGTTGGA)2 | 998251-998403 | hypothetical protein | 53°C | 139-152 | 2 | 0.204 |
Ta, annealing temperature of the primer pairs; H, Haploid genetic diversity
Each forward primer was labeled with FAM, NED, VIC fluorescent dyes at 5', respectively
Genotype and genetic diversity
A total of 117 genotypes (haplotypes) were identified (Additional file 1). Haplotypes identified within the sample population were restricted to the boundaries of their country of origin. The genetic diversity observed in different countries and locations are summarized in Table 2. Isolates from China possessed the largest number of alleles (5.7 alleles per locus), followed by India (5.4 alleles per locus). Overall haploid genetic diversities were the highest in Asian countries, followed by Brazil. The haploid genetic diversity of the Florida (USA) isolates was lowest among all the geographic groupings (Table 2).
Genetic structure
A UPGMA clustering analysis identified three major groups of 'Ca. L. asiaticus' (Figure 1). Isolates from India were clustered in a distinct group (group 3). Most of the isolates from China and other Asian countries, and Brazil were generally grouped in group 1. While some isolates from Florida occurred in group 1, most isolates from Florida were clustered in group 2 (Figure 1).
Figure 1.

UPGMA dendrogram showing the genetic relationships of 'Candidatus Liberibacter asiaticus' isolates from different locations within an individual country as well as from different countries (from Asia and Americas). Clone-corrected data were used for constructing the dendrogram based on DA distance [22]. Only bootstrap values > 25% are shown.
The STRUCTURE analysis based on Bayesian modeling further assessed the genetic structure of 'Ca. L. asiaticus'. This approach utilizes statistical methods to determine the relationships among the isolates without prior population information. In the analysis three different clusters (K) were identified based on the ad hoc statistic ΔK [23] (Figure 2). The membership of each individual isolate obtained from STRUCTURE analysis, can be estimated as (q), the ancestry coefficient, which varies on a scale between 0-1.0, with 1.0 indicating full membership in a population. Individuals can be assigned to multiple clusters (with values of q summing to 1.0) indicating they are admixed. Individual samples with q ≥ 0.90 (ancestry coefficient) were considered as having single lineage and individuals with q < 0.90 were considered as admixed lineages as followed by Williams et al. [24]. The result of STRUCTURE analysis is consistent with UPGMA in which isolates from India were grouped in a distinct cluster (Figure 2 in yellow). Brazilian and most east-southeast Asian isolates were clustered as a single lineage (q ≥ 0.90) (Figure 2, red). Some isolates taken from central Florida (Polk, Pasco, and Lake Counties) shared the same lineage with east-southeast Asian and Brazilian isolates (Figure 2, red). Most Florida isolates, however, grouped in a different cluster (Figure 2, green). Some admixed isolates (q <0.90) were found in Florida as well as in Baise and Nanning of Guangxi province in China, and in Cambodia.
Figure 2.

Individual assignments of 'Candidatus Liberibacter asiaticus' isolates obtained from nine different countries from Asia and Americas by STRUCTURE analysis. There were three clusters (K). Black lines within the squares distinguish geographic locations.
eBURST analysis with user-defined criteria (based on the analysis of haplotypes that shared identical genotypes for at least 5 of the 7 loci) predicted three founder haplotypes: haplotype-108 (Nanning, Guangxi province, China), haplotype-48 (São Paulo, Brazil) and haplotype-46 (Tirupati District, Andhra Pradesh, India) (Additiontal file 1 and Figure 3). The diagram generated by eBURST showed a primary network between haplotype-103 and 107 (Collier County, Florida) and predicted founder haplotype in China. A primary network was also identified with haplotype-51 (Pasco County, Florida) and the second predicted founder haplotype in Brazil. Haplotype-46 from Tirupati District, Andhra Pradesh, India) was predicted to be the third founder and hypothesized to be the founder haplotype of 'Ca. L. asiaticus' in India.
Figure 3.

Network diagram (based on nearly identical haplotypes that differed by two loci) from eBURST analysis. Solid blue circles in the diagram indicate three predicted founder haplotypes: China (Haplotye-108), Brazil (Haplotype-48) and India (Haplotype-46). A primary network was observed between haplotype-103 and 107 (Florida), and predicted founder haplotypes in China, and between haplotype-51 (Florida) with predicted founder haplotypes in Brazil, suggesting two separate introductions of 'Ca. L. asiaticus' into Florida.
Discussion
Characterization of worldwide and regional 'Ca. L. asiaticus' populations from HLB-affected plants can facilitate identification of introduction patterns and predict the possible relationships of HLB-associated Liberibacters found in different citrus growing regions. Multilocus microsatellite marker analysis can provide sufficient resolution for differentiating closely-related isolates and can be useful for tracking genotypes of interest; additionally, these markers may help identify the source of invasive strains.
In this study, seven microsatellite markers successfully genotyped 'Ca. L. asiaticus' from global populations. Sequence analysis indicated that three of the microsatellites appear to overlap with microsatellites recently developed by Katoh et al. [20]. Various microsatellite length variations were found in 'Ca. L. asiaticus' from worldwide collections, with some loci having as many as 30 alleles.
Historical evidence reviewed by da Graça [25] suggested that HLB was observed in Guangdong province, China in the late 19th century [26], and later spread to other parts of the country. It is assumed that HLB may have been introduced into China from India along sea trade routes [27]. The first record of HLB-like symptoms, referred to as 'dieback', was reported from India in the 18th century [28]; this was later suggested to be HLB [29]. As 'Ca. L. asiaticus' has been in Asian countries over a century, the genetic diversity in Asian populations was expected to be high, due to a longer period of mutation accumulation, population differentiation and natural selection. As hypothesized, a higher degree of genetic diversity for 'Ca. L. asiaticus' was observed in both China and India within the present study (Table 2). In contrast, the lower level of allelic and haploid genetic diversity of 'Ca. L. asiaticus' in Florida and Brazil populations are consistent with the hypothesis that 'Ca. L. asiaticus' populations in these regions have been derived from recent introductions [30].
Human movement of infected plant materials is probably the main cause of long distance dissemination of both 'Ca. L. asiaticus'-positive psyllids and HLB-affected plant material. The distributions of haplotypes observed in 'Ca. L. asiaticus' in this study did not detect any identical haplotypes from different continents or even from different countries within the same continent (Additional file 1). This result does not exclude the possibility of contemporary migration of 'Ca. L. asiaticus' among different countries through the movement of infected plant materials or by the migration of vector psyllids as rapid mutation and selection could lead to deviation of populations from their original sources. The vector, D. citri, has been in Brazil for over 60 years without any sign of HLB until its discovery on 2004 [4,25]. D. citri was discovered in Florida in Palm Beach, Broward and Martin counties in 1998 and has spread throughout the state since that time [7]. However, it is not clear when 'Ca. L. asiaticus' was introduced into Brazil and Florida. The rapid spread of HLB in Brazil and Florida after the first reports in 2004 and 2005, respectively, suggests that HLB was most likely introduced around that time, rather than evolving from an indigenous source.
As suggested by the historical evidence and review of the early literature related to HLB, the most ancient population of 'Ca. L. asiaticus' perhaps originated in India. From the 20th century onward, HLB spread through much of the citrus-growing regions of south and southeast Asia [2], the Arabian peninsula [31], East Timor and Papua New Guinea [32], and the western hemisphere (Brazil and the United States) [1]. It is difficult to precisely know when the disease entered each country and from where it was introduced. Frequent shipment of plant materials and unlawfully importation of plants has increased the risk of disseminating exotic plant pathogens around the world. The exact pathways responsible for introducing HLB and the Asian citrus psyllid into the United States and Brazil have not yet been determined.
The genetic relationships of the isolates in this study, as determined from the UPGMA based on Nei's genetic distance [22] and individual based clustering analysis by the STRUCTURE analyses, consistently identified three major genetic groups of 'Ca. L. asiaticus', with isolates from India included in a distinct genetic group (Figure 2 and Figure 3). The similar genetic makeup amongst most isolates from east-southeast Asia and South America (São Paulo, Brazil) support the hypothesis of the introduction of 'Ca. L. asiaticus' into South America from East Asia or Southeast Asia. While most isolates from Florida were clustered within a separate group, both UPGMA and STRUCTURE analyses showed that some isolates from central Florida overlapped with east-southeast Asian and Brazilian groups. The presence of two genetic groups in Florida suggests at least two introduction events are associated with the recent outbreak of HLB in Florida.
Based on the history of HLB, it could be predicted that populations of 'Ca. L. asiaticus' in Florida were most likely established following the introduction of HLB-affected plant materials or 'Ca. L. asiaticus'-carrying psyllid from Asia or other countries through human-mediated transport. The analyses in this study do not support the hypothesis of introduction of HLB into the Americas through biological materials sourced from India. Only a single isolate from India (Prakasam District, Andhra Pradesh) overlapped with the east-southeast Asian and Brazilian group (Figure 2, red). STRUCTURE analysis revealed that less-dominant clusters (Figure 2, red) in central Florida (Polk, Pasco, and Lake Counties) were observed in the same lineage (q ≥ 0.90) with east-southeast Asian and Brazilian clusters suggesting that the origin of members of this cluster in Florida might be derived from Asia or via Brazil. Moreover, some admixed (q < 0. 90) isolates between Florida and east-southeast Asia also support the hypothesis of introducing 'Ca. L. asiaticus' into Florida from Asia.
eBURST analysis provided further insights into the origin of 'Ca. L. asiaticus'. Founder haplotypes were identified from China, Brazil, and India. Based on their position within the eBURST network, these founders are predicted to have given rise to the three global genetic groups, consistent with prevailing theories of the geographic origins of HLB [1,2,4,7]. While one founder type was predicted in Brazil, the similar genetic makeup of Brazilian and east-southeast Asian isolates suggest that this founder could have been introduced into Brazil from any of these Asian countries. Consistent with the STRUCTURE analysis, the eBURST diagram also predicted the introduction of 'Ca. L. asiaticus' into Florida citrus groves through at least two separate introduction events. While a primary network was detected between a founder haplotype from China and two unique haplotypes in Florida, clear differentiation was observed between most isolates from China and Florida by Bayesian clustering and UPGMA analyses. Differences between the dominant groups found in Florida and China were also reported in a recent study using a single VNTR locus [21]. It is uncertain whether the dominant group of Florida isolates were introduced en masse or if a small population of nearly-identical 'Ca. L. asiaticus' haplotypes from China were introduced, evolved quickly, and established a large population. The recent discovery and rapid spread of HLB in Florida, along with wide distribution of dominant 'Ca. L. asiaticus' group observed in the present study suggests that isolates of this group have been directly introduced from an unknown location. Another recent study also indicated that some isolates of 'Ca. L. asiaticus' from Florida may have been introduced through two different events, and sources were unknown [21]. The analyses of microsatellites in the present study, however, suggest that the introduction of the less-dominant cluster was likely from a single source either Asia or Brazil. The low occurrence of less dominant group in some central counties in Florida suggests that the members of this group were perhaps introduced more recently (Figure 4). However, it is certainly plausible that these two haplotypes were introduced into Florida at nearly the same time. Isolates from one of the sources may have spread quickly due to selective advantage under a favorable set of biological or environmental conditions.
Figure 4.
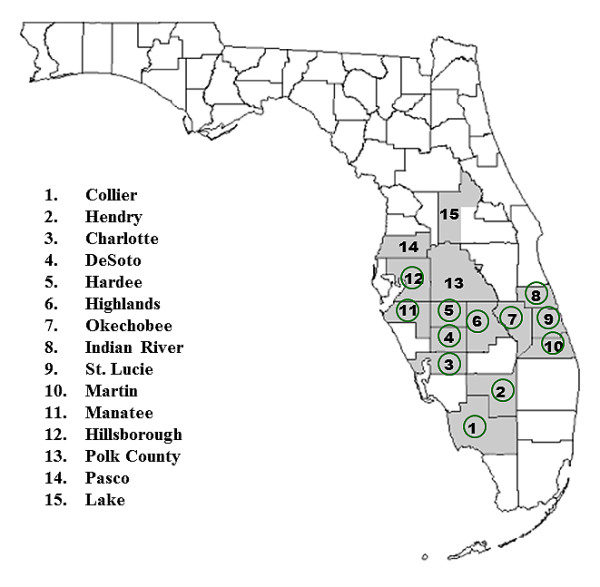
Sample distribution of 'Candidatus Liberibacter asiaticus' from 15 citrus-growing counties (gray highlighted) in Florida, USA. Green circles indicate the counties where only the dominant 'Ca. L. asiaticus' group were observed based on STRUCTURE analysis (green in Figure 2). Some isolates from Polk County (13), Pasco County (14) and Lake County (15) were included with the genetic group 2 (less dominant group) (see Figure 2).
Our analysis showed that a dominant group of 'Ca. L. asiaticus' genotypes are widely distributed in south-central Florida (Figure 4). HLB is highly invasive and spatial spread of the disease is rapid compared to other arboreal pathosystems [4]. Increased diversity of host, pathogen, vector, and environmental conditions likely influence the rates of HLB distribution. Moreover, the rates of HLB increase are directly related to increase and spread of the psyllid vector population: in June 1998, the Asian citrus psyllid was first detected in Palm Beach County; within two years of this discovery the disease occurred to 31 counties in Florida [33]. The vector is now present in nearly all citrus-growing areas of Florida [34]. In Florida, HLB was first discovered in Miami-Dade County in August 2005, seven years after detection of the vector in the same region [30]. By mid-October, the disease was found in many residential properties stretching northwards more than 250 km from Miami-Dade County to St. Lucie County and several commercial citrus groves were also affected in Palm Beach and Hendry Counties [1]. However, no epidemiological survey has clearly demonstrated HLB or 'Ca. L. asiaticus'-carrying psyllids being introduced into the southern part of Florida and then spreading northward through the continuous movement of psyllid vectors. Since 2005, HLB has spread to most citrus-producing counties in Florida [34,35]. The rapid and widespread distribution of this disease among citrus growing counties in Florida is most likely due to the result of the multiple secondary introductions of HLB-associated 'Ca. L. asiaticus'. Based on the present analyses, it appears that there were at least two 'Ca. L. asiaticus' introduction events in Florida. Moreover, the rapid distribution of HLB within Florida after 2005 is concomitant with the discovery of a dominant genetic cluster within south-central Florida. Taken together, this suggests that dominant 'Ca. L. asiaticus' haplotypes, possibly from different countries may have established a population within Florida through multiple introduction events.
Conclusions
The seven microsatellites developed in this study are useful for detection, isolate differentiation, and genetic analysis of 'Ca. L. asiaticus'. Our results showed that current 'Ca. L. asiaticus' populations in HLB-affected citrus in Asia and the Americas are comprised of three distinct genetic groups: (1) Indian, (2) predominantly east-southeast Asian and South American (Brazil) and (3) predominantly North American (Florida, USA). While regional differences were observed from the distribution of dominant clusters, the similar genetic makeup of east-southeast Asian and Brazilian isolates lead us to hypothesize that 'Ca. L. asiaticus' populations in Brazilian groves were most likely introduced from east or southeast Asia. The precise sources of the dominant genetic group of 'Ca. L. asiaticus' retrieved from Florida are not clearly resolved from the present analysis. However, less-pervasive groups may have been introduced directly from Asia or via Brazil. While the results here provide some insights into the origins and genetic structure of HLB-associated 'Ca. L. asiaticus', it should be noted that broader population analyses using a larger array of molecular markers will help resolved the questions on the origin and dissemination of HLB-associated 'Ca. L. asiaticus.
Methods
Sample collection/DNA extraction
DNA from HLB-affected samples from Asia (India, China, Cambodia, Vietnam, Thailand, Taiwan, and Japan), North America (Florida, USA) and South America (State of São Paulo, Brazil) were extracted from the respective sources and sent as microbially-sterile and non-infectious samples. HLB-associated Liberibacter-free DNA samples were used as negative controls. Basically, leaf samples were collected from citrus trees with blotchy mottle and blotchy mottle-like symptoms. Leaves were washed under running tap water and blotted dry with paper towels. The midribs were then excised from the leaf blade. Total genomic DNA was extracted from 4-5 midribs per sample. Samples were ground in liquid nitrogen and DNA was extracted using the CTAB method. Precipitated DNA was dissolved in 100 μl of TE buffer. The quality of DNA samples was checked by electrophoresis in 1.2% agarose gels. DNA samples were diluted 30 times with water for PCR.
Microsatellite marker development
To identify putative microsatellite regions in the 'Ca. L. asiaticus' genome, we used the program 'Tandem Repeats Finder' [36]. Following the identification of these regions, primers were designed (Eurofins-Operon) that flanked the prospective repeat sequence to generate a product of 150-400 base pairs. Over 100 primer sets were tested using multiple DNA samples obtained from HLB-affected plants from India, China, Brazil and Florida. We postulated that polymorphisms, if present, should be observed within this pilot sample due to their geographic separation. Following amplification of regions containing putative microsatellite using the test primers, the products of each reaction were then run on 5% of polyacrylamide gels. Silver staining was then used to visualize polymorphic alleles. This screening procedure identified seven loci with amplified sequence length variability. To facilitate high-throughput genotyping analysis, each of seven forward primers was labelled with a fluorescent dye (Table 1). Amplified products were analyzed by an ABI 3130 xl Genetic Analyser (Applied Biosystems, Foster City, CA).
PCR based genotyping
PCR was performed in 20 μl containing 2 μl of 10× reaction buffer, 1.5 mM MgCl2, 0.2 mM dNTPs, 0.25 U AmpliTaq Gold (Applied Biosystems, Foster City, CA), 2.5 pmole of each of SSR primer pairs and 2 μl of diluted DNA sample. PCR was conducted in the following conditions: 94°C for 4 minutes; 40 cycles consisting of 94°C for 45 seconds, annealing temperature (Table 1) for 45 seconds, and 72°C for 45 seconds; then a final extension at 72°C for 7 minutes. The successes of amplifications were checked running 5 μl of amplified products in agarose gel electrophoresis using 2.5% agarose-TBE gels. For multiplex analysis, 2 μl of each amplified product labeled with different fluorescent dye was pooled. From 8 μl of pooled product, 2.5 μl was mixed with 0.25 μl of GeneScan-500 Liz molecular size standard (Applied Biosystems Cat #4322682A) and 7.25 μl of Hi-Di Formamide (Applied Biosystems Cat. #4311320). The mixture of products was then loaded onto a Genetic Analyzer (Applied Biosystems, Foster City, CA) equipped with the 36 cm 16-capillary array filled with POP-7 polymer (Applied Biosystems, Foster City, CA). Data acquisition and fragment size determinations were carried out by GeneMapper v4.0 software (Applied Biosystems, Foster City, CA).
Genotypes and genetic diversity analysis
Genotypes were identified based on combination of allelic data from multiloucs microsatellite loci. A clone-corrected (removing repeated genotypes within a population) data set was built and used for the analysis of genetic diversity, linkage disequilibrium and genetic structure. GenAlEx Version 6.3 [37] was used to calculate the average number of alleles (Na) and haploid genetic diversity (H) at each locus as well as across all loci for each of the populations.
Linkage disequilibrium analysis
A global test (Fisher's method) implemented in GENEPOP web version 4.0.10 [38] was used to test for the genotyping linkage disequilibrium (LD) between all pair of loci across all samples in this study.
Genetic structure analysis
To determine the genetic relationships of 'Ca. L. asiaticus'isolates, a UPGMA dendrogram was constructed based on Nei's genetic distance [22]. The trees were calculated using POPULATION software package Version 1.2.31 (Olivier Langella, CNRS UPR9034, France found at web: http://bioinformatics.org/~tryphon/populations/) and graphically displayed with MEGA4 software [39]. Confidence in specific clusters of the resulting topology was estimated by bootstrap analysis with 1,000 replicates.
The program STRUCTURE 2.3.1 [40] was also used for a clustering algorithm based on a Bayesian model to assign individual isolate of 'Ca. L. asiaticus' to a specified number of clusters. This algorithm assumes a model in which there are K clusters (where K may be unknown), each of which is characterized by a set of allele frequencies at each locus. No linkage disequilibrium was detected between all pairs of loci across all samples with the clonal corrected data set. Therefore, the program STRUCTURE 2.3.1 [40] was rationally used to estimate the number of clusters (K) within 'Ca. L. asiaticus' where 10 independent runs of K = 1-10 were performed without any prior information as to the origin (location) of individual samples. For each run, a burn-in period of 25,000 iterations was used followed by a run length of 50,000 Markov chain Monte Carlo iterations, and a model with correlated allele frequencies and admixture among populations. The model was run with 10 independent simulations for each K. The number of clusters that best represents the observed data was determined by maximizing the estimated ln likelihood of the data for different values of K, and the ΔK index which is based on the rate of change in the ln likelihood of the data between successive K(1-10) [23]. The optimal probabilities for all individuals were estimated from 10 replicate runs at K = 3 with permutation analysis using CLUMPP version 1.1.2 [41], and the output of genetic clustering was visualized using software DISTRUCT version 1.1 [42].
To provide further insight into the relationships among 'Ca. L. asiaticus', the eBURST v3 program http://eburst.mlst.net/ was employed to identify putative founder types. For this analysis, user- defined group definition was set to include those haplotypes that shared identical genotypes for at least 5 of the 7 loci. The minimum single-locus variant count for subgroup definition was set to 3.
Authors' contributions
HL, MSI, JMG, YPD, HDC, GK and ELC coordinated the study, collected samples and provided preliminary data. HL, JMG, and YB carried out genotyping of HLB samples. MSI, JMG and HL analyzed results and wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
Sample and haplotype information for all isolates used in this study.
Contributor Information
Md-Sajedul Islam, Email: M.Sajed@gmail.com.
Jonathan M Glynn, Email: glynnjon@yahoo.com.
Yang Bai, Email: baictj@yahoo.cn.
Yong-Ping Duan, Email: yongping.duan@ars.usda.gov.
Helvecio D Coletta-Filho, Email: helvecio@centrodecitricultura.br.
Gopal Kuruba, Email: headcrs_tirupati@aphu.edu.in.
Edwin L Civerolo, Email: emciv@comcast.net.
Hong Lin, Email: hong.lin@ars.usda.gov.
Acknowledgements
We would like to thank Chuanwu Chen and Parminder Sahota for technical assistance. We also would like to thank Michael Irey for providing HLB samples from Florida. Funding for this project was provided by the Florida Citrus Production Research Advisory Council. USDA-ARS Project Number: 5302-22000-008-40T. Trade names or commercial products in this publication are mentioned solely for the purpose of providing specific information and does not imply recommendation or endorsement by the United States Department of Agriculture.
References
- Bové JM. Huanglongbing: A destructive, newly-emerging, century-old disease of citrus. J Plant Pathol. 2006;88(1):7–37. [Google Scholar]
- da Graça JV. Citrus greening disease. Annu Rev Phytopathol. 1991;29:109–136. doi: 10.1146/annurev.py.29.090191.000545. [DOI] [Google Scholar]
- Baldwin E, Plotto A, Manthey J, McCollum G, Bai J, Irey M, Cameron R, Luzio G. Effect of Liberibacter infection (huanglongbing disease) of citrus on orange fruit physiology and fruit/fruit juice quality: chemical and physical Analyses. J Agric Food Chem. 2009;58(2):1247–1262. doi: 10.1021/jf9031958. [DOI] [PubMed] [Google Scholar]
- Gottwald TR. Current epidemiological understanding of citrus huanglongbing. Annu Rev Phytopathol. 2010;48:119–139. doi: 10.1146/annurev-phyto-073009-114418. [DOI] [PubMed] [Google Scholar]
- Lin KH. Yellow shoot of citrus in Chinese. Acta Phytopathol Sin. 1956;2:1–12. [Google Scholar]
- Beattie GAC, Holford P, Mabberley DJ, Haigh AM, Broadbent P. On the origins of citrus, huanglongbing, Diaphorina citri and Trioza erytreae. Orlando, Florida, USA: International Conference of Huanglongbing Florida. 2008. pp. 25–57.
- Halbert SE, Manjunath KL. Asian citrus psyllids (Sternorrhyncha: Psyllida) and greening disease of citrus: a literature review and assessment of risk in Florida. Fla Entomol. 2004;87:330–353. doi: 10.1653/0015-4040(2004)087[0330:ACPSPA]2.0.CO;2. [DOI] [Google Scholar]
- Manjunath KL, Halbert SE, Ramadugu C, Webb S, Lee RF. Detection of 'Candidatus Liberibacter asiaticus' in Diaphorina citr and its importance in the management of citrus huanglongbing in Florida. Phytopathology. 2008;98(4):387–396. doi: 10.1094/PHYTO-98-4-0387. [DOI] [PubMed] [Google Scholar]
- Hodges A, Philippakos E, Mulkey D, Spreen T, Muraro R. Economic impact of Florida's citrus industry, 1999-2000. University of Florida IFAS Economic Information Report 01-2. 2001. pp. 1–14.
- Chung KR, Brlansky RH. Citrus diseases exotic to Florida: huanglongbing (citrus greening) Plant Pathology Department, Florida Cooperative Extension Service, Institute of Food and Agricultural Sciences, University of Florida. 2009. http://edis.ifas.ufl.edu PP-201.
- Duan Y, Zhou L, Hall DG, Li W, Doddapaneni H, Lin H, Liu L, Vahling CM, Gabriel DW, Williams KP. et al. Complete genome sequence of citrus huanglongbing bacterium, 'Candidatus Liberibacter asiaticus' Obtained Through Metagenomics. Mol Plant Microbe In. 2009;22(8):1011–1020. doi: 10.1094/MPMI-22-8-1011. [DOI] [PubMed] [Google Scholar]
- Nelson AJ, Elias KS, Arévalo GE, Darlington LC, Bailey BA. Genetic characterization by RAPD analysis of isolates of Fusarium oxysporum f. sp. erythroxyli associated with an emerging epidemic in Peru. Phytopathology. 1997;87(12):1220–1225. doi: 10.1094/PHYTO.1997.87.12.1220. [DOI] [PubMed] [Google Scholar]
- Wickert E, Machado MA, Lemos EGM. Evaluation of the genetic diversity of Xylella fastidiosa strains from citrus and coffee hosts by single-nucleotide polymorphism markers. Phytopathology. 2007;97(12):1543–1549. doi: 10.1094/PHYTO-97-12-1543. [DOI] [PubMed] [Google Scholar]
- Yuan X, Morano L, Bromley R, Spring-Pearson S, Stouthamer R, Nunney L. Multilocus sequence typing of Xylella fastidiosa causing Pierce's disease and oleander leaf scorch in the United States. Phytopathology. 2010;100(6):601–611. doi: 10.1094/PHYTO-100-6-0601. [DOI] [PubMed] [Google Scholar]
- Coletta-Filho HD, Bittleston LS, Almeida RPP. Spatial genetic structure of a vector-borne generalist pathogen. Appl Environ Microb. 2011;77(8):2596–2601. doi: 10.1128/AEM.02172-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes EJ III, Li W, Lewit Y, Ma H, Voelz K, Ren P, Carter DA, Chaturvedi V, Bildfell RJ, May RC. et al. Emergence and pathogenicity of highly virulent Cryptococcus gatti genotypes in the Northwest United States. PLoS Pathog. 2010;6(4):e1000850. doi: 10.1371/journal.ppat.1000850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomimura K, Miyata S, Furuya N, Kubota K, Okuda M, Subandiyah S, Hung TH, Su HJ, Iwanami T. Evaluation of genetic diversity among 'Candidatus Liberibacter asiaticus' isolates collected in Southeast Asia. Phytopathology. 2009;99(9):1062–1069. doi: 10.1094/PHYTO-99-9-1062. [DOI] [PubMed] [Google Scholar]
- Adkar-Purushothama CR, Quaglino F, Casati P, Ramanayaka JG, Bianco PA. Genetic diversity among 'Candidatus Liberibacter asiaticus' isolates based on single nucleotide polymorphisms in 16S rRNA and ribosomal protein genes. Ann Microbiol. 2009;59(4):681–688. doi: 10.1007/BF03179208. [DOI] [Google Scholar]
- Liu R, Zhang P, Pu X, Xing X, Chen J, Deng X. Analysis of a prophage gene frequency revealed population variation of 'Candidatus Liberibacter asiaticus' from two citrus-growing provinces in China. Plant Dis. 2011;95:431–435. doi: 10.1094/PDIS-04-10-0300. [DOI] [PubMed] [Google Scholar]
- Katoh H, Subandiyah S, Tomimura K, Okuda M, Su HJ, Iwanami T. Differentiation of "Candidatus Liberibacter asiaticus" isolates by variable-number tandem-repeat analysis. Appl Environ Microbiol. 2011;77(5):1910–1917. doi: 10.1128/AEM.01571-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Deng X, Sun X, Jones D, Irey M, Civerolo E. Guangdong and Florida populations of 'Candidatus Liberibacter asiaticus' distinguished by a genomic locus with short tandem repeats. Phytopathology. 2010;100(6):567–572. doi: 10.1094/PHYTO-100-6-0567. [DOI] [PubMed] [Google Scholar]
- Nei M, Tajima F, Tateno Y. Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J Mol Evol. 1983;19(2):153–170. doi: 10.1007/BF02300753. [DOI] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. 2005;14(8):2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- Williams DA, Overholt WA, Cuda JP, Hughes CR. Chloroplast and microsatellite DNA diversities reveal the introduction history of Brazilian peppertree (Schinus terebinthifoliu) in Florida. Mol Ecol. 2005;14(12):3643–3656. doi: 10.1111/j.1365-294X.2005.02666.x. [DOI] [PubMed] [Google Scholar]
- da Graça JV. Memorias del Taller Internacional sobre el Huanglongbing y el Psílido asiático de los cítricos. Hermosillo, Sonora. México; 2008. Biology, history and world status of huanglongbing. [Google Scholar]
- Zhao XY. Citrus yellow shoot (huanglongbing) in China: A review. Proceedings International Society of Citriculture. 1981;1:466–469. [Google Scholar]
- Beattie GAC, Mabberley DJ, Holford P, Broadbent P, de Barro P. Huanglongbing: its possible origins, collaborative research in Southeast Asia, and developing incursion management plans for Australia. Proceedings of the 2nd International Citrus Canker & Huanglongbing Research Workshop. 2005. p. 52.
- Capoor SP. Decline of citrus trees in India. Bull Natl Inst Sci India. 1963;24:48–64. [Google Scholar]
- Raychaudhuri SP, Nariani TK, Lele VC. Citrus die-back problem in India. Proceedings of the First International Citrus Symposium. 1969;3:1433–1437. [Google Scholar]
- Halbert SE. Proceedings of the 2nd International Citrus Canker and Huanglongbing Research Workshop. Orlando Florida, USA; 2005. The discovery of huanglongbing in Florida; p. 50. [Google Scholar]
- Bové JM, Garnier M. Citrus greening and psylla vectors of the disease in the Arabian peninsula. Proceedings of the 9th Conference of the International Organization of Citrus Virologist. 1984. pp. 109–114.
- Weinert MP, Jackson SC, Grimshaw JF, Bellis GA, Stephens PM, Gunea T, Kame MF, Davis RI. Detection of huanglongbing (citrus greening disease) in Timor-Leste (East Timor) and in Papua New Guinea. Aust Plant Pathol. 2004;33:135–136. doi: 10.1071/AP03089. [DOI] [Google Scholar]
- Halbert SE, Niblett CL, Manjunath KL, Lee RF, Brown LG. Proceedings of the International Society of Citriculture 9th Congress. Alexandria, Virgina, USA: ASHS Press; 2002. Establishment of two new vectors of citrus pathogens in Florida; pp. 1016–1017. [Google Scholar]
- Brlansky RH, Dewdney MM, Rogers ME. 2011 Florida citrus pest management guide: huanglongbing (citrus greening) Plant Pathology Department, Florida Cooperative Extension Service, Institute of Food and Agricultural Sciences, University of Florida. 2010;SP-43 http://edis.ifas.ufl.edu [Google Scholar]
- Spann TM, Atwood RA, Dewdney MM, Ebel RC, Ehsani R, England G, Futch SH, Gaver T, Hurner T, Oswalt C. et al. IFAS guidance for huanglongbing (greening) management. Horticultural Sciences Department, Florida Cooperative Extension Service, Institute of Food and Agricultural Sciences, University of Florida. 2010;HS1165 http://edis.ifas.ufl.edu [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucl Acids Res. 1999;27(2):573–58. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall R, Smouse P. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes. 2006;6:288–295. doi: 10.1111/j.1471-8286.2005.01155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond M, Rousset F. GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered. 1995;86:248–249. [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software Version 4.0. Mol Biol Evol. 2007;24(8):1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Pritchard J, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA. DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes (2004) 2004;4:137–138. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sample and haplotype information for all isolates used in this study.