

Abstract
Background
Plants respond to external stimuli through fine regulation of gene expression partially ensured by small RNAs. Of these, microRNAs (miRNAs) play a crucial role. They negatively regulate gene expression by targeting the cleavage or translational inhibition of target messenger RNAs (mRNAs). In Hevea brasiliensis, environmental and harvesting stresses are known to affect natural rubber production. This study set out to identify abiotic stress-related miRNAs in Hevea using next-generation sequencing and bioinformatic analysis.
Results
Deep sequencing of small RNAs was carried out on plantlets subjected to severe abiotic stress using the Solexa technique. By combining the LeARN pipeline, data from the Plant microRNA database (PMRD) and Hevea EST sequences, we identified 48 conserved miRNA families already characterized in other plant species, and 10 putatively novel miRNA families. The results showed the most abundant size for miRNAs to be 24 nucleotides, except for seven families. Several MIR genes produced both 20-22 nucleotides and 23-27 nucleotides. The two miRNA class sizes were detected for both conserved and putative novel miRNA families, suggesting their functional duality. The EST databases were scanned with conserved and novel miRNA sequences. MiRNA targets were computationally predicted and analysed. The predicted targets involved in "responses to stimuli" and to "antioxidant" and "transcription activities" are presented.
Conclusions
Deep sequencing of small RNAs combined with transcriptomic data is a powerful tool for identifying conserved and novel miRNAs when the complete genome is not yet available. Our study provided additional information for evolutionary studies and revealed potentially specific regulation of the control of redox status in Hevea.
Keywords: Gene expression, miRNA, MIR gene, Next-generation sequencing, Rubber tree, Transcription, Transcriptome, Abiotic stress, miRNA editing
Background
Hevea brasiliensis, the sole commercial source of natural rubber, is a tropical perennial species native to the Amazon basin. South-East Asian countries supplied 92% of the 10 million tons of natural rubber produced in 2010, but ever-growing worldwide demand calls for enhanced tree productivity. This goal is increasingly becoming a crucial challenge for the corresponding research activities in a context of global climate change and redistribution of land for food crops. Natural rubber is a cis-1,4 polyisoprene polymer biosynthesized in rubber particles located in specialized latex cells. Latex cells are periodically emitted from the cambium and then anastomosed to form latificer mantels [1]. Natural rubber is harvested by tapping. The cytoplasm of laticifers, containing 30-50% of rubber particles, is then expelled by high turgor pressure maintained in the soft bark tissues. An ethylene generator, 2-chloroethylphosphonic acid (ethephon), is applied to the tapping panel to stimulate latex production. Ethephon increases the duration of latex flow after tapping and its regeneration between two tappings [2]. Tapping Panel Dryness (TPD) is a physiological disease that causes 10-40% annual rubber production losses over the 30 years of a rubber cultivation cycle. This physiological disorder, triggered by oxidative stress, is partially induced by excessive environmental and harvesting stresses [3]. The generation of reactive oxygen species (ROS) in latex cells leads to in situ coagulation of rubber particles [4,5]. Research on ROS in plants initially focused on their cytotoxicity only, but nowadays ROS are also considered as signalling molecules [6,7]. A ROS signalling network emerges and is conserved in all aerobic organisms [8]. The ROS-scavenging system acts both to maintain redox homeostasis and to protect cell components from oxidative damage. Biotic and abiotic stress, such as drought, salinity, strong light, temperature, heavy metals, UV radiation, atmospheric contamination, mechanical wounding, nutrient starvation and pathogen attacks are major sources of ROS in plants [9].
MicroRNAs (miRNAs) particularly play a key function in responses to abiotic stress (for a review see [10-13]). Indeed, small RNAs are involved in fine-tuning gene expression in response to physiological, external and developmental stimuli [14,15]. Of these, miRNAs have been shown to be a large group of small endogenous RNAs that exist in animals, plants and viruses. Since their discovery in Caenorrhabditis elegans [16] and in Arabidopsis thaliana in 2002 [17], miRNAs have been described as negatively regulating gene expression by targeting specific messenger RNAs (mRNAs) for cleavage [18,19] or translational inhibition [20]. The biogenesis of miRNAs has been amply described in plants and animals [21-24]. Briefly, a MIR gene is first transcribed to primary miRNAs (pri-miRNA) by RNA polymerase II, the 70-500 bp long pri-miRNA forms an imperfectly paired hairpin which is processed by RNAse III-like DICER-like I (DCL1) to generate miR precursors (pre-miRNAs) [25,26]. Further cleavage of the pre-miRNA by DCLI releases a miRNA/miRNA* duplex. The duplex is then translocated into the cytoplasm by HASTY (plant ortholog to exportin 5 [27]), the canonical mature miRNA of 20-22 nucleotides is selectively incorporated into the RNA-induced silencing complex (RISC) associated with Argonaute 1 (AGO1). In the RISC complex, miRNAs bind to mRNA and inhibit gene expression through perfect or near-perfect complementarity between the miRNA and the mRNA [18]. In plants, the recent discovery of MIR genes generating both canonical miRNA (20-22 nucleotides) and long miRNA (lmiRNA, 23-27 nucleotides) suggests their bifunctionality [28]. The lmiRNAs derived from MIR genes are selectively processed by DCL3, can associate with AGO4 and guide DNA methylation at some of their target loci in trans in rice and moss [28,29]. The recent and major discovery of the role played by post-transcriptional modifications of miRNA precursors highlighted the complexity of miRNA biogenesis and the way miRNAs act [30,31]. Indeed, adding an oligo-uridine tail to the 3' end of the miRNA precursor is a mechanism that controls miRNA production in mice [31]. In plants, it has been reported that either a combination of 5' deletion and 3' uridylation of miRNA, or RNA editing events, alter the targeting and preference of Argonaute association [30]. Several miRNAs have been described as being up-regulated or down-regulated by high salinity, drought and low temperatures [32,33]. Furthermore, the miRNA-targeted gene copper zinc superoxide dismutase (CuZnSOD), which plays a major role in maintaining redox homeostasis, is cleaved by miR398 under stress-free conditions in Arabidopsis [34,35].
Currently, 15,172 mature miRNAs have been discovered and deposited in the public database, miRBase (Release 16, 2010, http://www.mirbase.org) [36,37], and 9,277 mature plant miRNAs in the Plant MicroRNA Database, PMRD http://bioinformatics.cau.edu.cn/PMRD[38]. Most miRNAs have been identified in model species such as Arabidopsis, Oryza [39,40], Populus [12,41], Physcomitrella [42], Vitis [43], whose genomes have been sequenced. Some miRNAs have also been identified in Glycine max [44], Arachis hypogaea [45], Solanum lycopersicum [46], Brassica napus [47], Phaseolus vulgaris [48] and in the euphorbiaceous species Ricinus communis [49]. Twenty-three miRNA families predicted in Ricinus communis were experimentally validated in four euphorbiaceous plants (Ricinus communis, Jatropha curcas, Manihot esculenta and Hevea brasiliensis) [49]. However, the strategy used did not allow exhaustive identification of miRNA families in Hevea.
The availability of large collections of Hevea Expressed Sequence Tags (ESTs; http://bassigny/cgi-bin/esttik_dev/quick_search.cgi) and next-generation sequencing techniques offers prospects for understanding the post-transcriptional regulation of key functions involved in responses to stress and in latex production in Hevea brasiliensis. This study involved the deep sequencing of small RNAs from Hevea plants grown under various conditions. We report here on the identification of 48 conserved miRNA families, sharing very high homology with that already known in other species. MiRNA precursor sequences were identified for nine families. In addition, ten putative novel miRNA families were also identified with their precursors. A scan of the Hevea EST databases with miRNA sequences revealed their putative targets. Of them, the predicted targets involved in responses to stimuli, the ROS-scavenging systems and transcription regulation are presented, and new insights into the control of redox status in Hevea are proposed.
Results
Classification of small RNAs
A total of 4,223,792 raw reads was generated by Solexa sequencing from the small RNA library prepared from pooled juvenile and mature plant materials subjected to various types of abiotic stress (Table 1 and Table 2). After removing adapters, cleaned reads amounted to 2,378,135 sequences corresponding to 670,645 unique sequences. The sequence lengths ranged from 17 to 32 nucleotides. Although some small RNAs were present about a thousand times in our dataset, most were sequenced only a few times. Small RNAs consisting of only one or two reads accounted for 32% and 53% of clean sequences, respectively.
Table 1.
Statistics of small RNA sequences from the Hevea brasiliensis small RNA library
| Stress small RNA library | Sequence number | Analysis |
|---|---|---|
| read sequences | 4,223,792 | |
| clean sequences | 2,378,135 | Solexa sequencing |
| unique clean sequences of small RNA | 670,645 | |
| no hit | 629,795 | |
| hit, unknown | 26,721 | |
| coding mRNA | 6,618 | |
| transposons/transposable element | 6,800 | |
| rRNA | 399 | Blast against Arabidopsis |
| pseudo mRNA | 138 | genome |
| ncRNA | 73 | |
| miRNA | 57 | |
| tRNA | 39 | |
| sn/snoRNA | 5 | |
| no hit | 652,215 | |
| hit | 18,430 | Blast against PMRD |
| conserved miRNA family | 48 | |
| putative novel miRNA family | 10 | LeARN pipeline |
Table 2.
List of the treatments applied to Hevea plants before small RNA isolation
| Treatments | Plant material | Tissue | Condition |
|---|---|---|---|
| Ethylene | in vitro plantlet (2 years old) | leaf, bark, root | 5 ppm for 4 hours |
| budded plant | leaf, bark | ||
| Methyl-jasmonate | in vitro plantlet (2 years old) | leaf, bark, root | 0.3 μM for 4 hours |
| budded plant | leaf, bark | ||
| Wounding | in vitro plantlet (2 years old) | leaf, bark, root | 2 hours |
| budded plant | leaf, bark | ||
| Drought | in vitro plantlet (2 years old) | leaf, bark, root | 10 days |
| budded plant | leaf, bark | 4 weeks | |
| Flooding | in vitro plantlet (2 years old) | leaf, bark, root | 4 days |
| budded plant | leaf, bark | ||
| Cold | in vitro plantlet (2 years old) | leaf, bark, root | 4°C for 8 hours |
| budded plant | leaf, bark | ||
| Light | in vitro plantlet (2 years old) | leaf, bark, root | PAR: 1000-1500 μmol/m2/s for |
| budded plant | leaf, bark | 4 hours | |
| NaCl | in vitro plantlet (2 years old) | leaf, bark, root | 300 mM for 4 days |
| budded plant | leaf, bark | ||
| in vitro plantlet (2 years old) | leaf, bark, root | ||
| Control | budded plant | leaf, bark | |
| in vitro plantlet (1 month old) | leaf, bark, root | ||
| callus | callus | ||
Annotation of the Hevea small RNAs was attempted by BLASTN on the Arabidopsis genome. Most of the sequences did not map onto the Arabidopsis genome sequence. Of the mapped sequences, 26,721 were not annotated. Annotated sequences predicted 6,618 products of mRNA degradation, 6,800 transposable elements, 399 rRNAs, 138 products of pseudogene degradation, 73 ncRNAs, 39 tRNAs, 5 sn/snoRNAs and 57 miRNAs corresponding to 14 families.
Further mapping was carried out with all the miRNA sequences available in the PMRD database. In all, 18,430 sequences matched (0 or 1 mismatch) to conserved miRNAs in other species. The accessions were classified into 48 miRNA families (Table 1). Since most sequences did not map against PMRD, the Hevea EST database was scanned using the LeARN pipeline. This strategy led to the identification of 10 additional miRNA families, based on the stem-loop structure, which were not described in other species (Table 1). The distribution of the 670,645 unique accessions showed that the most abundant accessions had in descending order 27, 19, 24, 26 and 17 nucleotides (Figure 1). Accessions with 17 and 19 nucleotides came from products of mRNA degradation and transposable elements according to the mapping results against the Arabidopsis genome sequence (data not shown).
Figure 1.

Length distribution of unique accessions in the small RNA dataset in Hevea brasiliensis.
Characteristics of conserved and putative novel miRNA families in Hevea brasiliensis
Reads of conserved and putative novel miRNAs were distributed according to their nucleotide lengths (Figure 2). The 48 conserved miRNA families mostly had 24 and 23 nucleotides (Figure 2A). For the 10 putative novel miRNA families, the most represented lengths were 24, 27 and 26 nucleotides (Figure 2B).
Figure 2.
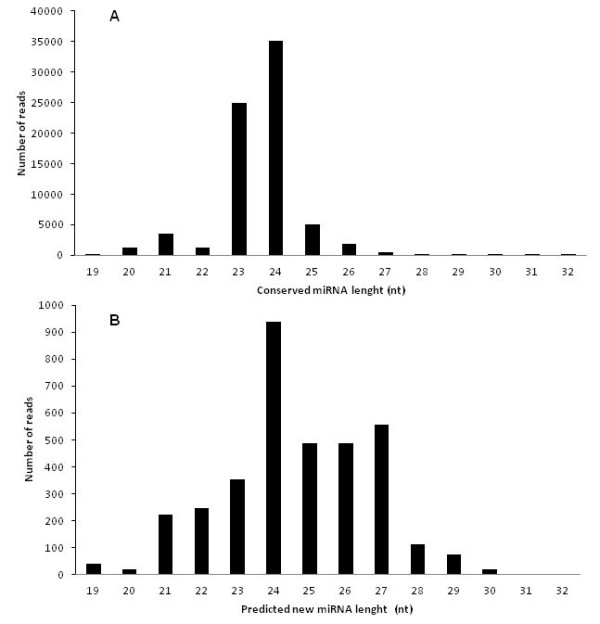
Length distribution of reads matching miRNA families in Hevea brasiliensis. (A) conserved miRNA (B) putatively novel families.
Of the 48 conserved miRNA families, 12 were predominant with more than 1,000 reads, including 3 families that displayed more than 20,000 reads. The latter corresponded to HbmiR159/319 and HbmiR408 (Figure 3A). By contrast, 36 families were less represented in the dataset with 18 families having fewer than 100 reads: HbmiR1310, HbmiR162, HbmiR168, HbmiR1863, HbmiR2118, HbmiR2910, HbmiR2914, HbmiR2915, HbmiR2916, HbmiR393, HbmiR394, HbmiR395, HbmiR399, HbmiR444, HbmiR476, HbmiR482, HbmiR828 and HbmiR845 (Figure 3B).
Figure 3.
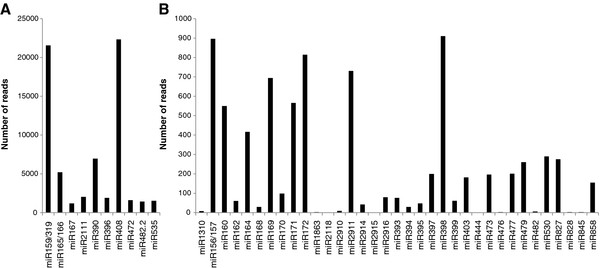
Abundance of the 48 conserved miRNA families in Hevea brasiliensis with 0 and 1 mismatch on a minimum length of 19 nucleotides. A) The sequencing frequency of a conserved miRNA family in the small RNA library above 1000 reads. B) The sequencing frequency of a conserved miRNA family in the small RNA library below 1000 reads.
A detailed analysis of length distribution within the Hevea miRNA families revealed some discrepancies (Figure 4 and Figure 5). Firstly, the 20-22 nucleotide and 23-27 nucleotide size classes were detected for both conserved and novel miRNA families. Secondly, the most abundant size for miRNAs was 21-24 nucleotides, except for HbmiR169 (27 nucleotides), HbmiR2911 (26 nucleotides), HbmiR482 (25 nucleotides), HbmiR472 (25 nucleotides), HbmiRn8 (25 nucleotides) and HbmiRn9 (27 nucleotides). Thirdly, the putative novel miRNAs occurred at low levels with fewer than 500 reads, and at extremely low levels for HbmiRn1 and HbmiRn2, with only three sequences (Figure 5).
Figure 4.

Number of reads according to the length distribution of accessions of conserved miRNA families in Hevea brasiliensis.
Figure 5.
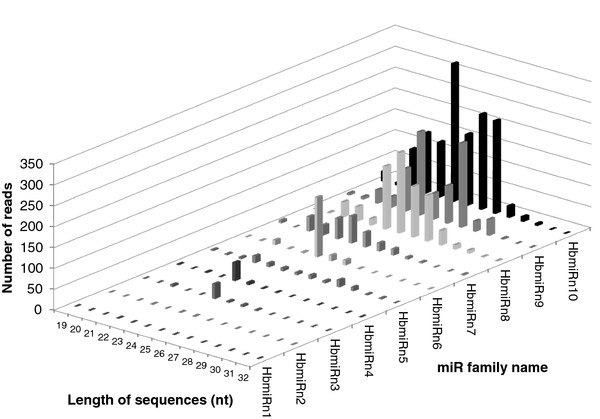
Number of reads according to the length distribution of accessions of putative novel miRNA families in Hevea brasiliensis.
Detection of single-nucleotide mutations in miRNA sequences
For all the conserved miRNA families, single-nucleotide modifications in the sequences were analysed in detail based on the data presented in [30]. In Arabidopsis, seventeen families are concerned by RNA editing events and nine families by 5' deletion and 3' uridylation modifications (called below -1 + UU modification). In Hevea, of the seventeen families, four families displayed single-nucleotide mutations at the sites of post-transcriptional RNA modifications described in Arabidopsis, namely AtmiR156, AtmiR159a, AtmiR164a, and AtmiR172a (Table 3). By contrast, no -1 + UU modification was observed in Hevea (Table 3).
Table 3.
Frequency of single-nucleotide mutation and of a combination of 5' deletion and 3' uridylation (-1 + UU) for four miRNA families from Hevea
| Family name in Arabidopsis | Single-nucleotide modification | -1 + UU modification | ||
|---|---|---|---|---|
| Arabidopsis | Hevea | Arabidopsis | Hevea | |
| AtmiR156 | G-A: 14.091% | A-U: 49.382% A-C: 3.571% |
yes | no |
| G-A: 10.407% | G-A: 2.127% | |||
| AtmiR159a | G-A: 8.597% | G-A: 4.166% | no | no |
| G-A: 5.882% | G-C: 2.127% G-U: 1.398% |
|||
| AtmiR164a | G-A: 3.967% | G-A: 7.692% | no | no |
| AtmiR172a | G-U: 0.599% | G-U: 6.153% A-U: 7.692% C-U: 7.692% |
no | no |
Sites of post-transcriptional modification identified in Arabidopsis [50] were analysed in the Hevea small RNA dataset. Only sequences present at > 0.5% are listed
Identification of precursor transcripts for conserved and putative novel miRNA families from the hevea brasiliensis transcriptome
Conserved and putative novel miRNA precursors were sought in the Hevea clone PB260 transcript sequence databases obtained from various organs using the LeARN pipeline [51]. Eight conserved miRNA families that mapped against RNA sequences displayed a stem-loop structure (Table 4 and Additional Table 1). A single miRNA precursor transcript was found for the HbmiR156, HbmiR159, HbmiR396, HbmiR476 and HbmiR2910 families. Two miRNA precursor transcripts were identified for the HbmiR166, HbmiR319 and HbmiR408 families. In addition, the LeARN pipeline predicted 10 putative novel miRNAs with their unique precursors (Table 5 and Additional Table 2). Mapping the sequences of the small RNA dataset on the precursor sequences led to the identification of miRNA*for all eight conserved families (HbmiR156, HbmiR159, HbmiR166, HbmiR319, HbmiR396, HbmiR408, HbmiR476 and HbmiR2910) and for the five putatively new families HbmiRn3, HbmiRn4, HbmiRn8, HbmiRn9 and HbmiRn10. Stem-loop reverse transcription polymerase chain reactions (RT-PCR) were successfully performed on seven precursors for the conserved families and on nine for the putatively new families (Figure 6).
Table 4.
List of precursors for conserved microRNAs identified in the Hevea clone PB260 transcriptome sequences
| New miR name | Accession Name | Mature sequence | Mature length | miRNA* (Accession No.) | Precursor length | EST |
|---|---|---|---|---|---|---|
| HbmiRn1 | acc_584359 | CAGGAACUGGUAUCAACCCAGC | 22 | nd | 130 | CL1Contig1853_r1 |
| HbmiRn2 | acc_495109 | UAUUGUAGAAAUUUUCAGGAUC | 22 | nd | 116 | CL1Contig490_r1 |
| HbmiRn3 | acc_185377 | UAAUGGGCUCUGCAUAGAUGG | 21 | Yes (3) | 108 | CL27Contig1_r1 |
| HbmiRn4 | acc_108425 | UUGCAUAUCUCAGGAGCUUCA | 21 | Yes (4) | 214 | CL19922Contig1_r1 |
| HbmiRn5 | acc_644211 | AAACGGCUACCACAUCCAA | 19 | nd | 87 | FYGXE6I01B755W_r1 |
| HbmiRn6 | acc_501419 | UAGGAUGUAGAAGAGCAUAA | 20 | nd | 90 | F5VNCTM02H9XL3_r1 |
| HbmiRn7 | acc_35715 | UAGUUUGUUUGAUGGUAUC | 19 | nd | 113 | FYGXE6I01EKFYA_r1 |
| HbmiRn8 | acc_246127 | GAUUGACAGACUGAGAGCUC | 20 | Yes (16) | 93 | FYGXE6I01B7GPL_r1 |
| HbmiRn9 | acc_103082 | UUUAUGAAAGACGAACAACUG | 21 | Yes (6) | 87 | CL1Contig2798_r1 |
The presence of the miRNA* sequence in the small RNA dataset is indicated. Pictures of the stem-loop structures are presented in Additional Table 1.
Table 5.
List of putatively new miRNA precursors identified in the Hevea clone PB260 transcriptome sequences
| MiRNA name | Accession Name | Mature sequence | Mature length | miRNA* (Accession No.) | Precursor length | EST |
|---|---|---|---|---|---|---|
| Hbmir156 | acc_462135 | UUGACAGAAGAUAGAGAGC | 19 | Yes (10) | 119 | FYGXE6I01DJH4X_r1 |
| Hbmir159 | acc_17403 | UUUGGAUUGAAGGGAGCUCUA | 21 | Yes (5) | 221 | GETTCZF01AWUSL_r1 |
| Hbmir166 | acc_28021 | UCGGACCAGGCUUCAUUCC | 19 | Yes (14) | 127 | CL2671Contig1_r1 |
| acc_150486 | UCGGACCAGGCUUCAUUCCCCC | 22 | Yes (14) | 120 | CL1Contig15359_r1 | |
| Hbmir319 | acc_19786 | UUGGACUGAAGGGAGCUCCCU | 21 | Yes (7) | 221 | FRZQES201D34W3_r1 |
| Hbmir396 | acc_112787 | UUCCACAGCUUUCUUGAACUG | 21 | Yes (15) | 154 | CL1Contig13811_r1 |
| Hbmir408 | acc_393516 | AAGACUGGGAACAGGCAGAGCA | 22 | Yes (294) | 128 | FYGXE6I01CVDV9_r1 |
| acc_39581 | ACUGGGAACAGGCAGAGCAUGA | 22 | Yes (294) | 120 | CL3908contig1_r1 | |
| Hbmir476 | acc_508145 | UAAUCCUUCUUUGCAAAGUC | 20 | Yes (1) | 126 | CL1Contig11471_r1 |
| Hbmir2910 | acc_251816 | GAGCGAUUUGUCUGGUUAAUC | 21 | Yes (5) | 126 | FYGXE6I01EDXGX_r1 |
The presence of the miRNA* sequence in the small RNA dataset is indicated (nd: not detected). Pictures of the stem-loop structures are presented in Additional Table 2.
Figure 6.
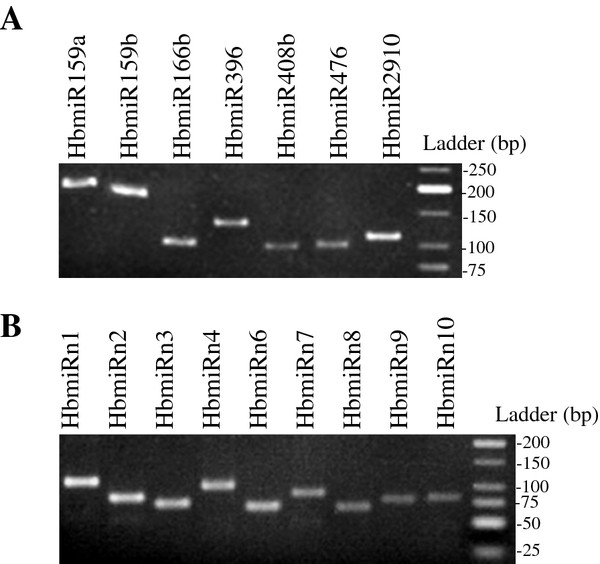
Gel electrophoresis of stem-loop RT-PCR. A) Stem-loop RT-PCR from seven precursors of conserved miRNA family. B) Stem-loop RT-PCR from nine precursors of putatively new miRNAs.
Comparison of the gene ontology classification of predicted targets for conserved and putative novel miRNA families
Computational prediction of miRNA targets led to the identification of 1,083 sequences for the 48 conserved families and 705 for the 10 putative novel miRNA families. Their involvement in biological processes and their molecular functions were attributed using Gene Ontology (Figure 7A, B, C and 7D). Most of the GO terms were represented in the same proportions for predicted targets from conserved and putative novel miRNA families. Target genes of the putative novel miRNA families all had GO terms of conserved miRNA families for biological processes and molecular functions. In biological process terms, the putative novel miRNA families had two additional terms (growth, cell wall organization or biogenesis). The number of miRNA target genes was smaller for putative novel than for conserved miRNAs for some GO terms: cellular process (25% as opposed to 27%), metabolic process (22% as opposed to 27%) and biological regulation (10% as opposed to 12%) (Figures 7A and 7C). This number was larger for putative novel miRNA targets compared to conserved targets for cellular component organization (5% as opposed to 3%), localization (7% as opposed to 4%) and response to stimulus (10% as opposed to 8%) (Figures 7A, B, C and 7D). As regards the GO terms for molecular function, although the distribution for antioxidant activity was the same for the 2 classes of miRNAs (1%), a decrease in the proportion of transcription regulator activity (4% as opposed to 7%) and an increase in structural molecule activity (4% as opposed to 2%) and transporter activity (7% as opposed to 4%) were observed for the putative novel miRNA families (Figures 7B and 7D).
Figure 7.
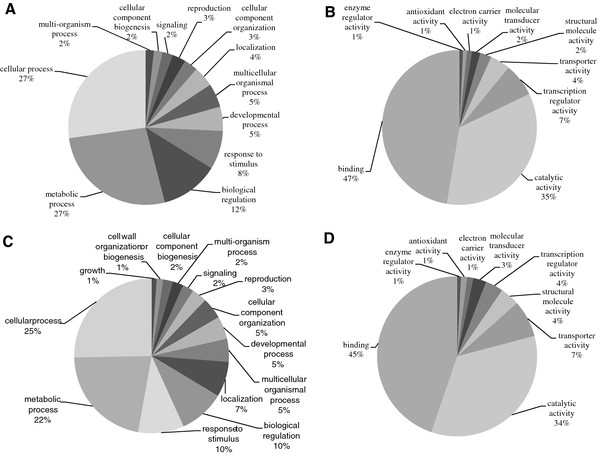
Pie chart representation of Gene Ontology terms at level 2 for putative targets of conserved (A and B) and novel miRNAs (C and D). A and C) Gene Ontology terms for biological processes, B and D) Gene Ontology terms for molecular functions.
Comparison of predicted target genes for conserved and putative novel miRNA families involved in "response to stimulus", and in "antioxidant" and "transcription regulation" activities
As the management of oxidative stress is crucial for rubber productivity, we set out to more effectively understand the mechanisms of redox homeostasis. We therefore gave priority to predicted miRNA-targeted genes involved in the "response to stimulus" (biological process) and in "antioxidant" and "transcription regulator" activities (molecular process) according to the GO annotations (Tables 6 and 7).
Table 6.
List of predicted targets for conserved miRNAs involved in response to stimulus, and in antioxidant and transcription regulator activities according to GO terms
| MiRNA family | GO term | Predicted target function | mfe kcal/mol | Target name |
|---|---|---|---|---|
| Transcription regulation activity | Squamosa promoter-binding protein | -45.50 | CL2120Contig2 | |
| HbmiR156/157 | APETALA2-like protein | -24.41 | hevea_454_rep_c24306 | |
| Antioxidant activity | flavonoid 3',5'-hydroxylase | -26.53 | CL2495Contig2 | |
| AKR (aldo keto reductase) | -32.90 | hevea_454_rep_c15059 | ||
| Transcription regulation activity | APETALA2-like protein | -27.80 | hevea_454_rep_c17780 | |
| sarcosine oxidase | -29.70 | CL11607Contig1 | ||
| serine/threonine protein kinase | -21.39 | CL11Contig15 | ||
| cinnamoyl-CoA reductase | -32.88 | CL1Contig7212 | ||
| CuZnSOD peroxysomal | -32.14 | CL1Contig3818 | ||
| HbmiR159 | Antioxidant activity | ABC transporter C family | -28.02 | CL1Contig3612 |
| cytochrome b6f complex | -36.18 | CL1Contig475 | ||
| TATA box binding protein | -29.38 | CL1Contig760 | ||
| oxidoreductase family protein | -32.88 | CL781Contig2 | ||
| ATP Synthase | -27.31 | CL1613Contig1 | ||
| HMG-CoA_reductase (HMGR) | -36.39 | CL8048Contig1 | ||
| 6-phosphogluconate dehydrogenase family protein | -33.44 | CL1Contig1588 | ||
| HbmiR319 | Antioxidant activity | ferritin putative | -35.86 | CL1Contig17258 |
| HbmiR160 | Transcription regulation activity | ARF | -52.20 | CL6582Contig1 |
| Antioxidant activity | FAD/NAD(P)-binding oxidoreductase-like protein | -23.10 | hevea_454_rep_c15774 | |
| HbmiR162 | ABC transporter C family | -26.47 | hevea_454_rep_c28843 | |
| Response to stimulus | zinc finger family protein | -31.40 | hevea_454_rep_c20758 | |
| HbmiR164 | Transcription regulation activity | NAC-domain protein | -44.12 | hevea_454_rep_c10543 |
| amine oxidase | -32.67 | CL21795Contig1 | ||
| Transcription regulation activity | NAC-domain protein | -32.43 | CL7990Contig1 | |
| PHAVOLUTA-like HD-ZIPIII protein | -46.68 | hevea_454_rep_c12680 | ||
| HbmiR165/166 | Antioxidant activity | LRR protein | -30.08 | hevea_454_c28164 |
| Antioxidant activity/Response to stimulus | malate deshydrogenase | -38.95 | hevea_454_rep_c94776 | |
| Response to stimulus | abscisic acid insensitive protein | -36.33 | hevea_454_rep_c69184 | |
| Hbmir167 | Response to stimulus | cap binding protein | -33.60 | hevea_454_c94262 |
| Transcription regulation activity | CCAAT-binding transcription factor | -32.79 | CL435Contig5 | |
| HbmiR169 | Laccase | -26.51 | CL1Contig13876 | |
| Antioxidant activity | aldehyde dehydrogenase | -26.56 | CL1Contig16398 | |
| annexin-like protein | -32.71 | CL1Contig2060 | ||
| HbmiR170 | Transcription regulation activity | GRAS domain-containing protein | -37.90 | hevea_454_rep_c11063 |
| Transcription regulation activity | GRAS domain-containing protein | -39.50 | CL3142Contig4 | |
| HbmiR171 | ribonucleoside-diphosphate reductase | -26.99 | CL1031Contig2 | |
| Antioxidant activity | oxophytodienoate reductase (OPR) | -27.23 | CL3790Contig2 | |
| glyceraldehyde-3-phosphate dehydrogenase | -24.88 | CL150Contig1 | ||
| Transcription regulation activity | APETALA2-like protein | -24.25 | hevea_454_rep_c45080 | |
| HD-ZIP protein | -33.63 | hevea_454_rep_c13497 | ||
| HbmiR172 | Antioxidant activity | NAD(P)H-quinone oxidoreductase | -24.54 | hevea_454_rep_c13256 |
| CLAVATA1 putative | -31.73 | hevea_454_rep_c2026 | ||
| Squalene monooxygenase | -29.04 | hevea_454_rep_c32943 | ||
| Response to stimulus | syntaxin | -31.00 | hevea_454_c68550 | |
| aspartate aminotransferase | -33.44 | hevea_454_rep_c4007 | ||
| HbmiR2111 | Antioxidant activity | laccase | -26.57 | hevea_454_rep_c5912 |
| HbmiR2910 | Antioxidant activity | 2OG-Fe(II) -dependent-oxygenase-like protein | -30.83 | hevea_454_rep_c1596 |
| ketol-acid reductoisomerase | -31.31 | hevea_454_rep_c5152 | ||
| HbmiR2914 | Antioxidant activity | FAD Binding domain-containing protein | -58.64 | hevea_454_rep_c15793 |
| Rboh | -31.88 | hevea_454_rep_c23550 | ||
| Transcription regulation activity | Heat shock protein | -30.89 | CL11591Contig1 | |
| Hbmir390 | WRKY transcription factor | -33.09 | CL338Contig4 | |
| Antioxidant activity | Phosphoenolpyruvate carboxylase | -19.81 | CL362Contig2 | |
| hydroxy-acid oxidase | -23.58 | CL8468Contig1 | ||
| HbmiR393 | Transcription regulation activity | APETALA2-like protein | -22.79 | hevea_454_c60993 |
| Transcription regulation activity | APETALA2-like protein | -24.47 | hevea_454_c37716 | |
| Hbmir395 | Antioxidant activity | 2OG-Fe(II) -dependent-oxygenase-like protein | -30.00 | hevea_454_rep_c21129 |
| Transcription regulation activity | APETALA2-like protein | -22.40 | hevea_454_rep_c13430 | |
| Transparent TESTA protein | -34.16 | hevea_454_rep_c14652 | ||
| GRAS domain-containing protein | -27.01 | hevea_454_rep_c8477 | ||
| HbmiR396 | Antioxidant activity | zinc finger family protein | -31.84 | hevea_454_c64598 |
| Lignin-forming anionic peroxidase | -26.08 | hevea_454_rep_c20896 | ||
| Antioxidant activity/Response to stimulus | Glutamyl-tRNA reductase | -35.76 | hevea_454_rep_c14169 | |
| fatty acyl-coenzyme A reductases (FAR) | -25.41 | hevea_454_rep_c16435 | ||
| cysteine protease | -33.18 | CL1Contig374 | ||
| HbmiR397 | Antioxidant activity | laccase | -40.97 | hevea_454_c73766 |
| Antioxidant activity | 4-coumarate-coa ligase | -37.60 | hevea_454_rep_c6270 | |
| HbmiR398 | CuZnSOD chloroplastique | -37.30 | CL4308Contig2 | |
| Response to stimulus | HVA22-like protein | -36.03 | hevea_454_rep_c27557 | |
| Transcription regulation activity | APETALA2-like protein | -28.43 | hevea_454_rep_c64305 | |
| serine/threonine protein kinase | -32.93 | CL23251Contig1 | ||
| Heat shock protein | -26.98 | CL530Contig7 | ||
| cytochrome P450 | -37.63 | CL1Contig8196 | ||
| HbmiR408 | enolase | -42.49 | CL5843Contig1 | |
| Antioxidant activity | ribonucleoside-diphosphate reductase | -37.34 | CL1031Contig6 | |
| thioredoxin reductase | -32.80 | CL1401Contig3 | ||
| gibberellin 20-oxidase | -37.63 | CL1Contig12612 | ||
| 4-hydroxy-3-methylbut-2-enyl diphosphate reductase | -30.00 | CL1Contig6885 | ||
| HbmiR444 | Transcription regulation activity | WRKY | -36.82 | hevea_454_rep_c7550 |
| Heat shock protein | -29.45 | CL2547Contig3 | ||
| HbmiR476 | Antioxidant activity | Rboh | -27.76 | CL1Contig1292 |
| cationic peroxidase 2 precursor | -31.63 | CL257Contig2 | ||
| HbmiR535 | Antioxidant activity | LRR protein | -29.18 | hevea_454_c18080 |
| HbmiR827 | Response to stimulus | fructose-bisphosphate aldolase | -26.90 | hevea_454_rep_c59114 |
| HbmiR828 | Transcription regulation activity | MYB class transcription factor | -26.76 | hevea_454_rep_c48615 |
Table 7.
List of predicted targets for putative novel miRNAs involved in response to stimulus, and in antioxidant and transcription regulator activities according to GO terms
| miRNA family | Accession number | GO term | Predicted target function | mfe kcal/mol | Target name |
|---|---|---|---|---|---|
| HbmiRn1 | Acc_584359 | Response to stimulus | Aquaporin like protein | -47.70 | CL1Contig2288 |
| Transcription regulation activity | Helicase like protein | -31.10 | CL18570Contig1 | ||
| HbmiRn2 | Acc_495109 | Response to stimulus | glycogen synthase kinase | -21.10 | CL1Contig2247 |
| Transcription regulation activity | TAZ zinc finger | -22.40 | CL5132Contig2 | ||
| HbmiRn3 | Acc_185377 | Transcription regulation activity | MYC | -27.14 | CL1Contig2024 |
| HbmiRn4 | Acc_108425 | Transcription regulation activity | bZIP domain trnscription factor | -29.10 | CL11387Contig1 |
| Peroxidase | -28.30 | CL1Contig15444 | |||
| Response to stimulus | gamma-glutamylcysteine synthetase | -27.80 | CL786Contig2 | ||
| HbmiRn5 | Acc_644211 | CTR1 | -29.18 | CL3295Contig2 | |
| Transcription regulation activity | Auxin-responsive protein | -29.20 | CL16079Contig1 | ||
| GRAS family protein | -33.17 | CL2606Contig1 | |||
| HbmiRn6 | Acc_501419 | Response to stimulus | ubiquitin fusion protein | -37.74 | CL1Contig4048 |
| Response to stimulus | heat shock protein | -27.50 | CL4213Contig3 | ||
| heat shock protein | -23.79 | CL1Contig6482 | |||
| glycerol kinase | -26.20 | CL6130Contig1 | |||
| HbmiRn7 | Acc_35715 | glycine-rich RNA-binding protein | -22.90 | CL2515Contig2 | |
| ubiquitin-protein ligase | -21.10 | CL7863Contig1 | |||
| Transcription regulation activity | NAC domain protein | -27.06 | CL678Contig1 | ||
| LRR receptor-like protein kinase | -26.16 | CL6991Contig1 | |||
| HbmiRn8 | Acc_246127 | Response to stimulus | SAUR family protein | -27.40 | CL3428Contig1 |
| HbmiRn9 | Acc_103082 | Response to stimulus | Peroxidase | -35.30 | CL1Contig7525 |
| HbmiRn10 | Acc_482742 | Response to stimulus | calcium-dependent protein kinase | -23.30 | CL1Contig9241 |
| longevity assurance factor | -22.15 | CL8027Contig1 | |||
Twelve members of the AP2/ERF domain-containing transcription factor family were putatively targeted by HbmiR156, HbmiR159, HbmiR172, HbmiR393, HbmiR395, HbmiR396, and HbmiR408. Two HD-ZIP III domain-containing protein members (HbmiR165/166, HbmiR172), a transcription factor with a bZIP domain (HbmiRn4), three NAC domain-containing proteins (HbmiR164, HbmiR165 and HbmiRn7), three Squamosa promoter binding proteins (HbmiR156/157), four WRKY transcription factors (HbmiR156/157, HbmiR390, HbmiR444), four Auxin Response Factor (ARF); HbmiR160 and HbmiRn5) and five GRAS family transcription factors (HbmiR170, HbmiR171, HbmiR396 and HbmiRn5) were also potentially targeted.
Sequences encoding protein involved in protein degradation, such as cysteine protease (HbmiR396) and ubiquitin protein ligase (HbmiRn6 and HbmiRn7), were identified for genes classified in the "response to stimulus" category.
Three sequences related to ABA signalling were also identified: an ABA-insensitive protein (HbmiR166, and HbmiR2910), and an HVA22-like protein (HbmiR398). In addition, one sequence encoding CTR1, a negative regulator of the ethylene signalling pathway, was targeted by HbmiRn5. MiRNA biogenesis was also highlighted by the presence of a cap binding protein predicted to be targeted by HbmiR167. Interestingly, two enzymes involved in natural rubber biosynthesis, HMG-CoA reductase and 4-hydroxyl-3-methylbut-2-enyl diphosphate reductase, were predicted to be targeted by HbmiR159 and HbmiR444, respectively. For the "antioxidant activity" category, several ROS-scavenging enzymes were identified. The chloroplastic and peroxisomal isoforms of CuZnSOD were putatively targeted by HbmiR398 and HbmiR159, respectively. Another enzyme involved in glutathione biosynthesis, gamma glutamyl cysteine ligase (GCL), was predicted to be targeted by HbmiRn5. Rboh (or NADPH oxidase) is an enzyme involved in ROS production, which is likely to be targeted by two miRNAs (HbmiR2914 and HbmiR476). In addition, targets that are subjected to or are involved in the redox status of proteins, such as genes encoding proteins involved in lignin synthesis, cynnamoyl CoA reductase, lignin forming anionic peroxidase precursor and 4-coumarate:CoA ligase, were also found to be targeted by HbmiR159, HbmiR408 and HbmiR398, respectively.
The cleavage site for three targets has been experimentally validated for a chloroplastic CuZnSOD, a Squamosa promoter binding protein and an ARF (Table 8). For these targets, the cleavage site is located at the canonical 10th nucleotide.
Table 8.
List of target genes experimentally cleaved by miRNA
| miRNA family |
Target name |
Hevea sequence name |
miRNA accession | mfe kcal/mol | Alignment | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Hbmir156 | Squamosa promoter binding protein (HbSquamosa) | CL2120Contig2 | acc_480780 | -45.5 |
miRNA Target |
23 766 |
CUACACGAGAGAGAGAAGACAGU : ::::::::::::::::::::: GUUGUGCUCUCUCUCUUCUGUCA ▲ 8/14 |
1 788 |
|||
| Hbmir160 | Auxin Response Factor (HbARF) | CL6582Contig1 | acc_370 | -52.2 |
miRNA Target |
21 524 |
ACCGUAUGUCCCUCGGUCCGU ::::::.:::::::::::::: UGGCAUGCAGGGAGCCAGGCA ▲ 27/30 |
1 544 |
|||
| HbmiR398 | Chloroplastic copper zinc superoxide dismutase (HbCuZnSODchl) |
CL4308Contig2 | acc_420 | -37.3 |
miRNA Target |
23 456 |
UAGU-C-CCCGCUGGACUCU-UGUGU .::: : :::.::::::.:: :::.: GUCAUGCGGGUGACCUGGGAAACAUA ▲ 6/14 |
1 480 |
|||
The number of sequences presenting the same cleavage site in indicated below the cleavage site located at the 10th nucleotide (arrow)
Discussion
Size distribution of small RNAs in Hevea may differ from others species
Next-generation high-throughput sequencing techniques are powerful tools for miRNA identification. The sequencing and analysis of the Hevea small RNA library generated more than 2 million sequences. In their vast majority (86%), the sequenced small RNAs were only represented by one or two reads. Similarly, 65% of unique miRNA sequences were found in Arabidopsis by 454 sequencing techniques [52]. Interestingly, we found that the size distribution of Hevea small RNAs differed from other species. The most abundant small RNAs had 17, 19 and 27 nucleotides in Hevea whereas they have 21 and 24 nucleotides in other species. Indeed, in Arabidopsis thaliana [52], Arachis hypogea [45] and Oryza sativa [53], the fraction of 24 nucleotides has proved to be predominant, accounting for 60%, 45% and 36% of unique small RNAs, respectively. The predominant size has been found to be 21 nucleotides in Pinus contorta [53] and Taxus chiniensis [54]. In Hevea, the high percentage of 17- and 19-nucleotide small RNAs resulted from the increased amount of RNA degradation products and transposable elements in response to abiotic treatments according to the mapping results against the Arabidopsis genome (data not shown), as previously observed in Brachypodium [55]. Indeed, a change in small RNA size distribution has been reported in Brachypodium in response to cold treatment. Under normal growth conditions, the 21- and 24-nucleotide classes were the most abundant small RNA families, whereas it was the 19-nucleotide class after cold treatment [55]. Our dataset was generated from pooled small RNAs of various tissues subjected to several types of abiotic stress. The size distribution may have been differentially affected by each type of abiotic stress and may explain the difference in size distribution in Hevea.
Long conserved and novel miRNAs of 23-27 nucleotides consisted of short miRNA sequences with an extension. This lmiRNA structure suggested that they were generated from the same MIR genes. Our observation suggests dual functions for 14 canonical MIR genes producing 20-22 nt and 23-27 nt lmiRNA in Hevea, namely HbMIR169, HbMIR2911, HbMIR482, HbMIR472, HbMIR408 and HbMIRn3-10. Across the plant kingdom, lmiRNA were previously classed as heterochromatic small interfering RNAs (hc-siRNA) [28]. In Arabidopsis, the hc-siRNA derived from the miRNA-generating site is dependent on DCL3, RDR2 and PolIV associated with AGO4 and direct DNA methylation for some target sites [28]. As regards the higher number of reads for the 24 nucleotide compared with the 20-22 nucleotide miRNAs, the 14 miRNA families producing 20-22 nt and 23-27 nt miRNAs might mostly regulate their targets through direct DNA methylation in response to abiotic stress. This may confirm the results of a recent study showing the existence of site-specific abiotic stress-induced DNA methylation for five genes in Hevea [56].
Ancient and recently differentiated Hevea brasiliensis miRNAs provide additional information for evolutionary studies
The 48 Hevea brasiliensis conserved miRNA families were more or less similar to those of other species. In Hevea, five miRNA families (HbmiR319, HbmiR156/157 and HbmiR165/166) have been found in more than forty plant species [57], and nine correspond to ancient miRNA already present in the common ancestor of Embryophytes (HbmiR156, HbmiR159/319, HbmiR160, HbmiR165/166, HbmiR171, HbmiR408 and HbmiR395), two in the common ancestor of Tracheophytes (HbmiR397 and HbmiR398) and nine in the common ancestor of Spermaphytes (HbmiR162, HbmiR164, HbmiR168, HbmiR169, HbmiR172, HbmiR393, HbmiR394, HbmiR399 and HbmiR827) [58]. Interestingly, the so-called monocotyledon-specific HbmiR444 family was identified in this study [58]. This suggests that HbmiR444 appeared before the speciation between monocotyledons and dicotyledons. Three woody species-specific miRNA families were also found in Hevea brasiliensis: HbmiR473 (Citrus sinensis and Populus trichocarpa), HbmiR476 (Populus trichocarpa) and HbmiR479 (Populus trichocarpa, Vitis vinifera, Gossypium hirsutum and Citrus sinensis) and were probably recruited independently by woody species during their evolution. The 10 novel miRNA families identified in this work are likely to be more recent, after Hevea diversification, as they have not been identified up to now in other euphorbiaceous plants. Furthermore, this information complements previous studies on euphorbiaceous species [49] in which only 25 conserved miRNAs were identified.
Our data could be used to study the evolution of MIR genes. We showed that 70% of the putatively new MIR genes were able to produce lmiRNA of 23-27 nucleotides, compared to 29% for conserved MIR genes. This corresponds to what has been observed in Arabidopsis, where it was previously suggested that young MIR genes consistently produced siRNAs, while ancient MIR genes produced predominantly canonical miRNAs, showing that the evolution of MIR genes is associated with changes in the use of DCL, resulting in specific miRNA size classes [59]. In addition, several so-called plant species-specific miRNAs were also found in Hevea, such as HbmiR1310 initially found only in Pinus taeda [53], HbmiR2910, HbmiR2914, HbmiR2915 and HbmiR2916 found in Populus euphratica [60] and HbmiR476 in Populus trichocarpa [61]. Our data suggest that those miRNAs should no longer be considered as species-specific miRNA.
Our study also revealed some conserved and divergent mechanisms at post-transcriptional level. Indeed, four families of miRNA were subjected to RNA editing events at the sites previously observed in Arabidopsis. However, for the thirteen families left, RNA editing was not observed under our experimental conditions. Under our experimental conditions, the -1 + UU modification was not observed in Hevea for the nine families selected from Ebhardt et al. [50]. Once the Hevea genome sequence is available, it will be possible to conduct an exhaustive analysis of the discrepancies between members of a multigenic family and RNA editing events.
Predicted target/miRNA pairs conserved between plant species and putative new ones in Hevea brasiliensis
In comparison with other plants, several regulation mechanisms seem to be conserved in Hevea brasiliensis, such as the regulation of HD-ZIP III protein by HbmiR166 [62], Squamosa promoter binding protein by HbmiR156 [63], NAC domain protein by HbmiR164 [64], APETALA2-like protein by HbmiR172 [65-67], CCAAT-binding transcription factor by HbmiR169 and cysteine proteinase by HbmiR396 [44]. These ancient miRNAs regulate ancestral transcription factors that coordinate highly conserved functions, such as organ polarity and separation, cell division, or hormonal control [68,69]. We predicted potentially new miRNA/target pairs in Hevea. New target/miRNA pairs were found for transcription factors, such as, for example, the NAC domain protein by HbmiR165 and HbmiRn7, the GRAS domain protein by HbmiR170, HbmiR396 and HbmiRn5, WRKY by HbmiR396 and HbmiR444, and the APETALA2-like transcription factor by HbmiR156, HbmiR159, HbmiR393, HbmiR396 and HbmiR408. (Duan et al., submitted). In addition, we also detected new pairs involved in protein degradation (ubiquitin protein ligase by HbmiRn6 and HbmiRn7), in response to hormones (ABA-insensitive by HbmiR166, ABA responsive element binding protein by HbmiR2910, and HVA22-like protein by HbmiR398, CTR by HbmiRn5) and in the regulation of miRNA (cap binding protein by HbmiR167). Further analyses are needed to validate the inhibition of these targets at transcriptional or translational level and to unravel specific regulation in Hevea brasiliensis regarding the response to stress in latex cells, and latex production.
Conserved and divergent regulation of redox homeostasis in Hevea brasiliensis
Several enzymes involved in ROS production or detoxification were found in our analysis. Two ROS-producing enzymes, Rboh, are likely to be targeted by HbmiR2914 and HbmiR476. They are key regulators in the ROS signalling network as they integrate different signal transduction pathways, such as calcium, protein phosphorylation and lipid signalling with ROS production [6]. Careful attention was also paid to targets classified in the GO term "antioxidant activity". For conserved miRNAs, two CuZnSOD chloroplastic and peroxisomal isoforms were predicted to be targeted by HbmiR398 and HbmiR159 respectively. Surprisingly, the CuZnSOD cytosolic isoform was not predicted by the LeARN pipeline and, even manually, no correct alignment was possible with HbmiR398. So far, validation by 5' RACE PCR has failed under our experimental conditions. By contrast, previous studies in Arabidopsis showed that HbmiR398 targeted both chloroplastic and cytosolic isoforms [34].
Some miRNA families have been reported to be H2O2-responsive in rice, such as OsmiR169, OsmiR397, OsmiR528, OsmiR827, OsmiR319 and OsmiR408 [70]. In rice, OsmiR397 targets laccase genes involved in lignin biosynthesis and those targets were also found for HbmiR397 in Hevea.
Conclusion
A library of small RNAs was deeply sequenced to identify conserved and novel miRNAs in Hevea brasiliensis. Combined with the Hevea transcript database, this led to the identification of 48 miRNA families and 10 novel miRNAs. Our study revealed new insights for evolutionary studies on woody plants and the potentially specific regulation of redox homeostasis in Hevea. All the putative targets of interest for Hevea stress biology and latex production will be experimentally validated and the target genes and their corresponding miRNAs will undergo co-expression analysis.
Methods
Production of in vitro plantlets
Embryogenic friable callus from line CI05538 of clone PB 260 was maintained on MM medium in the culture room at 27°C, with 60% humidity in the dark [71]. In vitro plants were regenerated according to the procedure described in [72]. Briefly, somatic embryogenesis was initiated by sub-culturing 1 gram of callus in 250 mL flasks containing 50 mL of a semi-solid Induction Medium. Pro-embryos were then developed in a temporary immersion system (RITA®, CIRAD, France) with 1 min of immersion per day in the liquid development media DEV1 and DEV2. For plant regeneration, only well-shaped mature embryos were collected after 8 weeks of culturing [73]. They were transferred to glass tubes on a semi-solid DEV3 medium for one month under a light intensity of 60 μmol m-2 s-1 and a 12 h light/dark photoperiod for embryo conversion into plants. Plantlets were then acclimatized in the greenhouse at 28°C with 60% relative humidity.
Plant material cultivation and treatments
Budded plants and two-year-old in vitro plants of clone PB260 were grown in the greenhouse at 27°C, with 50% humidity under natural light conditions. Budded plants and two-year-old in vitro plants of clone PB260 were exposed to cold at 4°C for 8 h, light (PAR = 1000-1500 μmol/m2/s for 4 h), drought (4 weeks for young budded plants and 10 days for two-year-old in vitro plants without watering), flooding (covered with water for 4 days), salinity (300 mM NaCl for 4 days) and wounding for 4 h (Table 2). Hormonal treatments were applied in hermetically sealed boxes, either with 5 ppm of ethylene for 4 h or 0.3 μM of methyl jasmonate for 4 h as described in [74,75].
Isolation of small RNAs
For all treatments, leaves, bark and roots were sampled in liquid nitrogen and then frozen at -80°C pending small RNA isolation. Small RNAs were purified with the mirPremier microRNA Isolation Kit (Sigma-Aldrich, St. Louis, MO, USA), following the manufacturer's instructions for plant tissues. Briefly, 100 mg of the plant tissue powder was mixed with a lysis mix that released small RNA and, at the same time, inactivated ribonucleases and interfering secondary metabolites that might exist in plant tissues. Large RNA and genomic DNA remained insoluble and were removed from the lysate along with other cellular debris by brief centrifugation. Small RNA was then captured on a silica binding column in the presence of alcohol. Residual impurities were removed by wash solutions, and purified small RNA was eluted in RNase-free water.
Deep sequencing of small RNAs
Small RNA samples from callus and from control and treated plants were pooled. A Hevea small RNA library was constructed by the SKULTECH Company (France). Briefly, small RNAs were separated on a denaturing 15% polyacrylamide gel. The gel slices containing RNA with a size of about 15-50 nucleotides were excised and the RNA was eluted. Small RNAs were ligated to 5' and 3' adapters using T4 RNA ligase. Products from the second ligation were gel purified then reverse transcribed and amplified using PCR to produce cDNA. The PCR was performed with two primers that annealed to the ends of the adapters. PCR products were separated on 15% polyacrylamide gel with ethidium bromide staining. The gel slices containing DNA with a size of about 92 nucleotides were excised and the DNA was eluted. The resulting cDNA was sequenced using Solexa/Illumina technology (Illumina Inc., San Diego, CA, USA). Raw reads were trimmed using Trim_Adaptor application (non Open source application) with the minimum clip length fixed at 6 nucleotides.
Bioinformatic analyses
This work was supported by the high performance cluster of the SouthGreen Bioinformatics platform (southgreen.cirad.fr - UMR AGAP - CIRAD) comprising 26 compute nodes (208 Nehalem cores) and a 16 to storage capacity connected through a low latency Infiniband network. Sun Grid Engine was used as the workload manager to schedule jobs on the different compute nodes.
Identification of the conserved hevea miRNAs
Adapters and low-quality sequences were removed from the raw reads obtained from Illumina sequencing using cross-match software http://www.phrap.org/phredphrpconsed.html; the non-redundant dataset was produced using WU nrdb software. The unique small RNA sequences were then compared with BlastN [76] against the Arabidopsis genome (Phytozome V6) and the PMRD database [38]http://bioinformatics.cau.edu.cn/PMRD in order to classify the small RNAs and identify mature miRNAs in Hevea already known in several species.
Identification of Hevea miRNA precursors
Conserved miRNAs and putative novel miRNA precursors were identified in the PB260 transcriptome http://bassigny/cgi-bin/esttik_dev/quick_search.cgi using the LeARN pipeline http://symbiose.toulouse.inra.fr/LeARN/release 1;5.0[50], a platform for detecting, clustering and annotating non-coding RNAs. Novel miRNAs were separated into 4 classes according to their position on the stem-loop structure. Class 1 precursors only produced small RNAs corresponding to the predicted miR and miR* (i.e., small RNAs in a 25-bp region around the miR:miR* predicted region to be considered with two nucleotide overhangs linked to DCL action); class 2 comprised hairpins containing both predicted miR and miR* and additional small RNAs, of lower abundance, outside this region; class 3 involved precursors with reads on the defining small RNA region only (3a, at least two reads; 3b, only one read); class 4 consisted of precursors containing the defining small RNA plus reads of lower abundance outside the predicted miR:miR* duplex. Finally, class 5 contained hairpins that generated small RNAs of greater abundance than the defining RNA outside the predicted miR:miR* region [77]. Each miRNA precursor was checked manually following the rules described in [45]. Specifically, we considered dominant, mature sequences residing in the stem region of the stem-loop structure and ranging between 20-22 nt with a maximum free-folding energy of -25 kcal mol-1. A maximum of six unpaired nucleotides between the miRNA and miRNA* was allowed. The distance between the miRNA and miRNA* was fixed between 5 and 240 nucleotides.
Identification of putative Hevea miRNA targets
Computational identification of miRNA targets was performed with the Miranda toolbox included in the pipeline with the default parameters (gap_value = 2, mm_value = 1, gu_value = 0.5, score_threshold = 3, min_length_alignment = 20 and no_mismatch_positions = 10;11) for conserved miRNA and secondly checked with psRNAtarget http://plantgrn.noble.org/psRNATarget/(length_alignment = 18), [78]. For the putative novel miRNAs, psRNAtarget server (length_alignment = 18) and Miranda (gap_value = 2, mm_value = 1, gu_value = 0.5, score_threshold = 3, min_length_alignment = 18 and no_mismatch_positions = 10; 11) were used to scan the Hevea EST databases (Duan et al., to be submitted, http://bassigny/cgi-bin/esttik_dev/quick_search.cgi). A first assembly set was generated from reads of leaves, bark, latex, embryogenic tissues, and roots separately to create tissue-specific transcript databases with sequence names CLxxcontigxx. Then, reads from all tissues were used to generate one transcript sequence database for the Hevea clone PB260 with the sequence name hevea_454_xxx.
Gene Ontology analyses were performed with the Blast2GO program [79,80]. In detail, sequence analyses, such as alignments and BLAST, were performed with Geneious software [81].
Total RNA extraction
Total RNAs from leaves, bark and roots subjected to abiotic stress treatments were extracted using the caesium chloride cushion method adapted from Sambrook [82] by Duan et al. [75]. One gram of fresh matter was ground and transferred to a tube containing 30 ml of extraction buffer consisting of 4 M guanidium isothiocyanate, 1% sarcosine, 1% polyvinylpyrrolidone and 1% ß-mercapto-ethanol. After homogenization, the tubes were kept on ice and then centrifuged at 10,000 g at 4°C for 30 minutes. The supernatant was transferred to a new tube containing 8 ml of 5.7 M CsCl. Ultracentrifugation in a swinging bucket was carried out at 89,705 g at 20°C for 20 hours. The supernatant and caesium cushion were discarded, whilst the RNA pellet was washed with 70% ethanol. After 30 minutes of air drying, the pellet was dissolved in 200 μl of sterile water. RNAs were conserved at -80°C.
Experimental validation of miRNA targets
The GeneRacer kit (Invitrogen, Carlsbad, CA, USA) was used to ligate a 5'RNA adapter to pooled RNAs prior to reverse transcription. The resulting cDNAs were used as a template for PCR amplification using one Gene-Specific Primer (GSP) designed downstream of the predicted miRNA:target binding site and one GeneRacer 5' forward primer. The primers used were HbCuZnSODchl R: GGGTAACCAGCAAATGCAAGCAGC, HbARF R: CTTGTGTTGAGTTGCAGCGCG and HbSquamosa R: TGGAGCCTAATTGGCTTCCTTCTGC. The first PCR product was then used for nest amplification performed with a nested GSP primer and a nested GeneRacer 5' forward primer. The nested primers used were HbCuZnSODchl nested R: GCAGGGAACAATGGCTGCC, HbARF nested R: ATGTTCAGCTCAGTGGACACGG, and HbSquamosa nested R: TTGTTTGGCACCACGCTTGTGGAAG. The PCR products were gel purified, cloned into PCR2.1 TOPO TA vector (Invitrogen, Carlsbad, CA, USA) and sequenced.
Complementary DNA (cDNA) synthesis and stem-loop RT-PCR
We checked for the absence of contaminating genomic DNA prior to cDNA synthesis. If genomic DNA was detected, a DNAse treatment was performed using TurboDNAse (Ambion, Texas, USA) following the manufacturer's instructions. Four micrograms of DNA-free RNAs was used for cDNA in a 40 μL reaction mixture using a RevertAid™ M-MuLV reverse transcriptase following the manufacturer's instructions (MBI, Fermentas, Canada). The primers used for the stem-loop amplification are presented in Table 9. RT-PCR cycling conditions comprised one denaturation cycle at 95°C for 2 min, followed by 35 amplification cycles (95°C for 20 s, 58°C for 30 s, and 72°C for 30 s). All primers were also validated by real-time PCR. The real-time PCR reaction mixtures consisted of 2 μL of RT product cDNA, 1 μL of 5 μM of each primer, and 3 μL 2 × SYBR green PCR master mix (LightCycler® 480 SYBR Green I Master, Roche Applied Sciences) in a 6 μL volume. PCR cycling conditions comprised one denaturation cycle at 95°C for 10 min, followed by 45 amplification cycles (95°C for 5 s and 60°C for 20 s). Melting curves were analysed to check the specificity of the PCR amplification (Table 9). The standard curve was generated using a two-fold dilution series of 10 points in triplicate from a mixed cDNA sample. This standard curve allowed the calculation of primer efficiencies (Table 9).
Table 9.
Primer list for the stem-loop RT-PCR and the real-time PCR and their efficiencies
| Precursor name | Forward primer | Reverse primer | Unique melting pick | PCR efficiency |
|---|---|---|---|---|
| Hbpre-miR156 | TGGTGATGTTGTTGACAGAAGATAGAGAGC | GCACAAAGGAGTGAGATGCAGAGTCC | Yes | 1,790 |
| Hbpre-mir159a | GGTTAAGAAGTGGAGCTCCTTGAAGTC | GCTCCCTTCAATCCAAACAAGGATC | Yes | 1,958 |
| Hbpre-mir159b | GTGGAGCTCCTTGAAGTCCAATAGAGG | AGAGCTCCCTTCAATCCAAACAAGG | Yes | 1,881 |
| Hbpre-mir166b | GGGGAATGTTGTCTGGTTCGATG | TCAAATCAAACCCTGTTGGGGG | Yes | 1,738 |
| Hbpre-mir396 | TGACCCTCTTCGTATTCTTCCACAGC | CCCACAGCTTTATTGAACCGCAAC | Yes | 1,782 |
| Hbpre-mir408b | GACATACAAAGACTGGGAACAGGCAG | GCCACAAGCCAGGGAAGAGGC | Yes | 1,792 |
| Hbpre-mir476 | GCCTTGTATGTTTCATTTAGTAATCCTTCT | GATAATCCTTCTATGCAAAGTCTTTTATGC | Yes | 1,732 |
| Hbpre-mir2910 | CAGGTCCAGACATAGTAAGGATTGACAG | CGTTAACGGATTAACCAGACAAATCGC | Yes | 1,728 |
| Hbpre-miRn1 | ACCAGGAACTGGTATCAACCCAGC | TGCTACCAATGAATCGGACCCACC | Yes | 1,837 |
| Hbpre-miRn2 | CAGTAAATAGCAGTATCGTGGATAGGG | GTCCAATCATTGATCCTGAAAATTTCTAC | Yes | 1,828 |
| Hbpre-miRn3 | TGGATTGGAGCCCAATACTGTGAC | CTGCTCCATTGATTTTACCATCTATGC | Yes | 1,873 |
| Hbpre-miRn4 | TGCGTGGGTAGATTGAGCTGC | GCTCCTGAGATATGCAAGCCACAAG | Yes | 1,863 |
| Hbpre-miRn6 | ACCTAGGATGTAGAAGAGCATAAC | ACTACATGAGTGGATATATAGGAATCC | Yes | 1,787 |
| Hbpre-miRn7 | ATCTGCTACTCGGATAACCGTAG | GCAGCAAATCTTCCATAGCATCC | Yes | 1,925 |
| Hbpre-miRn8 | TTTCTTGATTCTAGTGGGTGGTG | GGAATTAACCAGACAAATCGCTC | Yes | 1,905 |
| Hbpre-miRn9 | AGTCGGGGGCATTCGTATTTC | GCAAATACTTTCGCAGTTGTTCG | Yes | 1,956 |
| Hbpre-miRn10 | TGGATTTATGAAAGACGAACAACTGCG | TTCGAGCCCCCCAATTTCG | Yes | 1,712 |
Abbreviations
miRNAs: microRNAs; TPD: Tapping Panel Dryness; ROS: Reactive Oxygen Species; PMRD: Plant microRNA database; DCL1: RNAse III-like DICER-like I; HASTY: plant ortholog to exportin 5; RISC: RNA-induced silencing complex; AGO1: Argonaute 1; EST: Expressed Sequence Tag
Authors' contributions
GV, AX, AW and PB carried out the bioinformatics studies. AX carried out the EST assembly and managed work with the LeARN pipeline. PB installed the cluster and made all the IT changes to adapt the LeARN program to the cluster. DC provided AP2/ERF sequences from Hevea. GV, MP and LJ drafted the manuscript. MP and LJ devised the research project and took part in its design and coordination. All the authors read and approved the final manuscript.
Contributor Information
Virginie Gébelin, Email: virginie.gebelin@cirad.fr.
Xavier Argout, Email: xavier.argout@cirad.fr.
Worrawat Engchuan, Email: worrawat.bank@gmail.com.
Bertrand Pitollat, Email: bertrand.pitollat@cirad.fr.
Cuifang Duan, Email: cuifang.duan@cirad.fr.
Pascal Montoro, Email: pascal.montoro@cirad.fr.
Julie Leclercq, Email: julie.leclercq@cirad.fr.
Acknowledgements
This work was supported by the Institut Français du Caoutchouc. The authors thank Gérald Oliver and Maryannick Rio for technical assistance. We are grateful to Dr Kuswanhadi and Tetty Chaidamsari for RNA isolation from the latex and bark of trees grown at the Sembawa Research Centre (Indonesia). The authors thank Peter Biggins for his revision of the English.
Notes: All precursors and mature miRNA sequences will be submitted to the MiRbase registry after acceptance of the manuscript for publication
References
- De Faÿ E, Jacob JL, D'Auzac J, Jacob JL. In: Physiology of rubber tree latex. Chrestin H, editor. Boca Raton (FL): CRC Press; 1989. Anatomical organization of the laticiferous system in the bark; pp. 4–14. [Google Scholar]
- Lacote R, Gabla O, Obouayeba S, Eschbach JM, Rivano F, Dian K, Gohet E. Long-term effect of ethylene stimulation on the yield of rubber trees is linked to latex cell biochemistry. Field Crop Res. 2010;115:94–98. doi: 10.1016/j.fcr.2009.10.007. [DOI] [Google Scholar]
- Venkatachalam P, Thulaseedharan A, Raghothama K. Molecular identification and characterization of a gene associated with the onset of tapping panel dryness (TPD) syndrome in rubber tree (Hevea brasiliensis Muell.) by mRNA differential display. Mol Biotechnol. 2009;41(1):42–52. doi: 10.1007/s12033-008-9095-y. [DOI] [PubMed] [Google Scholar]
- Chrestin H. In: Physiology of rubber tree latex. D'Auzac-J JJL, Chrestin H, editor. Boca Raton (FL): CRC Press; 1989. Biochemical aspects of bark dryness induced by overstimulation of rubber trees with Ethrel; pp. 432–439. [Google Scholar]
- Chrestin H, Bangrantz J, D'Auzac J, Jacob JL. Role of the lutoidic tonoplast in the senescence and degeneration of the laticifer of Hevea brasiliensis. Zeitschrift-für-Pflanzenphysiologie. 1984;114(3):261–268. [Google Scholar]
- Suzuki N, Miller G, Morales J, Shulaev V, Torres MA, Mittler R. Respiratory burst oxidases: the engines of ROS signaling. Curr Opin Plant Biol. 2011;14(6):691–699. doi: 10.1016/j.pbi.2011.07.014. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Koussevitzky S, Mittler R, Miller G. ROS and redox signalling in the response of plants to abiotic stress. Plant Cell Environ. 2012;35(2):259–270. doi: 10.1111/j.1365-3040.2011.02336.x. [DOI] [PubMed] [Google Scholar]
- Mittler R, Vanderauwera S, Suzuki N, Miller G, Tognetti VB, Vandepoele K, Gollery M, Shulaev V, Van Breusegem F. ROS signaling: the new wave? Trends Plant Sci. 2011;16(6):300–309. doi: 10.1016/j.tplants.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Jaspers P, Kangasjarvi J. Reactive oxygen species in abiotic stress signaling. Physiol Plant. 2010;138(4):405–413. doi: 10.1111/j.1399-3054.2009.01321.x. [DOI] [PubMed] [Google Scholar]
- Phillips JR, Dalmay T, Bartels D. The role of small RNAs in abiotic stress. FEBS Lett. 2007;581(19):3592–3597. doi: 10.1016/j.febslet.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Sunkar R, Chinnusamy V, Zhu J, Zhu JK. Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 2007;12(7):301–309. doi: 10.1016/j.tplants.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Lu S, Sun YH, Chiang VL. Stress-responsive microRNAs in Populus. Plant J. 2008;55(1):131–151. doi: 10.1111/j.1365-313X.2008.03497.x. [DOI] [PubMed] [Google Scholar]
- Shukla LI, Chinnusamy V, Sunkar R. The role of microRNAs and other endogenous small RNAs in plant stress responses. Biochim Biophys Acta. 2008;1779(11):743–748. doi: 10.1016/j.bbagrm.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Nonogaki H. MicroRNA gene regulation cascades during early stages of plant development. Plant Cell Physiol. 2010;51(11):1840–1846. doi: 10.1093/pcp/pcq154. [DOI] [PubMed] [Google Scholar]
- De-Kang L, Bai X, Li Y, Ding XD, Ge Y, Cai H, Ji W, Wu N, Zhu YM. Profiling of cold-stress-responsive miRNAs in rice by microarrays. Gene. 2010;459(1-2):39–47. doi: 10.1016/j.gene.2010.03.011. [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–854. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- Llave C, Kasschau KD, Rector MA, Carrington JC. Endogenous and silencing-associated small RNAs in plants. Plant Cell. 2002;14(7):1605–1619. doi: 10.1105/tpc.003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Carrington JC, Ambros V. Role of microRNAs in plant and animal development. Science. 2003;301(5631):336–338. doi: 10.1126/science.1085242. [DOI] [PubMed] [Google Scholar]
- Lanet E, Delannoy E, Sormani R, Floris M, Brodersen P, Crete P, Voinnet O, Robaglia C. Biochemical evidence for translational repression by Arabidopsis microRNAs. Plant Cell. 2009;21(6):1762–1768. doi: 10.1105/tpc.108.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Pan X, Cobb GP, Anderson TA. Plant microRNA: a small regulatory molecule with big impact. Dev Biol. 2006;289(1):3–16. doi: 10.1016/j.ydbio.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Chen X. MicroRNA biogenesis and function in plants. FEBS Lett. 2005;579(26):5923–5931. doi: 10.1016/j.febslet.2005.07.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet E, Van de Peer Y, Rouze P. The small RNA world of plants. New Phytol. 2006;171(3):451–468. doi: 10.1111/j.1469-8137.2006.01806.x. [DOI] [PubMed] [Google Scholar]
- Kim VN. Small RNAs: classification, biogenesis, and function. Mol Cells. 2005;19(1):1–15. doi: 10.1016/j.molcel.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Xie Z, Kasschau KD, Carrington JC. Negative feedback regulation of Dicer-Like1 in Arabidopsis by microRNA-guided mRNA degradation. Curr Biol. 2003;13(9):784–789. doi: 10.1016/S0960-9822(03)00281-1. [DOI] [PubMed] [Google Scholar]
- Schauer SE, Jacobsen SE, Meinke DW, Ray A. DICER-LIKE1: blind men and elephants in Arabidopsis development. Trends Plant Sci. 2002;7(11):487–491. doi: 10.1016/S1360-1385(02)02355-5. [DOI] [PubMed] [Google Scholar]
- Bollman KM, Aukerman MJ, Park MY, Hunter C, Berardini TZ, Poethig RS. HASTY, the Arabidopsis ortholog of exportin 5/MSN5, regulates phase change and morphogenesis. Development. 2003;130(8):1493–1504. doi: 10.1242/dev.00362. [DOI] [PubMed] [Google Scholar]
- Chellappan P, Xia J, Zhou X, Gao S, Zhang X, Coutino G, Vazquez F, Zhang W, Jin H. siRNAs from miRNA sites mediate DNA methylation of target genes. Nucleic Acids Res. 2010;38(20):6883–6894. doi: 10.1093/nar/gkq590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu C, Qi Y. DNA Methylation Mediated by a MicroRNA Pathway. Mol Cell. 2010;38(3):465–475. doi: 10.1016/j.molcel.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Ebhardt HA, Tsang HH, Dai DC, Liu Y, Bostan B, Fahlman RP. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res. 2009;37(8):2461–2470. doi: 10.1093/nar/gkp093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman MA, Mani V, Hammond SM. Deep sequencing of microRNA precursors reveals extensive 3' end modification. RNA. 2011;17(10):1795–1803. doi: 10.1261/rna.2713611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16(8):2001–2019. doi: 10.1105/tpc.104.022830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khraiwesh B, Zhu JK, Zhu J. Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim Biophys Acta. 2012;1819(2):137–138. doi: 10.1016/j.bbagrm.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkar R, Kapoor A, Zhu JK. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell. 2006;18(8):2051–2065. doi: 10.1105/tpc.106.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagadeeswaran G, Saini A, Sunkar R. Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta. 2009;229(4):1009–1014. doi: 10.1007/s00425-009-0889-3. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:140–144. doi: 10.1093/nar/gkj430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Saini HK, Van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:154–158. doi: 10.1093/nar/gkn221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yu J, Li D, Liu F, Zhou X, Wang T, Ling Y, Su Z. PMRD: plant microRNA database. Nucleic Acids Res. 2006;38:806–813. doi: 10.1093/nar/gkp818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archak S, Nagaraju J. Computational prediction of rice (Oryza sativa) miRNA targets. Genomics Proteomics Bioinformatics. 2007;5(3-4):196–206. doi: 10.1016/S1672-0229(08)60007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunkar R, Girke T, Jain PK, Zhu JK. Cloning and characterization of microRNAs from rice. Plant Cell. 2005;17(5):1397–1411. doi: 10.1105/tpc.105.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat A, Wall PK, Diloreto S, Depamphilis CW, Carlson JE. Conservation and divergence of microRNAs in Populus. BMC Genomics. 2007;8:481. doi: 10.1186/1471-2164-8-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arazi T. MicroRNAs in the moss Physcomitrella patens. Plant Mol Biol. DOI: 10.1007/s11103-011-9761-5. [DOI] [PubMed]
- Mica E, Piccolo V, Delledonne M, Ferrarini A, Pezzotti M, Casati C, Del Fabbro C, Valle G, Policriti A, Morgante M. et al. High throughput approaches reveal splicing of primary microRNA transcripts and tissue specific expression of mature microRNAs in Vitis vinifera. BMC Genomics. 2009;10:558. doi: 10.1186/1471-2164-10-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Pan X, Stellwag EJ. Identification of soybean microRNAs and their targets. Planta. 2008;229(1):161–182. doi: 10.1007/s00425-008-0818-x. [DOI] [PubMed] [Google Scholar]
- Zhao CZ, Xia H, Frazier TP, Yao YY, Bi YP, Li AQ, Li MJ, Li CS, Zhang BH, Wang XJ. Deep sequencing identifies novel and conserved microRNAs in peanuts (Arachis hypogaea L.) BMC Plant Biol. 2010;10:3. doi: 10.1186/1471-2229-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zeng R, Chen J, Liu X, Liao Q. Identification of conserved microRNAs and their targets from Solanum lycopersicum Mill. Gene. 2008;423(1):1–7. doi: 10.1016/j.gene.2008.05.023. [DOI] [PubMed] [Google Scholar]
- Xie FL, Huang SQ, Guo K, Xiang AL, Zhu YY, Nie L, Yang ZM. Computational identification of novel microRNAs and targets in Brassica napus. FEBS Lett. 2007;581(7):1464–1474. doi: 10.1016/j.febslet.2007.02.074. [DOI] [PubMed] [Google Scholar]
- Arenas-Huertero C, Perez B, Rabanal F, Blanco-Melo D, De la Rosa C, Estrada-Navarrete G, Sanchez F, Covarrubias AA, Reyes JL. Conserved and novel miRNAs in the legume Phaseolus vulgaris in response to stress. Plant Mol Biol. 2009;70(4):385–401. doi: 10.1007/s11103-009-9480-3. [DOI] [PubMed] [Google Scholar]
- Zeng C, Wang W, Zheng Y, Chen X, Bo W, Song S, Zhang W, Peng M. Conservation and divergence of microRNAs and their functions in Euphorbiaceous plants. Nucleic Acids Res. 2010;38(3):981–995. doi: 10.1093/nar/gkp1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebhardt HA, Fedynak A, Fahlman RP. Naturally occurring variations in sequence length creates microRNA isoforms that differ in argonaute effector complex specificity. Silence. 2009;1(1):12. doi: 10.1186/1758-907X-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noirot C, Gaspin C, Schiex T, Gouzy J. LeARN: a platform for detecting, clustering and annotating non-coding RNAs. BMC Bioinformatics. 2008;9:21. doi: 10.1186/1471-2105-9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006;20(24):3407–3425. doi: 10.1101/gad.1476406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Aksay G, Dolgosheina E, Ebhardt HA, Magrini V, Mardis ER, Sahinalp SC, Unrau PJ. Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res. 2008;18(4):571–584. doi: 10.1101/gr.6897308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu D, Pan X, Wilson IW, Li F, Liu M, Teng W, Zhang B. High throughput sequencing technology reveals that the taxoid elicitor methyl jasmonate regulates microRNA expression in Chinese yew (Taxus chinensis) Gene. 2009;436(1-2):37–44. doi: 10.1016/j.gene.2009.01.006. [DOI] [PubMed] [Google Scholar]
- Zhang J, Xu Y, Huan Q, Chong K. Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genomics. 2009;10:449. doi: 10.1186/1471-2164-10-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uthup TK, Ravindran M, Bini K, Thakurdas S. Divergent DNA Methylation Patterns Associated with Abiotic Stress in Hevea brasiliensis. Mol Plant. 2011. DOI: 10.1093/mp/ssr039. [DOI] [PubMed]
- Sunkar R, Jagadeeswaran G. In silico identification of conserved microRNAs in large number of diverse plant species. BMC Plant Biol. 2008;8:37. doi: 10.1186/1471-2229-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuperus JT, Fahlgren N, Carrington JC. Evolution and functional diversification of MIRNA genes. Plant Cell. 2011;23(2):431–442. doi: 10.1105/tpc.110.082784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Blevins T, Ailhas J, Boller T, Meins F Jr. Evolution of Arabidopsis MIR genes generates novel microRNA classes. Nucleic Acids Res. 2008;36(20):6429–6438. doi: 10.1093/nar/gkn670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Yin W, Xia X. Identification of microRNAs and their targets from Populus euphratica. Biochem Biophys Res Commun. 2009;388(2):272–7. doi: 10.1016/j.bbrc.2009.07.161. [DOI] [PubMed] [Google Scholar]
- Lu S, Sun YH, Shi R, Clark C, Li L, Chiang VL. Novel and mechanical stress-responsive MicroRNAs in Populus trichocarpa that are absent from Arabidopsis. Plant Cell. 2005;17(8):2186–2203. doi: 10.1105/tpc.105.033456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Wang H, Li H, Yuan Z, Li F, Yang L, Huang H. Two types of cis-acting elements control the abaxial epidermis-specific transcription of the MIR165a and MIR166a genes. FEBS Lett. 2009;583(22):3711–3717. doi: 10.1016/j.febslet.2009.10.076. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhang Z, Liu D, Zhang K, Li A, Mao L. SQUAMOSA promoter-binding protein-like transcription factors: star players for plant growth and development. J Integr Plant Biol. 2010;52(11):946–951. doi: 10.1111/j.1744-7909.2010.00987.x. [DOI] [PubMed] [Google Scholar]
- Guo HS, Xie Q, Fei JF, Chua NH. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for arabidopsis lateral root development. Plant Cell. 2005;17(5):1376–1386. doi: 10.1105/tpc.105.030841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science. 2004;303(5666):2022–2025. doi: 10.1126/science.1088060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmann H, Mica E, Todesco M, Long JA, Weigel D. On reconciling the interactions between APETALA2, miR172 and AGAMOUS with the ABC model of flower development. Development. 2010;137(21):3633–3642. doi: 10.1242/dev.036673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu QH, Helliwell CA. Regulation of flowering time and floral patterning by miR172. J Exp Bot. 2011;62(2):487–495. doi: 10.1093/jxb/erq295. [DOI] [PubMed] [Google Scholar]
- Allen E, Xie Z, Gustafson AM, Sung GH, Spatafora JW, Carrington JC. Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat Genet. 2004;36(12):1282–1290. doi: 10.1038/ng1478. [DOI] [PubMed] [Google Scholar]
- Axtell MJ. Evolution of microRNAs and their targets: are all microRNAs biologically relevant? Biochim Biophys Acta. 2008;1779(11):725–734. doi: 10.1016/j.bbagrm.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Li T, Li H, Zhang YX, Liu JY. Identification and analysis of seven H2O2-responsive miRNAs and 32 new miRNAs in the seedlings of rice (Oryza sativa L. ssp. indica) Nucleic Acids Res. 2011;39(7):2821–2833. doi: 10.1093/nar/gkq1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carron M-P, Etienne H, Lardet L, Campagna S, Perrin Y, Leconte A, Chaine C. In: Somatic embryogenesis in woody plants. Jain SGP, Newton R, editor. Dordrecht: Kluwer Academic Publishers; 1995. Somatic embryogenesis in rubber (Hevea brasiliensis Müll. Arg.) pp. 117–136. [Google Scholar]
- Lardet L, Martin F, Dessailly F, Carron MP, Montoro P. Effect of exogenous calcium on post-thaw growth recovery and subsequent plant regeneration of cryopreserved embryogenic calli of Hevea brasiliensis (Mull. Arg.) Plant Cell Rep. 2007;26(5):559–569. doi: 10.1007/s00299-006-0278-3. [DOI] [PubMed] [Google Scholar]
- Lardet L, Piombo G, Oriol F, Dechamp E, Carron M. Relation between biochemical characteristics and conversion ability in Hevea brasiliensis zygotic and somatic embryos. Can J Bot. 1999;77:1168–1177. [Google Scholar]
- Kuswanhadi, Leclercq J, Rio M, Tregear J, Ducamp-Collin MN, Montoro P. Isolation of three members of the multigene family encoding ACC oxidases in Hevea brasiliensis and investigation of their responses to ethylene stimulation and wounding. Journal of rubber research. 2010;13(3):185–205. [Google Scholar]
- Duan C, Rio M, Leclercq J, Bonnot F, Oliver G, Montoro P. Gene expression pattern in response to wounding, methyl jasmonate and ethylene in the bark of Hevea brasiliensis. Tree Physiol. 2010;30(10):1349–1359. doi: 10.1093/treephys/tpq066. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Lelandais-Briere C, Naya L, Sallet E, Calenge F, Frugier F, Hartmann C, Gouzy J, Crespi M. Genome-wide Medicago truncatula small RNA analysis revealed novel microRNAs and isoforms differentially regulated in roots and nodules. Plant Cell. 2009;21(9):2780–2796. doi: 10.1105/tpc.109.068130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39(2):W155–W159. doi: 10.1093/nar/gkr829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Götz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36(10):3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Heled J, Kearse M, Moir R, Stones-Havas S, Sturrock S, Geneious v5.3. 2010. Available from http://www.geneious.com/
- Sambrook J, Fritsch EF, Maniatis T. In: A laboratory manual. CS H, editor. New York: Cold Spring Harbor Laboratory Press; 1989. Molecular cloning. [Google Scholar]