

Abstract
Background
We conducted a pilot study of reproducibility and associations of microbial diversity and composition in fecal microbial DNA.
Methods and results
Participants (25 men, 26 women, ages 17–65 years) provided questionnaire data and multiple samples of one stool collected with two Polymedco and two Sarstedt devices pre-loaded with RNAlater. 16S rRNA genes in each fecal DNA aliquot were amplified, sequenced (Roche/454 Life Sciences), and assigned to taxa. Devices were compared for ease of use and reproducibility [intraclass correlation coefficient (ICC)] between duplicate aliquots on diversity and taxonomic assignment. Associations were tested by linear regression. Both collection devices were easy to use. Both alpha diversity (Shannon index) and beta diversity (UniFrac) were higher between than within duplicates (P≤10−8) and did not differ significantly by device (P≥0.62). Reproducibility was good (ICC ≥0.77) for alpha diversity and taxonomic assignment to the most abundant phyla, Firmicutes and Bacteroidetes (71.5% and 25.0% of sequences, respectively), but reproducibility was low (ICC≤0.48) for less abundant taxa. Alpha diversity was lower with non-antibiotic prescription medication (P=0.02), younger age (P=0.03) and marginally with higher body mass index (P=0.08).
Conclusions
With sampling from various parts of a stool, both devices provided good reproducibility on overall microbial diversity and classification for the major phyla, but not for minor phyla. Implementation of these methods should provide insights on how broad microbial parameters, but not necessarily rare microbes, affect risk for various conditions.
Keywords: Microbiome, alpha diversity, beta diversity, bacterial phylogenetics, medications, body mass index
Introduction
The community of intestinal microbes, which is known as the microbiota (see Glossary), includes hundreds of bacterial species, thousands of variants, and approximately 1014 organisms/gr of feces, which is 10-fold more than the number of cells in a human body [1]. Through its complexity and mass, the microbiota could affect human health by way of multiple mechanisms. These include blocking pathogens, detoxifying potential carcinogens, digesting food to produce energy, synthesizing nutrients, conditioning and maintaining mucosal and systemic immunity, and facilitating homeostasis [2–4]. How the microbiota exerts these effects is poorly understood, as most of the organisms cannot be grown in culture. Animal models and culture-independent genomic methods have started to provide insight, particularly with respect to broad functional activities of the intestinal microbiota [5–7].
Thus far, microbiome research has been largely exploratory. There are only a few reports on the accuracy and especially the reproducibility of human microbial diversity and taxonomy [8–11]. In particular, advantages and disadvantages of methods to collect feces for microbiome research have not been described. Collection methods could affect the validity of data to be obtained and the interpretations of epidemiologic and clinical associations.
We describe herein, for the first time in a population of human volunteers, highly reproducible estimates of fecal microbial diversity and taxonomy obtained with a strict protocol that included self-collection and careful handling of specimens, as well as state-of-the-art 16S rRNA amplification and pyrosequencing methods, all of which are consistent with procedures recently recommended by others [12]. Terms are briefly defined in the Glossary. As a secondary aim, we evaluated associations of fecal microbiome parameters with questionnaire data provided by the participants.
Methods
All procedures for this study were consistent with the protocols of the Human Microbiome Project, which are available at http://www.hmpdacc.org/tools_protocols/tools_protocols.php.
Participants
Fifty-one healthy volunteers (25 male, ages 17–63; and 26 female, ages 24–65; Table 1), employees of the Division of Cancer Epidemiology and Genetics, National Cancer Institute (NCI), were recruited to assess the reproducibility of microbial measures in self-collected fecal specimens. The study was approved by the NCI Special Studies Institutional Review Board. Following face-to-face instructions and signed informed consent, participants were provided a toilet-attached pouch (Protocult, Rochester, MN), from which they collected multiple aliquots of an early or mid-morning stool at home. After sample collection, they completed a brief self-administered questionnaire on demographics, broad dietary categories, ease-of-use of the devices, and factors potentially related to gut microbiota. To increase confidentiality, data on race and ethnicity were not collected.
Table 1.
Characteristics of the 51 study participants.*
| Variable | Males | Females | P-value | |
|---|---|---|---|---|
| Age | N, mean (±SD) | 25, 38.8 (±12.27) | 26,40.2 (±12.4) | 0.79 |
| Range (min, max) | (17, 63) | (24, 65) | ||
| Body mass index (BMI) category n(%) | Normal (BMI 18.5–25) | 14 (56) | 16 (61) | |
| Overweight (BMI 25–30) | 11 (44) | 9(35) | ||
| Obese (BMI>30) | 0 | 1 (4) | 0.56 | |
| Weight change n(%) | Gain >10lb | 2 (8) | 1 (3) | |
| Gain 5–10lb | 2 (8) | 2 (8) | ||
| Maintained | 17 (68) | 19 (73) | ||
| Lost 5–10lb | 4 (16) | 3 (12) | ||
| Lost >10lb | 0 | 1 (4) | 0.64 | |
| Antibiotic use n(%) | <6 months | 4 (16) | 9 (36) | |
| 6–12 months | 9 (36) | 4 (16) | ||
| >12 months | 12 (48) | 9 (36) | ||
| Never | 0 | 3 (12) | 0.08 | |
| Non-antibiotic prescription n(%) | <6 months | 13 (52) | 15 (60) | |
| 6–12 months | 4 (16) | 3 (12) | ||
| >12 months | 5 (20) | 6 (24) | ||
| Never | 3 (12) | 1 (4) | 0.59 | |
| Food allergy n(%) | No | 22 (88) | 22 (88) | |
| Yes | 3 (12) | 3 (12) | 1.00 | |
| Vegan n(%) | No | 25 (100) | 24 (100) | |
| Vegetarian n(%) | No | 22 (88) | 24 (96) | |
| Yes | 3 (12) | 1 (4) | 0.61 | |
| Probiotic supplement use n(%) | No | 18 (72) | 16 (62) | |
| Yes | 7 (28) | 10 (38) | 0.56 | |
| Ever smoker n(%) | No | 22 (88) | 21 (81) | |
| Yes | 3 (12) | 5 (19) | 0.70 | |
| Ease of use, Polymedco device n(%) | Very easy | 7 (28) | 9 (36) | |
| Rather easy | 10 (40) | 8 (32) | ||
| Neither easy nor difficult | 6 (24) | 6 (24) | ||
| Difficult/very difficult | 2 (8) | 2 (8) | 0.50 | |
| Ease of use, Sarstedt device n(%) | Very easy | 12 (48) | 8 (32) | |
| Rather easy | 7 (28) | 13 (52) | ||
| Neither easy nor difficult | 6 (24) | 4 (16) | ||
| Difficult/very difficult | 0 | 0 | 0.19 |
Totals may not sum to 51 due to missing values.
Fecal Specimens
Each participant collected four aliquots from various parts of a stool with Polymedco OC-auto collection devices that were pre-loaded with 2ml RNAlater (QIAGEN Inc., Valencia, CA). Polymedco is a leak-proof device with a snap-cap-attached probe that is widely used for colon cancer screening (FOBT-CHECK®). For comparison, four aliquots were collected with Starstedt feces collection containers (SARSTEDT, Nümbrecht, Germany), a 10mL tube with a screw-cap-attached scoop, each of which was pre-loaded with 5ml RNAlater (QIAGEN Inc., Valencia, CA). The Sarstedt device holds approximately 20-fold more fecal matter (~1gm) than does the Polymedco device. All specimens were immediately chilled (frozen gel packs), frozen in liquid nitrogen within 1–3 hours, and stored at −80°C until thawing for DNA extraction. Four aliquots from each participant (duplicates with each device) were used for the current study. The remaining aliquots were reserved for future studies. Additional details are provided elsewhere [13].
Fecal DNA Extraction
Genomic DNA from stool samples preserved and transported in RNAlater was extracted from 300 mg of feces. The samples were mixed with 350 μL of lysis buffer composed of 0.05 M potassium phosphate buffer containing 50 μL lyzosyme (10 mg/mL), 6 μL of mutanolysin (25,000 U/ml; Sigma-Aldrich) and 3 μL of lysostaphin (4,00 U/mL in sodium acetate; Sigma-Aldrich, St. Louis, MO). The mixture was incubated for 1 hour at 37°C, following which 10 μL proteinase K (20 mg/ml), 100 μL 10% SDS, and 20 μL RNase A (20 mg/ml) were added. This mixture was incubated for 1h at 55°C. To further lyse microbial cells, mechanical disruption (bead beating) was performed on the mixture using a FastPrep instrument (MP Biomedicals, Solon, OH) set at 6.0 m/s for 30 sec. The lysate was processed using the ZR Fecal DNA extraction kit (ZYMO Research, Irvine, CA) according to the manufacturer's recommendation omitting the lysis steps (steps 1–3). The DNA was eluted into 100 μL of storage buffer [QIAsymphony reagent buffer AVE (0.04% sodium azide), Qiagen], pH 8.0. DNA was quantified by Quant-iT PicoGreen (Molecular Probes, Inc., Eugene, OR) in a SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA).
454 Pyrosequencing of barcoded 16S rRNA Genes
Universal primers 27F and 338R were used for PCR amplification of the V1–V2 hypervariable regions of 16S rRNA genes (3). The 338R primer included a unique sequence tag to barcode each sample. The primers were as follows: 27F-5'-GCCTTGCCAGCCCGCTCAGTCAGAGTTTGATCCTGGCTCAG-3' and 338R-5'-GCCTCCCTCGCGCCATCAGNNNNNNNNCATGCTGCCTCCCGTAGGAGT-3', where the underlined sequences are the 454 Life Sciences® FLX sequencing primers B and A in 27F and 338R, respectively, and the bold font denotes the universal 16S rRNA primers 27F and 338R. The 8-bp barcode within primer 338R is denoted by 8 Ns. Using 96 barcoded 338R primers, the V1–V2 regions of 16S rRNA genes were amplified in 96 well microtiter plates as follows: 5.0 μL 10× PCR buffer II (Applied Biosystems, Foster City, CA), 3.0 μL MgCl2 (25 mM; Applied Biosystems), 2.5 μL Triton X-100 (1%), 2.0 μL deoxyribonucleoside triphosphates (10 mM), 1.0 μL each of primer 27F and 338R (20 pmol/μL each), 0.5 μL AmpliTaq DNA polymerase (5U/μL; Applied Biosystems), and 50 ng of template DNA in a total reaction volume of 50 μL. Reactions were run in a PTC-100 thermal controller (MJ Research Inc., Waltham, MA) using the following cycling parameters: 5 min of denaturation at 95°C, followed by 20 cycles of 30 sec at 95°C (denaturing), 30 sec at 56°C (annealing) and 90 sec at 72°C (elongation), with a final extension at 72°C for 7 minutes. Non-template controls (NTC) were used as negative controls for each set of barcoded primers. The presence of amplicons was confirmed by gel electrophoresis on a 2% agarose gel and staining with SYBRGreen (Applied Biosystems, Foster City, CA). Equimolar amounts (~100 ng) of the PCR amplicons were mixed in a single tube, and amplification primers and reaction buffer were removed by processing the mixture with the Agencourt AMPure XP Kit (Beckman Coulter Genomics, Danvers, MA). The purified amplicon mixtures were sequenced by 454 FLX Titanium pyrosequencing (Roche Diagnostics Corp., Indianapolis, IN) with 454 Life Sciences® primer A by the Genomics Resource Center at the Institute for Genome Sciences, University of Maryland School of Medicine using protocols recommended by the manufacturer.
Classification of Operational Taxonomic Units
Sequence read quality used the Institute of Genome Sciences bioinformatics pipeline that complies with standard operating procedures of the National Institutes of Health Human Microbiome Project and recent recommendations [12;14]. Terms are defined in the Glossary. Briefly, after trimming the primer and barcode sequences, raw sequence reads were filtered using the QIIME pipeline (http://qiime.sourceforge.net) [15] with the following criteria to optimize the quality and integrity of the data: 1) minimum and maximum read length of 300 bp and 500 bp; 2) no ambiguous base calls; 3) no homopolymeric runs longer than 8 bp; 4) average quality value >q25 within a sliding window of 50 bp; 5) 60% match to a previously determined 16S rRNA gene sequence; and 6) chimera-free using the UCHIME software (http://www.drive5.com/uchime/) [16]. Sequence reads with the same barcode were binned by sample. Operational taxonomic units (OTUs) were defined using QIIME as sequences with at least 97% similarity, and sequences were classified at the genus level using the Ribosomal Database Project (RDP) naïve Bayesian classifier [17].
Statistical Analysis
To assess taxonomic and phylogenetic reproducibility, we used one duplicate fecal DNA sample per device per participant (n=2×2×51 participants = 204 samples). Intraclass correlation coefficients (ICC) between each device's duplicate samples were calculated for each microbial parameter, separately for each device. Relative abundance of each OTU, alpha diversity, and beta diversity were computed using QIIME [15]. Alpha diversity was estimated by the Shannon index (SI), which adjusts the number of taxa (OTUs) detected for their relative abundance (proportions). SI is calculated as minus the sum over OTUs of the proportion of a given OTU times the logarithm of that proportion in each sample. Beta diversity, which is a measure of separation of the phylogenetic structure of the OTUs in one sample, compared to all other samples, was estimated by unweighted Unifrac distances (http://qiime.sourceforge.net)]. The pseudo F-statistic was calculated using PERMANOVA to assess the reproducibility of beta diversity given the unweighted UniFrac distance matrix for each device [18]. Differences in ICCs and pseudo F-statistics between the devices were tested by comparison against null distributions obtained by randomly permuting the two device labels within individuals with 10,000 replicates (Supplementary Methods). The Visualization and Analysis of Microbial Population Structures (VAMPS, Marine Biology Laboratories, Woods Hole, MA) pipeline was used for presentation of beta diversity of RDP-classified taxa, with the Bray-Curtis dissimilarity index for genus [19]. Linear regression was carried out to test the association of each microbial parameter with age, sex, body mass index, and use of antibiotics or other prescription medications. Age and body mass index were mutually adjusted in the regression model. All P-values are two-sided. Analyses were conducted using the statistical software R (http://www.r-project.org/).
Results
Study Participants
The questionnaire data provided by the 51 participants are summarized in Table 1. Four participants were vegetarian; none was vegan. Thirteen participants had taken an antibiotic within six months of enrollment, including three who had taken an antibiotic within one month. A non-antibiotic prescription had been used within one month of enrollment by 17 participants and 2–6 months before enrollment by 11 additional participants. Four participants reported that collection of multiple samples of stool was difficult or very difficult with the Polymedco device, compared to none with the Sarstedt device, but difference in ease-of-use was not significant (P=0.11).
Reproducibility of Fecal Microbial Diversity and Taxonomy
Pyrosequencing of the V1–V2 region of the 16S rRNA gene yielded 1,270,073 raw reads that were edited and de-noised to 764,698 final reads (mean 3805 reads per participant) for analysis. These were assigned to 6914 OTUs (mean 170 OTUs per sample and 431 OTUs per participant). Quality criteria were fully met for 101 (99%) of the Sarstedt samples and 100 (98%) of the Polymedco samples.
Alpha diversity of the OTUs, as estimated by the Shannon index (SI) was markedly and significantly greater between than within participants (P=1.6×10−13 for Polymedco, P=1.1×10−11 for Sarstedt). Intraclass correlation coefficients (ICCs) of alpha diversity did not differ significantly between the two collection devices: ICCPolymedco=0.794 and ICCSarstedt=0.790 (P=0.62, Figure 1A).
Figure 1.
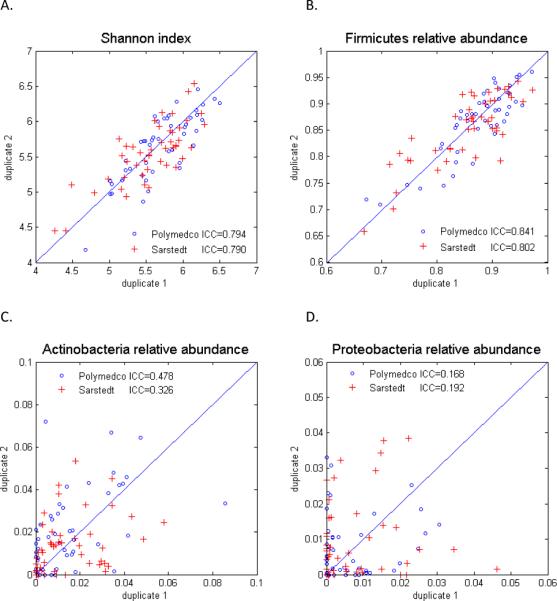
Reproducibility of 16S rRNA alpha diversity and relative abundance in fecal specimens obtained with two different collection devices. Shannon index of alpha diversity (A). Relative abundance of Firmicutes (B), Actinobacteria (C) and Proteobacteria (D), as aligned to the Ribosomal Data Project. The intraclass correlation coefficient (ICC) did not differ significantly between the two devices.
With both collection devices, taxonomic assignment of OTUs was highly reproducible for the most abundant colonic phyla, specifically Firmicutes [mean relative abundance 71.5%, ICCPolymedco=0.841 and ICCSarstedt=0.802 (P=0.62), Figure 1B] and Bacteroidetes [25.0%, ICCPolymedco=0.801 and ICCSarstedt=0.772 (P=0.69)]. In contrast, reproducibility in estimated relative abundance was low for Actinobacteria (1.6%, ICC≤0.48, Figure 1C) and Proteobacteria (0.8%, ICC≤0.19, Figure 1D). As summarized in Table 2, with either collection device, the differences between the duplicates and the standard deviations were as large, or even larger, than the relative abundance mean values for the uncommon phyla.
Table 2.
Mean and standard deviation (SD) relative abundance of bacterial phyla, by collection device.
| Firmicutes |
Bacteroidetes |
Actinobacteria |
Proteobacteria |
Other bacteria |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Device* | Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD |
| PA | 73.1% | 11.9% | 23.9% | 12.3% | 1.5% | 1.7% | 0.6% | 0.8% | 0.8% | 1.0% |
| PB | 71.6% | 13.2% | 24.8% | 13.9% | 2.0% | 1.9% | 0.7% | 0.9% | 0.9% | 1.4% |
| |PA – PB| difference | 5.6% | 4.0% | 5.8% | 4.6% | 1.3% | 1.3% | 0.8% | 0.8% | 0.8% | 1.1% |
| SA | 70.4% | 12.7% | 26.1% | 13.0% | 1.4% | 1.4% | 0.8% | 1.1% | 0.7% | 1.0% |
| SB | 70.2% | 11.5% | 26.2% | 12.0% | 1.3% | 1.3% | 1.0% | 1.3% | 0.8% | 1.3% |
| |SA – SB| difference | 5.6% | 5.0% | 5.6% | 5.0% | 1.2% | 1.1% | 1.1% | 1.0% | 0.6% | 0.8% |
Mean values, with their standard deviations, of individual Polymedco (PA, PB) and Sarstedt (SA, SB) samples, as well as mean absolute differences |A minus B| between duplicate samples, with their standard deviations.
As for alpha diversity, beta diversity was significantly greater between than within participants (P<10−16 for both devices), and it did not differ by collection device (P=0.96). This is illustrated for the genus level in Figure 2, in which color indicates pair-wise Bray-Curtis dissimilarity. Duplicate samples within each participant are paired in adjacent rows and columns in Figure 2 and are hardly distinguishable, indicating good reproducibility within individuals. The lower right insert in Figure 2 shows the relative abundance of the genera found within participant 59 and between participants 59 and 60.
Figure 2.
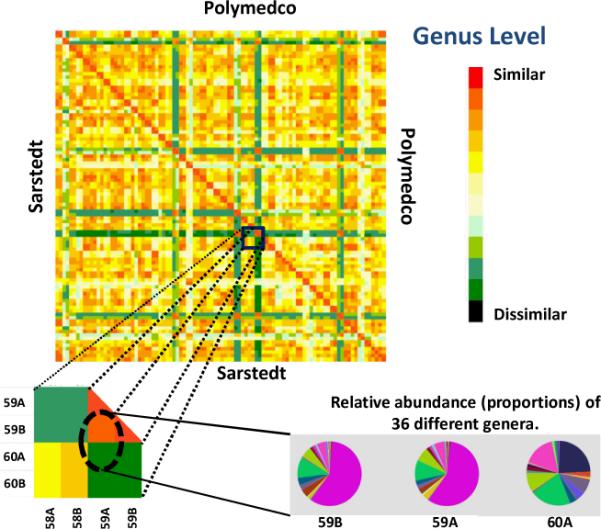
Beta diversity of 16S rRNA pyrosequence data, at the genus level, by collection device. For all pairs of samples, each cell indicates Bray-Curtis 2-way dissimilarity [minimum red, maximum black]. Red diagonal (no dissimilarity) is a sample with itself. Each participant is presented as two adjacent rows (or columns) for his or her sample pair. Several participants (blue/green rows and columns) had highly reproducible beta diversity that was distinctive from other participants' data. Lower left is a magnification of 2-way dissimilarity for selected pairs. Lower right presents relative abundance of 36 genera, with low dissimilarity in participant 59's A and B duplicates (Bray-Curtis 0.003) and high dissimilarity between A samples from participants 59 and 60 (Bray-Curtis 0.911).
Relationship of Fecal Microbial Diversity and Taxonomy to Questionnaire Data
As shown in Figure 3, in a bivariate linear regression model, alpha diversity was higher with older age (βSI=0.01, Ptrend=0.03) and tended to be higher with lower body mass index (βSI=−0.04, Ptrend=0.08). Alpha diversity did not differ by sex (P=0.54) or use of probiotic dietary supplements (P=0.86). In contrast, alpha diversity tended to be lower in participants who reported antibiotic use within one month (N=3, βSI=−0.46, P=0.10), and it was significantly lower in those who reported taking a non-antibiotic prescription medication within one month (N=17, βSI=−0.30, P=0.02). Antibiotic use was associated with lower relative abundance of Firmicutes genus Clostridiales (mean 66.8%, β=−0.21, P=0.002) and higher relative abundance of Bacteroidetes genus Bacteroidales (24.7%, β=0.24, P=0.0005).
Figure 3.
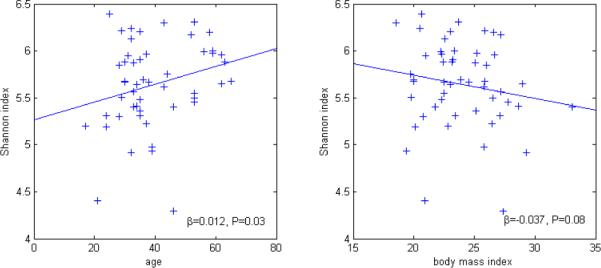
Association of 16S rRNA alpha diversity with mutually adjusted age (left) and body mass index (right).
Discussion
Microbiome research relevant to human health will depend on methods that yield reproducibly accurate exposure data, as well as an understanding of the limits of such methods. This is especially important if specimens are to be self-collected. With our protocol, we found that either of two fecal collection devices, pre-loaded with a preservative (RNAlater) and used by volunteers at home, yielded high quality sequences of the universal 16S rRNA bacterial gene, as well as highly reproducible estimates of alpha diversity, beta diversity, and the major phyla of the fecal microbiome. Conversely, reproducibility was low for the minor phyla, probably due to heterogeneity within a stool and the small number of minor-phyla OTUs (mean n=6 per sample). We formally tested and thereby replicated previous observations of significantly higher fecal microbial diversity in samples between versus within individuals [20–22]. These findings suggest that our protocol could yield valid data for analytic epidemiologic research. However, fecal collection procedures may prove to be more challenging and potentially inconsistent in populations that are less educated and less motivated than our epidemiology colleagues.
Given the reproducibility that we found, our second objective considered microbial associations of likely relevance to several aspects of human health. Specifically, our data support prior observations of lower alpha diversity of the fecal microbiome related to antibiotics and obesity [22–27]. In addition, we found evidence of lower alpha diversity with other prescription medications and with younger age. Much more research will be needed to determine whether alpha diversity (the diversity within a specimen), beta diversity (the ratio of unique taxa to shared taxa across specimens), or even individual taxa are related to particular diseases.
Plottel and Blaser have proposed that cancer risk could relate to the microbiota, or even to individual microbes, in three ways: direct effects on susceptible cells, alterations of immunity, and dysregulation of metabolism, nutrition or hormones [4]. Regarding direct effects, two groups recently compared the microbiomes of colon cancer tissue to normal colon epithelium from the same patient, discovering that colon cancers were frequently and significantly associated with Fusobacterium nucleatum [28;29], a highly invasive bacterium that is not commonly found in the colon. Based on taxonomic analysis, none of our fecal specimens contained F. nucleatum above our relative abundance cutoff of 0.1%.
Two bacterial phyla, Bacteroidetes and Firmicutes, predominate in the human colon. The relative abundance of Bacteroidetes may be reduced with obesity, a deficit that may resolve during a year of successful dieting [23]. More globally, Arumugam et al proposed that individuals have one of three relatively stable gut microbial communities (“enterotypes”)[30], and a recent study suggested that these may reflect dietary differences in carbohydrates (with Prevotella predominating) versus protein and animal fat (with Bacteroides predominating) [31]. In a small feeding study, changes in fecal microbial taxa were seen within 24 hours of switching between high-fat/low-fiber and low-fat/high-fiber diets, even though enterotypes appeared to persist [31]. Our sample size, questionnaire data, and sequencing methods were insufficient to address these observations, but we did observe a trend of lower alpha diversity with higher body mass index, even with rather few overweight or obese participants. This supports a prior association of obesity with reduced alpha diversity [21].
Antibiotic use has an abrupt impact on microbial composition that largely but incompletely returns to baseline after discontinuation [24–27]. Because our primary objective was to assess reproducibility, we did not exclude participants who had used antibiotics or other prescription medications. We note that the effect can be profound, as the three participants who reported antibiotic use within one month of enrollment had a highly significant shift in relative abundance from Firmicutes to Bacteroidetes. Earlier antibiotic use (not presented) had no apparent effect. Perhaps of more interest, we found that use of a non-antibiotic prescription medication was associated with significantly reduced alpha diversity. This has not been noted previously [30]. Indeed, one study found no association between non-antibiotic prescriptions and composition (relative abundance) of the fecal microbiome [22]. Although we lacked data on the specific non-antibiotic medications, our observation is fully compatible with the accumulating evidence that commonly used proton-pump inhibitors and H2 blockers increase the risk of clinically significant colonic infection with Clostridium difficile [32]. Despite the effects of antibiotics and diet, major metabolic and other functions of the colonic microbiota appear to be uniform among individuals [22;33].
Older participants in our study appeared to have higher alpha diversity, which is a novel observation. Several investigations have reported age-associated differences in the composition of the fecal microbiota [22;30;34–36]. Surprisingly, differences in alpha or beta diversity have not been associated with aging in adults. Even the large MetaHIT consortium study of European adults reported “The only significant correlation between a host property and a taxonomic group is a negative one between age and the abundance of an unknown Clostridiales genus ….” [30]. It is noteworthy that alpha diversity had the opposite association (lower) with medication use and body mass index, two variables that might otherwise have been confounders of older age.
The primary limitations of our study are its small size and cross-sectional design which did not allow us to evaluate changes in the fecal microbiome that occur over time [20]. In addition, we did not study a representative sample of adults, but rather volunteers who were highly motivated, highly educated and generally in good health. Although our volunteers included several from Asia, Latin America, and Europe, as well as the United States, we did not collect race and ethnicity data, as these were deemed irrelevant to assessment of the two fecal collection devices and the reproducibility of the 16S rRNA sequences. The associations that we found with medications, age and body mass index must be viewed with caution, because they may have arisen by chance due to multiple comparisons. Independent replication of our findings is needed. Finally, we observed low reproducibility for rare taxa, probably attributable to sampling various parts of heterogeneous stool. The implications are that statistical power to detect cancer associations with rare taxa will be low unless studies are much larger or collect homogenized feces or many more samples.
Given the paucity of understanding of the functions of the gut microbiota and its transient or sustained effects on the human host, our study, despite its limitations, makes a useful contribution by describing methods to effectively and reproducibly collect fecal specimens at home and to generate data on the diversity and composition of the fecal microbiome. Methods like these, applied to analytic epidemiologic research, will clarify how the gut microbiota affects myriad aspects of human health.
Supplementary Material
Acknowledgements
We thank Mr. Andrew Para for help with collection of the specimens and data; and Drs. Charles Rabkin, Eric Engels and Christian Abnet for helpful comments on the manuscript. We are especially grateful to the study participants.
Financial support: This project (Z01-CP010214) was funded by the Intramural Research Program of the National Cancer Institute, National Institutes of Health.
Glossary. Terms used in this study
- 16S rRNA
A universal obligate gene present in all bacteria, but not in other microbes.
- 16S rRNA amplicons
Numerous DNA copies of a targeted part of the 16S rRNA gene, obtained by polymerase chain reaction.
- Alpha diversity
The biodiversity within a specified ecologic niche, starting with richness, which is the number of taxa in the niche.
- Barcode
A unique sequence of DNA used to link each 16S rRNA amplicon to the sample from which it came.
- Beta diversity
A comparison of biodiversity between and among ecologic niches, particularly the ratios of unique taxa to shared taxa between and among niches.
- Microbe
A microscopic organism, usually including bacteria, archea, fungi, protozoa, and sub-microscopic organisms (e.g., viruses).
- Microbiota
The collection or community of all microbes in a specified ecologic niche.
- Microbiome
The collection of all genes in a microbiota.
- Operational taxonomic unit (OTU)
A terminal node in a phylogenetic analysis; implies an organism.
- Participant
A human volunteer for the research study.
- Sample
A portion of the niche (stool specimen). Each participant provided four samples of the stool specimen.
- Specimen
A stool that was sampled by the participant. The specimen was the ecologic niche for this study.
- Pyrosequencing
A proprietary method for sequencing single strands of DNA in a complex mixture.
- Relative abundance
The proportion (prevalence) of a taxon in an ecologic niche.
- Sequence reads
DNA sequences, either before editing (raw) or after editing (final) for quality specifications.
- Shannon index
A measure of alpha diversity in which richness is adjusted by each taxon's relative abundance.
- Taxonomy
The practice and science of classification.
- Taxon (pleural, taxa)
A distinct group of organisms.
Footnotes
Potential conflicts of interest: None.
References
- 1.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Relman DA. Microbial genomics and infectious diseases. N Engl J Med. 2011;365(4):347–357. doi: 10.1056/NEJMra1003071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474(7351):327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Plottel CS, Blaser MJ. Microbiome and malignancy. Cell Host Microbe. 2011;10(4):324–335. doi: 10.1016/j.chom.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu B, Wang X, Li L. Human gut microbiome: the second genome of human body. Protein Cell. 2010;1(8):718–725. doi: 10.1007/s13238-010-0093-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turnbaugh PJ, Quince C, Faith JJ, McHardy AC, Yatsunenko T, Niazi F, et al. Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc Natl Acad Sci U S A. 2010;107(16):7503–7508. doi: 10.1073/pnas.1002355107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McNulty NP, Yatsunenko T, Hsiao A, Faith JJ, Muegge BD, Goodman AL, et al. The impact of a consortium of fermented milk strains on the gut microbiome of gnotobiotic mice and monozygotic twins. Sci Transl Med. 2011;3(106):106ra106. doi: 10.1126/scitranslmed.3002701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lauber CL, Zhou N, Gordon JI, Knight R, Fierer N. Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol Lett. 2010;307(1):80–86. doi: 10.1111/j.1574-6968.2010.01965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu GD, Lewis JD, Hoffmann C, Chen YY, Knight R, Bittinger K, et al. Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol. 2010;10:206. doi: 10.1186/1471-2180-10-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salonen A, Nikkila J, Jalanka-Tuovinen J, Immonen O, Rajilic-Stojanovic M, Kekkonen RA, et al. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J Microbiol Methods. 2010;81(2):127–134. doi: 10.1016/j.mimet.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 11.Maukonen J, Simoes C, Saarela M. The currently used commercial DNA extraction methods give different results of clostridial and Actinobacterial populations derived from human fecal samples. FEMS Microbiol Ecol. 2011 doi: 10.1111/j.1574-6941.2011.01257.x. [DOI] [PubMed] [Google Scholar]
- 12.Kuczynski J, Lauber CL, Walters WA, Parfrey LW, Clemente JC, Gevers D, et al. Experimental and analytical tools for studying the human microbiome. Nat Rev Genet. 2012;13(1):47–58. doi: 10.1038/nrg3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flores R, Shi J, Gail MH, Ravel J, Goedert JJ. Assessment of the human fecal microbiota: I. Measurement and reproducibility of selected enzymatic activities. 2011. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, et al. The NIH Human Microbiome Project. Genome Res. 2009;19(12):2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, et al. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009;37(Database issue):D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecology. 2001;26:32–46. [Google Scholar]
- 19.Bray JR, Curtis JT. An ordination of upland forest communities of southern Wisconsin. Ecological Monographs. 1957;27:325–349. [Google Scholar]
- 20.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326(5960):1694–1697. doi: 10.1126/science.1177486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Claesson MJ, Cusack S, O'Sullivan O, Greene-Diniz R, de WH, Flannery E, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4586–4591. doi: 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 24.Jakobsson HE, Jernberg C, Andersson AF, Sjolund-Karlsson M, Jansson JK, Engstrand L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One. 2010;5(3):e9836. doi: 10.1371/journal.pone.0009836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manichanh C, Reeder J, Gibert P, Varela E, Llopis M, Antolin M, et al. Reshaping the gut microbiome with bacterial transplantation and antibiotic intake. Genome Res. 2010;20(10):1411–1419. doi: 10.1101/gr.107987.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reeves AE, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile Infection. Gut Microbes. 2011;2(3):145–158. doi: 10.4161/gmic.2.3.16333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2011 doi: 10.1101/gr.126516.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2011 doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arumugam M, Raes J, Pelletier E, Le PD, Yamada T, Mende DR, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loo VG, Bourgault AM, Poirier L, Lamothe F, Michaud S, Turgeon N, et al. Host and pathogen factors for Clostridium difficile infection and colonization. N Engl J Med. 2011;365(18):1693–1703. doi: 10.1056/NEJMoa1012413. [DOI] [PubMed] [Google Scholar]
- 33.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312(5778):1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tiihonen K, Ouwehand AC, Rautonen N. Human intestinal microbiota and healthy ageing. Ageing Res Rev. 2010;9(2):107–116. doi: 10.1016/j.arr.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 35.Agans R, Rigsbee L, Kenche H, Michail S, Khamis HJ, Paliy O. Distal gut microbiota of adolescent children is different from that of adults. FEMS Microbiol Ecol. 2011;77(2):404–412. doi: 10.1111/j.1574-6941.2011.01120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biagi E, Nylund L, Candela M, Ostan R, Bucci L, Pini E, et al. Through ageing, and beyond: gut microbiota and inflammatory status in seniors and centenarians. PLoS One. 2010;5(5):e10667. doi: 10.1371/journal.pone.0010667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.