

Abstract
Objective
To report the first case of surgical treatment for severe kyphoscoliosis associated with respiratory disorder in a patient with congenital neuromuscular disease with uniform type 1 fibers (CNMDU1), including management of the possible onset of malignant hyperthermia (MH) in general anesthesia.
Summary of background data
CNMDU1 is rare among congenital neuromuscular diseases, and surgery for spinal deformity in CNMDU1 has not been described. Onset of MH in general anesthesia is a concern in this disease.
Methods
A 13-year-old female with motor retardation, suspected myopathy, and severe spinal deformity was followed at another pediatric hospital before referral to Meijo Hospital. Symptoms at the initial consultation were mild general muscular weakness and muscular atrophy. The rib hump was 60° and trunk balance was poor. The tendon reflex showed hyporeflexia, and blood tests were normal. Vital capacity was 0.69 L and forced expiratory volume percentage in 1 s was 75.5%, showing a restrictive and obstructive ventilatory defect. A plain radiograph showed severe kyphoscoliosis with thoracic scoliosis of 130° (T5–L1) and thoracic kyphosis of 110° (T2–T12) with almost no flexibility in bending or traction film.
Results
After preoperative halo traction for 2 months, one-stage anterior and posterior correction and fusion from T2 to L3 was conducted. MH did not occur, but recovery of respiratory function required 8 days by intubation after surgery. Postoperatively, thoracic kyphosis improved to 25° and thoracic scoliosis was 66° (correction rate: 49%). Pathological results of an intraoperative muscle biopsy from the paraspinal muscles confirmed the diagnosis of CNMDU1. At 6 years after surgery, the patient has no problems in daily life and no respiratory difficulty.
Conclusion
Spinal deformity in CNMDU1 has a risk of severe progression, which makes early diagnosis by biopsy important. The surgery may be recommended before severe progression of spinal deformity and respiratory disorder. Perioperative MH is a concern, but can be managed by appropriate procedures.
Keywords: Congenital neuromuscular disease with uniform type 1 fibers, Congenital neuromuscular disease, Severe kyphoscoliosis, Surgical correction, Malignant hyperthermia
Introduction
Congenital neuromuscular disease with uniform type 1 fibers (CNMDU1) is an extremely rare disease that was first described by Une et al. [1]. In 1983, Oh and Danon described CNMDU1 as a distinct nonprogressive congenital neuromuscular disease with features of delayed motor development with early onset, mild proximal muscle weakness, hyporeflexia, normal levels of serum muscle enzymes, including creatine kinase (CK), uniform type 1 fibers, and nonprogression of the disease course [2]. The etiology is unknown, but mutation of the ryanodine receptor gene one (RYR1) may be involved in CNMDU1 [3].
Fewer than 20 cases of CNMDU1 have been reported in the literature. Mental retardation in CNMDU1 has been reported [4, 5], but most cases are normal. CNMDU1 may also be complicated with high palate, torticollis, joint contracture, congenital hip dislocation, and scoliosis [6–11]. Since the disease is rare and most case reports are written from the perspective of pediatricians, the details and treatment of scoliosis in CNMDU1 have not been described.
In this report, we describe the first case of surgical treatment for CNMDU1 with severe kyphoscoliosis and preoperative respiratory impairment, including management of the possible onset of malignant hyperthermia (MH) in general anesthesia.
Case report
The patient was a female aged 13 years. At birth, she had no abnormality other than neonatal asphyxia. Bilateral congenital hip dislocation was found at 3 months after birth and torticollis was discovered at 1 year. The patient began to walk at the age of 1.5 years and walked independently at 2 years; however, mild motor retardation was subsequently observed. She had normal language development and no mental retardation. In her family, her mother and aunt had slight muscle weakness, but no definitive diagnosis. The patient was suspected to have myopathy, but was followed up without treatment or diagnosis by biopsy at another pediatric hospital.
At 3 years of age, mild scoliosis was found, but the patient was not treated with a brace due to her parents’ request. She was diagnosed with progression of spinal deformity at another hospital, but surgery was considered to be difficult due to risks in the surgical outcome and with general anesthesia. At 13 years old, the patient developed respiratory disorder and was referred to Meijo Hospital for consultation regarding surgery.
Symptoms at the initial consultation were mild general muscular weakness (muscle manual test: 4/5) and muscular atrophy. Her height and weight were 136 cm and 22 kg, respectively. The rib hump was 60°, and trunk balance was poor. The tendon reflex showed hyporeflexia, but the patient did not have other neurological abnormalities, fasciculation, pseudohypertrophy, Gowers sign or joint laxity. She could walk independently, but with a slouch due to her kyphoscoliosis. The results of blood tests including CK were within normal ranges. In a respiratory function test, vital capacity (VC) was 0.69 L (percentage of expected value: 28.2%) and forced expiratory volume percentage in 1 s (FEV 1.0%) was 75.5% (percentage of expected value: 85.1%), showing a restrictive and obstructive ventilatory defect.
Plain radiography showed severe kyphoscoliosis with thoracic scoliosis of 130° (T5–L1) and thoracic kyphosis of 110° (T2–T12) (Fig. 1a, 1b), with almost no flexibility in bending or traction film (Fig. 1c, 1d, 1e). Preoperative CT showed marked right thoracic deformity associated with severe scoliosis, leading to respiratory disorder (Fig. 2). Whole spine MRI showed no medullar abnormality.
Fig. 1.

Preoperative plain radiographs, a plain radiograph showing severe scoliosis of 44° (T1–T5), 130° (T5–L1), and 43° (L1–L5); b lateral radiograph showing severe thoracic kyphosis of 110° (T2–T12) and 64° (T12–L5); c–e bending (c, d) and traction (e) radiographs showing rigid thoracic scoliosis that could be corrected only to a limited extent
Fig. 2.

Preoperative CT findings. deformity, and narrowing of the thorax were apparent due to severe spinal deformity. Vital capacity was 0.69 L, indicating ventilatory impairment
The patient with kyphoscoliosis complained of respiratory difficulty, and therefore surgery was considered. Her parents also wanted her to undergo surgery. A preoperative biopsy for definitive diagnosis was proposed due to the possibility of congenital neuromuscular disease (CMD); however, the patient and her parents did not want an invasive preoperative examination, and preferred an intraoperative biopsy. The surgery was scheduled after the perioperative risks were explained to her parents, since there was a concern that surgery could be associated with long fusion and that general anesthesia may cause MH-associated myopathy. Parental informed consent was obtained after this explanation was given.
Preoperative halo traction using six pins was performed for 2 months. There were no problems associated with halo traction and no additional treatment was needed, such as changing the pin position due to infection or loosening. After halo traction for 2 months, thoracic scoliosis improved to 84° (T5–L1) and thoracic kyphosis to 72° (T2–T12). VC was 0.75 L (percentage of expected value: 30.0%) and FEV 1.0% was 80.6% (percentage of expected value: 91.1%), showing slight improvement of respiratory function after halo traction. Intra- and postoperative respiratory control and MH were thoroughly discussed with an anesthesiologist, who recommended use of intravenous propofol and fentanyl, instead of an inhalational anesthetic and depolarizing muscle relaxants, for prevention of MH. Consequently, a procedure of one-stage anterior release and posterior correction and fusion was designed. After surgery, respiratory control using intubation was planned to prevent atelectasis caused by sputum.
In the surgery, anterior thoracotomy was first performed, in which anterior discs from T6 to T12 were dissected and fused with rib grafts. Subsequently, posterior T2 to L3 were corrected and fixed with implants (TSRH-RP, Medtronic Sofamor Danek) and posterior iliac bone grafting. Pedicle screws, hooks, and sublaminar wires were used, but pedicle screws around the thoracic apex could not be used due to the small diameter of the thoracic pedicles. For surgical correction, Ponte osteotomies from T5 to T12 were first performed, but severe rigid kyphosis restricted it to slight correction. Therefore, the cantilever technique was applied for the severe kyphoscoliosis. During use of the cantilever technique, the right T2 pedicle hook made a small crack at the right T2 lamina, and the right T2 residual lamina was fixed with sublaminar wire instead of a pedicle hook. Intraoperative motor-evoked potentials indicated no concerns regarding paralysis. The total surgical time was 9 h 10 min and the estimated blood loss was 383 mL. General anesthesia was induced with vecuronium and propofol and maintained during surgery with air, oxygen, propofol, and fentanyl. There were no problems with anesthesia and no development of MH.
After surgery, respiratory control was performed by endotracheal intubation, as planned, and the patient was weaned 8 days after surgery. This long period before weaning was due to high levels of sputum. The patient wore a hard corset and began ambulation 2 weeks after surgery. She was subsequently discharged from hospital walking independently.
A postoperative plain radiograph showed that thoracic kyphosis improved from 110° to 25°; however, postoperative thoracic scoliosis was 66° and the correction rate was only 49% (Fig. 3a, 3b). At 6 years after surgery, the patient has no specific problems in daily life and can walk independently without a slouch. Her VC is 0.82 L (percentage of expected value: 32.9%) and FEV 1.0% is 87.8% (percentage of expected value: 99.5%), which indicate an improved respiratory outcome compared to her preoperative status.
Fig. 3.
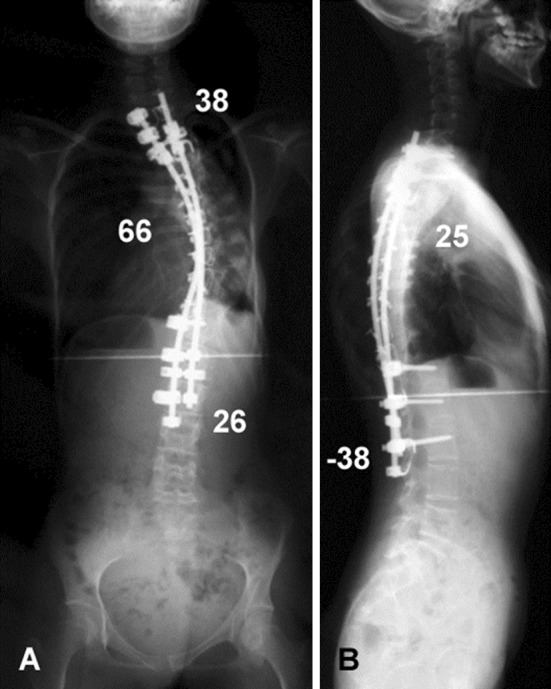
Postoperative plain radiographs. a Frontal radiograph; b lateral radiograph. Thoracic kyphosis was corrected to 25°, but the postoperative thoracic curve was 66° and the percentage correction was only 49%
The pathological results of muscle biopsy sampling from several points in the right and left paraspinal muscles in the surgical area showed that all fibers were type 1 with slightly different sizes. No type 2 fibers, nemaline rods, central core, core structure, ragged red fibers, peripheral halo, or rounding cells were found. These results led to a final diagnosis of CNMDU1 (Fig. 4).
Fig. 4.

Pathological findings. Almost 100% of fibers were type 1 with slightly different sizes. There were no type 2 fibers. Nemaline rods, central core, ragged red fibers, peripheral halo, and rounding cells were not found. a NADH-TR; b ATPase, pH 4.5, ×200)
Discussion
CNMDU1 is an extremely rare CMD. Most cases develop at an early age, but the disease is not progressive and has a good prognosis. Mild muscular weakness and motor retardation are observed. CMDs include nemaline myopathy [12], central core disease [13], myotubular myopathy, [14], and congenital fiber-type disproportion [15]. Nemaline myopathy and central core disease are often complicated by scoliosis and MH [16–18]. CMDs are classified by the characteristics of the muscle fiber structure in a pathological examination. In most CMDs, there is a type 1 predominance with relatively small type 1 fibers. In normal children, about one-third of the fibers are type 1 [10], whereas biopsies revealing more than 55% type 1 fibers are considered to show type I predominance. In CNMDU1, more than 99% of the muscle fibers are type 1, and characteristic muscle fiber structures such as nemaline rods, central core, and core structures are absent [2, 19].
This is the first case report of kyphoscoliosis associated with CNMDU1. It is unclear why spinal deformity is a complication of CMD; however, studies have shown type 1 fiber predominance in the paraspinous muscles on the convex side of the scoliosis [20, 21]. In our case, there was no pathological difference between the convex and concave sides of the scoliosis and no relationship between the muscle fibers and spinal deformity.
The issue of spinal deformity was important in our patient, since surgery was performed only after she had developed rigid and severe kyphoscoliosis associated with respiratory disorder. Kyphosis was corrected well by surgery, but scoliosis was corrected less sufficiently, and it took 8 days for recovery of respiratory function after surgery. These results show that early diagnosis by biopsy is important before progression to advanced scoliosis. Previous cases of central core disease with mild muscle weakness and motion retardation have shown rapid progression, while CMD was not diagnosed [16, 22]. Similarly, CNMDU1 complicated with spinal deformity may be followed up incorrectly as idiopathic scoliosis, with resulting rapid progression. Therefore, it is important for doctors to understand the pathogenesis of CNMDU1 and make a definite diagnosis. Achievement of good correction of spinal deformity and facilitation of postoperative respiratory control may require surgery before advancement of spinal deformity by early diagnosis of CNMDU1. Biopsy from paraspinal muscle only may not be sufficient for a definite diagnosis [18], and biopsies from areas such as the biceps, triceps, quadriceps, or gastrocnemius may be required. If CMD (including CNMDU1) is suspected based on findings such as mild muscle weakness and complicates with even mild spinal deformity, biopsy may be recommended in the early stage.
A further concern is development of MH under general anesthesia. The cause of MH may be associated with mutation of RYR1, a calcium release channel in the sarcoplasmic reticulum [23–25]. Central core disease may also be related to RYR1 mutation and is likely to be complicated with MH [26–28]. An RYR1 mutation has also been suggested to be involved in CNMDU1 [3]. Therefore, perioperative MH is a concern in patients with suspected CNMDU1, which is another reason that preoperative definite diagnosis by biopsy is essential. Since inhalation anesthetics and depolarizing muscle relaxants can induce MH [29], these agents were not used in our patient. Similarly to a case report describing the use of general anesthesia in cranioplasty in a CNMDU1 patient [30], we carefully chose agents for general anesthesia and avoided the onset of MH and prolonged anesthesia due to muscle relaxants. For a patient with a history of MH, a provocation test may also be useful. Miyamoto used this test in patients with a central core disease with an actual history of MH, and surgical correction of scoliosis under general anesthesia was successfully performed without MH [17]. Using these approaches, we suggest that surgery for spinal deformity associated with CNMDU1 can be performed safely and with a good outcome.
Conclusion
This is the first description of surgery for severe spinal deformity associated with respiratory disorder in a patient with CNMDU1. Though a single case report does not permit broad treatment recommendations to be made, spinal deformity in CNMDU1 may have a risk of severe progression, and therefore early diagnosis by biopsy may be important. Perioperative MH is a concern, but can be avoided with appropriate management of general anesthesia.
Acknowledgments
The authors thank Ms. Keiko Ogura for her assistance throughout this study.
Conflict of interest
None of the authors has any potential conflict of interest.
References
- 1.Une Y, Haraguchi H. Congenital myopathy with type 2 fiber deficiency and without specific structural abnormalities. No To Hattatsu. 1980;12:554–556. [Google Scholar]
- 2.Oh SJ, Danon MJ. Nonprogressive congenital neuromuscular disease with uniform type 1 fiber. Arch Neurol. 1983;40:147–150. doi: 10.1001/archneur.1983.04050030041007. [DOI] [PubMed] [Google Scholar]
- 3.Sato I, Wu S, Ibarra MC, Hayashi YK, Fujita H, Tojo M, Oh SJ, Nonaka I, Noguchi S, Nishino I. Congenital neuromuscular disease with uniform type 1 fiber and RYR1 mutation. Neurology. 2008;70:114–122. doi: 10.1212/01.wnl.0000269792.63927.86. [DOI] [PubMed] [Google Scholar]
- 4.Giometti CS, Danon MJ. The expression of myosin light chains and tropomyosin in human muscle biopsies with histochemical type 1 and type 2 fiber deficiency. Muscle Nerve. 1990;13:209–214. doi: 10.1002/mus.880130307. [DOI] [PubMed] [Google Scholar]
- 5.Jong YJ, Huang SC, Liu GC, Chiang CH. Mental retardation in congenital nonprogressive myopathy with uniform type 1 fibers. Brain Dev. 1991;13:444–446. doi: 10.1016/S0387-7604(12)80046-5. [DOI] [PubMed] [Google Scholar]
- 6.Huang ML, Tsai CH, Lee CC. Congenital myopathy with uniform type 1 fiber predominance and type 2 fiber hypoplasia: report of one case. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi. 1998;39:62–64. [PubMed] [Google Scholar]
- 7.Kitagawa Y, Hashimoto K, Kuriyama M. Hypertrophic branchial myopathy with uniform predominance of type 1 fibres. Case report. Scand J Plast Reconstr Surg Hand Surg. 2000;34:391–396. doi: 10.1080/028443100750059200. [DOI] [PubMed] [Google Scholar]
- 8.Korematsu S, Imai K, Sato K, Maeda T, Suenobu S, Kojo M, Izumi T. Congenital neuromuscular disease with uniform type-1 fibers, presenting early stage dystrophic muscle pathology. Brain Dev. 2006;28:63–66. doi: 10.1016/j.braindev.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 9.Na SJ, Kang SW, Lee KO, Lee KY, Kim TS, Choi YC. A case of congenital neuromuscular disease with uniform type 1 fiber. Yonsei Med J. 2004;45:150–152. doi: 10.3349/ymj.2004.45.1.150. [DOI] [PubMed] [Google Scholar]
- 10.Sakamoto HM, Yoshioka M, Tsuji M, Kuroki S, Higuchi Y, Nonaka I, Nishino I. A case of congenital neuromuscular disease with uniform type 1 fibers. Brain Dev. 2006;28:202–205. doi: 10.1016/j.braindev.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 11.Tojo M, Ozawa M, Nonaka I. Central core disease and congenital neuromuscular disease with uniform type 1 fibers in one family. Brain Dev. 2000;22:262–264. doi: 10.1016/S0387-7604(00)00108-X. [DOI] [PubMed] [Google Scholar]
- 12.Shy GM, Engel WK, Somers JE, Wanko T. Nemaline myopathy. A new congenital myopathy. Brain. 1963;86:793–810. doi: 10.1093/brain/86.4.793. [DOI] [PubMed] [Google Scholar]
- 13.Magee KR, Shy GM. A new congenital non-progressive myopathy. Brain. 1956;79:610–621. doi: 10.1093/brain/79.4.610. [DOI] [PubMed] [Google Scholar]
- 14.Blondeau F, Laporte J, Bodin S, Superti-Furga G, Payrastre B, Mandel JL. Myotubularin, a phosphatase deficient in myotubular myopathy, acts on phosphatidylinositol 3-kinase and phosphatidylinositol 3-phosphate pathway. Hum Mol Genet. 2000;9:2223–2229. doi: 10.1093/oxfordjournals.hmg.a018913. [DOI] [PubMed] [Google Scholar]
- 15.Clarke NF, North KN. Congenital fiber type disproportion–30 years on. J Neuropathol Exp Neurol. 2003;62:977–989. doi: 10.1093/jnen/62.10.977. [DOI] [PubMed] [Google Scholar]
- 16.Mertz KD, Jost B, Glatzel M, Min K. Progressive scoliosis in central core disease. Eur Spine J. 2005;14:900–905. doi: 10.1007/s00586-005-0938-y. [DOI] [PubMed] [Google Scholar]
- 17.Miyamoto K, Shimizu K, Matsumoto S, Sumida H, Iida H, Hosoe H. Surgical treatment of scoliosis associated with central core disease: minimizing the effects of malignant hyperthermia with provocation tests. J Pediatr Orthop B. 2007;16:239–242. doi: 10.1097/BPB.0b013e3280107014. [DOI] [PubMed] [Google Scholar]
- 18.Nagai T, Tsuchiya Y, Maruyama A, Takemitsu M, Nonaka I. Scoliosis associated with central core disease. Brain Dev. 1994;16:150–152. doi: 10.1016/0387-7604(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 19.Pellegrini G, Barbieri S, Moggio M, Cheldi A, Scarlato G, Minetti C. A case of congenital neuromuscular disease with uniform type I fibers, abnormal mitochondrial network and jagged Z-line. Neuropediatrics. 1985;16:162–166. doi: 10.1055/s-2008-1059533. [DOI] [PubMed] [Google Scholar]
- 20.Bylund P, Jansson E, Dahlberg E, Eriksson E. Muscle fiber types in thoracic erector spinae muscles. Fiber types in idiopathic and other forms of scoliosis. Clin Orthop Relat Res. 1987;214:222–228. [PubMed] [Google Scholar]
- 21.Ford DM, Bagnall KM, McFadden KD, Greenhill BJ, Raso VJ. Paraspinal muscle imbalance in adolescent idiopathic scoliosis. Spine (Phila Pa 1976) 1984;9:373–376. doi: 10.1097/00007632-198405000-00008. [DOI] [PubMed] [Google Scholar]
- 22.Merlini L, Mattutini P, Bonfiglioli S, Granata C. Non-progressive central core disease with severe congenital scoliosis: a case report. Dev Med Child Neurol. 1987;29:106–109. doi: 10.1111/j.1469-8749.1987.tb02114.x. [DOI] [PubMed] [Google Scholar]
- 23.Esteve E, Eltit JM, Bannister RA, Liu K, Pessah IN, Beam KG, Allen PD, Lopez JR. A malignant hyperthermia-inducing mutation in RYR1 (R163C): alterations in Ca2+ entry, release, and retrograde signaling to the DHPR. J Gen Physiol. 2010;135:619–628. doi: 10.1085/jgp.200910328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Girard T, Suhner M, Levano S, Singer M, Zollinger A, Hofer CK. A fulminant malignant hyperthermia episode in a patient with ryanodine receptor gene mutation p.Tyr522Ser. Anesth Analg. 2008;107:1953–1955. doi: 10.1213/ane.0b013e3181857903. [DOI] [PubMed] [Google Scholar]
- 25.Kaufmann A, Kraft B, Michalek-Sauberer A, Weigl LG. Novel ryanodine receptor mutation that may cause malignant hyperthermia. Anesthesiology. 2008;109:457–464. doi: 10.1097/ALN.0b013e318182a93b. [DOI] [PubMed] [Google Scholar]
- 26.Foster RN, Boothroyd KP. Caesarean section in a complicated case of central core disease. Anaesthesia. 2008;63:544–547. doi: 10.1111/j.1365-2044.2007.05411.x. [DOI] [PubMed] [Google Scholar]
- 27.Quinlivan RM, Muller CR, Davis M, Laing NG, Evans GA, Dwyer J, Dove J, Roberts AP, Sewry CA. Central core disease: clinical, pathological, and genetic features. Arch Dis Child. 2003;88:1051–1055. doi: 10.1136/adc.88.12.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Treves S, Vukcevic M, Jeannet PY, Levano S, Girard T, Urwyler A, Fischer D, Voit T, Jungbluth H, Lillis S, Muntoni F, Quinlivan R, Sarkozy A, Bushby K, Zorzato F. Enhanced excitation-coupled Ca(2+) entry induces nuclear translocation of NFAT and contributes to IL-6 release from myotubes from patients with central core disease. Hum Mol Genet. 2011;20:589–600. doi: 10.1093/hmg/ddq506. [DOI] [PubMed] [Google Scholar]
- 29.Denborough M. Malignant hyperthermia. Lancet. 1998;352:1131–1136. doi: 10.1016/S0140-6736(98)03078-5. [DOI] [PubMed] [Google Scholar]
- 30.Okutani R, Arashi D, Tsujii K. Anesthetic case in a child with congenital neuromuscular disease with uniform type 1 fibers (CNMDU1) J Anesth. 2010;24:797–800. doi: 10.1007/s00540-010-0977-3. [DOI] [PubMed] [Google Scholar]