

Abstract
The Wiskott–Aldrich syndrome (WAS) is an X-linked disorder characterized by eczema, thrombocytopenia and immunodeficiency. Hematopoietic cell transplantation can cure the disease and gene therapy is being tested as an alternative treatment option. In this study, we assessed the use of foamy virus (FV) vectors as a gene transfer system for WAS, using a Was knockout (KO) mouse model. Preliminary experiments using FV vectors expressing the green fluorescent protein under the transcriptional control of the endogenous WAS promoter or a ubiquitously acting chromatin opening element allowed us to define transduction conditions resulting in high (>40%) and long-term in-vivo marking of blood cells after transplantation. In following experiments, Was KO mice were treated with FV vectors containing the human WAS complementary DNA (cDNA). Transplanted animals expressed the WAS protein (WASp) in T and B lymphocytes, as well as platelets and showed restoration of both T-cell receptor-mediated responses and B-cell migration. We also observed recovery of platelet adhesion and podosome formation in dendritic cells (DCs) of treated mice. These data demonstrate that FV vectors can be effective for hematopoietic stem cell (HSC)-directed gene correction of WAS.
Introduction
The Wiskott–Aldrich syndrome (WAS) is an X-linked immunodeficiency, characterized by thrombocytopenia, eczema, recurrent infections, and high incidence of autoimmunity and malignancies.1 The disease is caused by mutations of the WAS gene, which result in abnormal expression and function of the WAS protein (WASp).2 WASp is a cytosolic adaptor molecule expressed exclusively in cells of hematopoietic origin where it plays a major role in the regulation of cytoskeleton reorganization. A number of cellular binding partners of WASp have been identified, suggesting its broad implication in signal transduction and various cellular processes.3
Patients affected with WAS can be cured by allogeneic hematopoietic cell transplantation. In the absence of HLA-matched donors, however, high incidence of hematopoietic cell transplantation-related complications is observed.4 Gene therapy has therefore been proposed and developed as a highly desirable alternative treatment option for patients lacking suitable donors.
Previous in-vivo preclinical work has demonstrated that corrective gene transfer into bone marrow (BM) hematopoietic stem cells (HSC), using both gammaretrovirus- and lentivirus-based vectors, can achieve restoration of the immunological defects in Was knockout (KO) mice,5,6,7,8 and supported the recent clinical debut of gammaretrovirus- and lentivirus-mediated gene therapy applications.9,10
Vectors based on simian foamy virus (FV) present several advantages over gammaretrovirus and lentivirus vectors in that FV are not human pathogens11 and their integrations in the genome of HSCs of the nonhuman primate natural host is not associated with insertional oncogenesis.12 In addition, recent studies have shown that FV vectors efficiently transduce murine13 and human hematopoietic progenitor cells,14,15,16 and can correct HSC defects in large animal models17 often following short transduction protocols.17,18,19 Furthermore, it has been shown that FV vectors have more random genomic integration patterns compared to gammaretrovirus and lentivirus vectors,20 thus making these constructs less prone to the risk of insertional oncogenesis. Altogether, FV vectors represent a class of gene transfer vectors with very appealing safety and efficacy characteristics for clinical gene transfer into HSC.
For these reasons, we set out to assess FV vectors as an alternative gene transfer system for WAS, and to evaluate their efficacy in a Was KO mouse gene therapy model.
Results
Production of FV vectors for WAS gene expression
FV vectors were constructed to express the human WAS or enhanced green fluorescent protein (EGFP) complementary DNAs (cDNAs) under the control of two different promoters (Figure 1a). To obtain physiological regulation of WAS expression, the endogenous promoter sequence (Wa0.5) was isolated from the 5′ flanking region of the WAS genomic sequence. This promoter is known to be tissue specific and less likely to transactivate genes flanking vector insertion sites compared to strong retroviral long-terminal repeats.8 As an exogenous promoter, a 631-bp fragment (UCOE631) was isolated from the 2.6 kb A2UCOE sequence,21 which consists of a methylation-free CpG island without classic enhancer activity. UCOE631 was able to promote expression of EGFP at comparable levels to the original 2.6 kb sequence in K562 cells (data not shown).
Figure 1.

Structure and in vitro analysis of foamy virus vectors.(a) The structure of the foamy virus vectors. Two different internal promoters we used. Wiskott–Aldrich syndrome (WAS) endogenous promoter (Wa0.5) was derived from the 0.5-kb 5′-flanking sequence of the WAS gene for physiological expression. A 631-bp fragment of the UCOE from human HNRPA2B1-CBX3 locus was characterized for completely stable transgene expression. Desired promoter sequences and transgenes are inserted into vector plasmid. (b) WAS protein (WASp) expression in a FV vector-transduced T-cell line established from a WAS patient. Western blot analysis was performed 4 weeks after transduction. CMV, cytomegalovirus; LTR, long-terminal repeat.
A human lymphotropic virus type 1-transformed T-cell line established from patient WAS16 was transduced with the wW and uW vectors and analyzed for WASp expression. Expression of WASp became detectable by western blot (WB) analysis around 10 days after transduction and intensified gradually over time in culture, becoming increasingly stronger 2–3 weeks after transduction (Figure 1b). These data demonstrate that the wW and uW FV vectors can effectively transfer and express the WAS cDNA in lymphoid cells.
Multiple exposures increase FV transduction efficiency in hematopoietic progenitors
In preliminary experiments, Lin-BM cells were prestimulated with growth factors for 48 hours and then transduced for 17–24 hours with FV vectors encoding EGFP under the transcriptional control of either the Wa0.5 endogenous promoter (wE) or the UCOE631 ubiquitous promoter (uE) at a multiplicity of infection of 10. To assess the transduction efficiency of hematopoietic progenitors, cells were then plated in methylcellulose medium. After 7 days, the proportion of EGFP+ colonies was <40% by flow cytometry (FACS) analysis (Figure 2a), with no significant differences noted between wE and uE vectors. Transduced cells were also intravenously injected into irradiated mice (2.5 × 105/mouse). Five to six months after transplantation, the percentage of peripheral blood T-cells expressing EGFP was 8–12%. These levels of expression were maintained for >12 months (Figure 2b).
Figure 2.

Transduction efficiency in hematopoietic progenitors and expression level of transgene in vivo by single or repeated infection. Lineage marker depleted (Lin-) and Sca-1+ (Lin-/Sca-1+) cells were purified from Was knockout (KO) mice, and transduced with foamy virus (FV) vectors encoding enhanced green fluorescent protein (EGFP) (wE and uE). (a) The percentage of colony-forming unit in culture (CFU-C) colonies positive for EGFP after single transduction [multiplicity of infection (MOI) = 10] and double transduction (MOI = 20) is shown. Flow cytometry (FACS) analysis for EGFP expression was performed in CFU-C colonies after 7-day culture. (b) EGFP expression in peripheral blood T-cells 6 and 12 months after single transduction. (c) Transduction efficiency of uE in Lin-/Sca-1+ cells after repeated infection (MOI = 20 at each cycle). Peripheral blood was analyzed 8 months after transduction. EGFP expression of T cells (CD3), B-cells (B220), granulocytes (Gr1), and platelets is shown.
To increase the efficacy of the procedure, we decided to expose Lin-/Sca-1+ BM hematopoietic stem/progenitor cells to two rounds of transduction at a higher multiplicity of infection (multiplicity of infection = 20 every 12 hours) after an overnight prestimulation. We also transplanted a greater number of cells in recipient mice (5 × 105 cells/mouse). These changes in the transduction protocol resulted in a transduction efficiency of >50% by colony-forming unit in culture assay and translated to improved in vivo transduction of transplanted mice with >40% of peripheral blood T cells expressing EGFP and long-term (>8 months) transgene expression in all peripheral blood lineages (Figure 2c). These data support the use of multiple transduction cycles for efficient gene transfer of HSCs with FV vectors.
FV vector-mediated expression of WASp in vivo
To assess the efficacy of FV vectors for WASp gene transfer in vivo, we transduced Lin-/Sca-1+ cells isolated from Was KO mice using the wW and uW vectors and the double exposure protocol. Transduction efficiency in transplanted cells was assessed by real-time PCR analysis of DNA from colonies generated in colony-forming unit in culture assays. The proportion of vector-positive colonies was >50% for both vectors (Figure 3a). WB analysis of transduced BM cells showed that WASp expression was reconstituted in Was KO cells, although at lower levels compared to wild-type (WT) BM cells (Figure 3b). Six to eight months after transplantation of transduced Lin-/Sca1+ cells, the expression of human WASp was confirmed in splenocytes from mice treated with either vector. WASp expression, however, was lower than that observed in Was KO mice transplanted with WT BM (Figure 3c). The UCOE631 promoter expressed WASp at slightly higher levels than the endogenous Wa0.5 promoter in these cells. T-cells isolated from treated mice showed high levels of WASp expression, possibly due to selective advantage pressure. We also detected WASp in splenic B-cells and platelets from peripheral blood samples. Densitometry analysis showed that splenocytes, T-cells and B-cells from mice treated with wW or UW FV vectors expressed WASp at 44–75%, 37–39%, and 60–66% of WT levels, respectively. Platelets were found to express WASp at 30–33% of levels found in platelets from animals transplanted with WT Lin-/Sca-1+ cells (Figure 3c). These results indicate that FV vector-transduced hematopoietic stem/progenitor cells can give rise to peripheral multilineage hematopoietic cells with restored expression of WASp.
Figure 3.

Expression of human Wiskott–Aldrich syndrome protein (WASp) in hematopoietic cells after transduction. (a) Proportion of colony-forming unit in culture (CFU-C) colonies positive for the integrated foamy virus (FV) vector. Transduction efficiency in Lin-/Sca-1 cells was assessed by real-time-PCR before transplantation. Means ± SD of three independent experiments are shown. (b) WASp expression in transduced Lin-/Sca-1 cells. Western blot (WB) analysis was performed 5 days after transduction. (c) WASp expression in spleen cells and platelets from peripheral blood. WB analysis of whole splenocytes and spleen T and B-cells is shown. Platelets from peripheral blood were also analyzed for WASp expression. Results of densitometry analysis of band intensity are also shown.
Restoration of T- and B-cell function after FV-mediated gene therapy
In WASp deficiency, responses to T-cell receptor stimulation are remarkably reduced.22,23,24 To assess whether FV vector-mediated gene transfer can achieve restoration of WASp function, we measured T-cell proliferation and interleukin-2 (IL-2) production after T-cell receptor stimulation.
When purified CD90+ T cells were stimulated with plate-bound anti-CD3 monoclonal antibody, both T-cell proliferation and IL-2 production were markedly reduced in Was KO mice. Mice treated with FV vectors showed significant correction of both these defects (Figure 4a,b). T-cell proliferation in response to anti-CD3 in treated mice reached a Stimulation Index equal to 60–62% of that of WT mice. Similarly, the level of IL-2 production in treated mice was 57–59% of that observed in WT controls. No significant differences were observed between mice treated with the wW and uW FV vectors. As expected, mice treated with FV vectors expressing EGFP did not show recovery of T-cell function. Altogether, these results indicate that FV vector-mediated WASp gene transfer can lead to restoration of T-cell receptor-driven responses in vivo.
Figure 4.

Restoration of T- and B-cell function after gene therapy. (a) Splenic T cells were purified with immunomagnetic beads and stimulated with 2 µg/ml anti-CD3 monoclonal antibody (mAB). T-cell receptor (TCR)-driven proliferation was measured by 3H-thymidine incorporation. Results are expressed by the stimulation index (SI), the ratio between cpm of stimulated and nonstimulated cells. Wt n = 7, KO n = 7, wW n = 6, uW n = 6, enhanced green fluorescent protein (EGFP) n = 5. (b) Interleukin (IL)-2 level in conditioned supernatant was measured by ELISA technique. Wt n = 9, KO n = 11, wW n = 6, uW n = 6, EGFP n = 5. (c) Splenic B-cells were incubated in 5-µm transwell and allowed to migrate in response to medium only or medium with 50 µg/ml CXCL12. Results are expressed as the percentage of input cells that migrated. Five mice were analyzed in each group except EGFP (n = 3). Error shown is ±1 SD. *P < 0.05 as compared to KO group, Student t-test.
We also studied the effects of FV vector treatment on B-cell chemotaxis which is also known to be defective in Was KO mice.25,26 Isolated splenic B-cells were tested in a Transwell migration assay toward the chemokine, CXCL12. Basal B-cell migration in the absence of chemokine was comparable in all groups. When exposed to CXCL12, B-cells from Was KO mice untreated or treated with EGFP-transduced BM cells showed impaired migration compared to WT control cells, while B-cells from mice treated with WASp-expressing FV vectors showed significant recovery of chemotaxis (Figure 4c). Similar to the results of the T-cell function studies, no difference between the wW and uW FV vectors was observed.
Functional effects of Was gene transfer on platelets and DCs
In contrast to the human condition, Was KO mice have only modestly reduced platelet counts. Also unlike humans with WAS, platelets from Was KO mice are not characterized by reduced volume.23 These animals, therefore, are not considered good models for the thrombocytopenia feature of WAS. While we did observe higher platelet numbers in mice treated with WASp-expressing FV vectors, we could not document a significant increase (data not shown) compared to untreated controls. Because we did observe WASp expression in platelets from treated mice (Figure 3c), we set out to assess the functional relevance of such finding. In platelets, WASp binds to the calcium- and integrin-binding protein that associates with the αIIb cytoplasmic tail of platelet integrin αIIbβ3.27 Since the WASp–integrin-binding protein complex has been shown to be important for αIIbβ3-mediated cell adhesion, we assessed the ability of platelets to bind to fibrinogen, a ligand of αIIbβ3. To this aim, platelets from experimental mice were stimulated with thrombin and incubated on fibrinogen-coated culture dishes, which resulted in increased surface area in platelets from WT control mice, but not in platelets isolated from Was KO animals. Importantly, platelets from mice treated with wW and uW FV vectors showed a significant increase in average surface area, as compared to Was KO mice (Figure 5a,b), thus indicating that reconstitution of WASp expression by FV vectors can restore a functional WASp–integrin-binding protein complex in platelets of treated mice.
Figure 5.

Improvement of platelet spreading and podosome formation in bone marrow (BM)-derived dendritic cells (DCs) after gene therapy. (a) Morphology of wild-type (WT), KO, and gene therapy- treated (wW, uW) murine platelets. Purified platelets were placed on fibrinogen-coated plate for 45 minutes in the presence of 1 U/ml thrombin and imaged. Results are representative of three experiments in each group. (b) The mean surface area of adherent platelets in WT, KO, and gene therapy-treated mice, was quantified using ImageJ software. *P < 0.05 as compared to KO group, Student t-test. (c) Bone marrow cells, harvested from WT, KO, and gene therapy-treated mice, were cultured in the presence of mGM-CSF and interleukin (IL)-4 for the induction of DCs. Actin is represented in red, vinculin in green and DAPI in blue. (a) Podosome structure in DC from WT mice. (b) Absence of podosome in DC from KO mice. (c,d) Reconstitution of podosomes in gene therapy-treated mice. Arrows indicate podosomes in DCs.
Podosomes are adhesion structures principally found in monocyte-derived cells that are necessary for directional movement and that require WASp expression for optimal assembly.3,28 To assess the podosome formation in mice treated with FV vectors, BM-derived dendritic cells (BM-DCs) were differentiated from WT control, Was KO, and FV vector-treated mice, 6–8 months after transplantation. After 12 days of culture in the presence of mGM-CSF and IL-4, >90% of cells were CD11c+ in FACS analysis (data not shown). Podosome formation was assessed in two mice from each group and revealed large podosome clusters in BM-DCs from control mice and the expected, marked defect in podosome formation in cells from Was KO animals. Mice treated with Was gene transfer showed partial restoration of podosome formation (Figure 5c and Supplementary Table S1). These results show that functional defects in cytoskeleton regulation can be corrected in myeloid cells by WASp expression with FV vectors.
Molecular analysis of FV vector integrations
The number of provirus integrations in hematopoietic cells from mice treated with FV vectors was assessed 7–8 month after transplantation (Figure 6a). The average copy numbers observed in vivo in our study was consistent with previous data in the literature17,18,19,20 and showed increased marking in unfractionated splenocytes (0.84 copy/cell) and CD90+ T cells (1.17 copy/cell) compared to BM cells (0.57 copy/cell). These results are consistent with a selective advantage of WASp-expressing cells, particularly among T cells.
Figure 6.

Profile of provirus integration in transduced mice. (a) Foamy virus (FV) provirus copy number in transduced mice. Genomic DNA was extracted from whole spleen cells, splenic T cells and bone marrow (BM) of FV-treated mice 6–10 month after infusion. Copy number was determined by quantitative-PCR. (b) Position of FV integration sites. Percentages of all integration sites within 15 kb of transcriptional start sites, within genes, and within 30 kb of oncogenes is shown for FV vector-treated mice (FV) and computer-generated random sites (RND). (c) Position of FV vector integration sites relative to RefSeq gene transcription start sites.
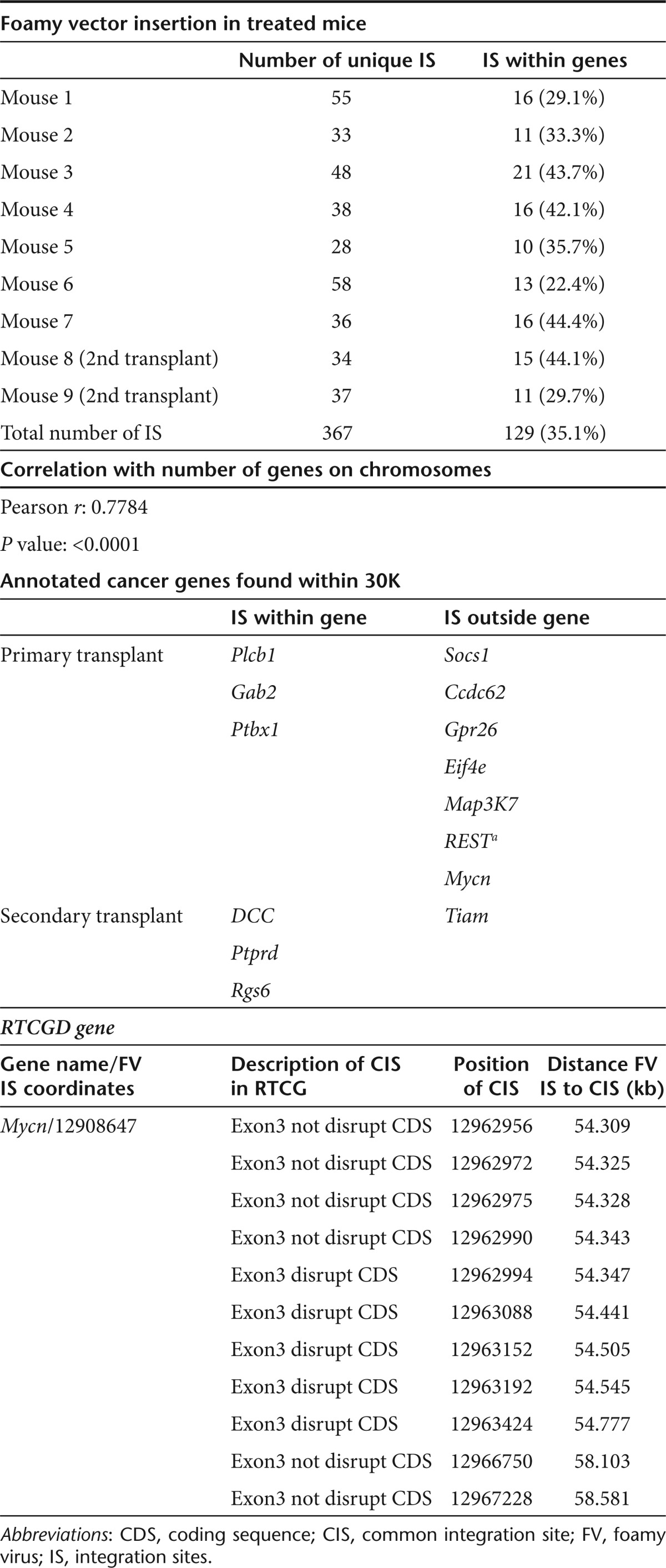
We next identified the provirus integration sites (IS) by standard linker-mediated PCR analysis.29 Genomic DNA was extracted from splenocytes of seven primary transplant recipient and two secondary recipient mice, 6–10 months after transplantation and 367 unique IS were identified. Characterization of the IS revealed that their chromosomal location correlated highly with gene density on mouse chromosomes (Supplementary Figure S1). Analysis also showed that ~35% of IS were located inside gene transcriptional units, and that 5.0% of them were located in or near oncogenes, a frequency consistent with previous studies18,20 (Figure 6b and Table 1). Of note, no integration events near the MDS-Evi1 gene or the LMO2 locus were detected and only one of the oncogenes located near the recovered IS (Mycn) was found in the RTCGD database. The location of the latter IS was outside of the Mycn gene transcriptional unit and >50 kb from those reported as common IS in RTCGD that are all located within Mycn exon 3. Finally, similar to the previously described integration pattern of FV vectors, the IS recovered from our experimental animals showed only modest preference for regions near transcriptional starts sites (Figure 6c).
Table 1. Foamy vector insertion site analysis.
Was gene transfer and expression in secondary recipients mice
BM cells harvested from three male mice treated with FV vectors were transplanted into three female recipient Was KO mice. Five to six months post-transplant recipient animals showed ~60–70% engraftment of donor cells in the spleen and BM that contained an average of 0.543 and 0.135 foamy VCN, respectively (Figure 7a–b). Lin-cells sorted from BM cells of secondary recipient animals showed detectable WASp expression (Figure 7c). These data indicate that repopulating HSCs were successfully transduced with WASp-expressing FV vectors.
Figure 7.

Vector integration in hematopoietic cells of secondary recipients. (a) Engraftment of donor cells in spleen and bone marrow cells. Percentage of donor cells was determined by Y chromosome-specific real-time PCR. (b) FV vector copy number in secondary recipient animals. Vector copy number in hematopoietic cells was determined by real-time PCR and normalized by the percentage of donor cells. n = 3. (c) Western blot analysis of Wiskott–Aldrich syndrome protein (WASp) expression in lysates of lin- bone marrow cells of secondary recipients. As control, WASp expression in lin- cells from wild-type (wt) is depicted.
Discussion
Corrective gene transfer into HSCs has long been proposed as an alternative form of treatment for WAS and the first successful results of a clinical trial have recently been reported.10 The occurrence of insertional leukemia in this trial (C Klein, personal communication, ASH annual meeting 2010) substantiates the need for development and testing of gene transfer tools alternative to gammaretroviral vectors. In this study, we investigated the efficacy of FV vectors for genetic correction of a murine model of WAS.
While requiring mitosis for transduction, the stability of the FV preintegration complex makes FV vectors attractive for transduction of quiescent cells that are stimulated to proliferate, such as HSCs.30,31,32 Several studies have shown that efficient FV vector transduction can be obtained after short exposure to vectors following brief or no prestimulation.17,18,19 Our findings that repeated transductions of cells exposed to short overnight prestimulation result in efficient gene transfer into HSCs are consistent with these data.
We observed that repeated transduction procedures roughly doubled the vector copy number in transduced cells of treated animals from 0.4–0.7 copy/cell to 0.7–1.2 copy/cell in mice treated with WASp-expressing vectors and to 1.7 copy/cell in animals treated with EGFP-expressing FV vectors, suggesting that a gene transfer protocol based on multiple exposures to FV vectors can increase overall transduction efficiency without markedly affecting the average copy number in transduced cells.
The characteristics of FV vector genomic integration are attractive in that, unlike gammaretroviral and lentiviral vectors, they do not show preference for integrating within genes. In addition, compared to gammaretroviral vectors, FV vectors have lower tendency to integrate near transcriptional start sites and CpG island and within or near oncogenes.20 Our analysis of FV IS in seven treated mice confirmed the existing notions in terms of slight preference for transcriptional start sites and low incidence of integration involving oncogenes (~5%). In comparison with the analysis of the IS a WAS lentiviral vector,33 the IS recovered from FV-treated mice appear to have targeted genomic sequences inside transcriptional units with significant less frequency (22–44% versus 46–72%) and common IS listed in the RTCGD database with similar low frequency (1 versus 2 RTCGD genes). Of note, no integrations within 500 kb of LMO2 oncogene were recovered. One mouse was found to carry an FV vector IS 630 kb from the EVI1 locus, a common IS for gammaretroviral vector-mediated myeloid malignancy in mice.34,35 These findings, together with the fact that no untoward outcomes were attributed to insertional oncogenesis in any of the treated animals, support the safety of FV as a gene transfer tool for gene therapy of WAS.
Success of a gene therapy approach for primary immunodeficiencies has been facilitated by a selective advantage of gene-corrected cells in vivo. Previous investigations have shown that selective advantage can be expected in WASp-expressing lineages, including T-, B-, and NKT cells.36,37 Mice treated with FV vectors showed expression of WASp in spleen mononuclear cells, lymphocytes (T- and B-cells) and platelets long-term after transplantations (6–8 months). WB analysis of T-cells from treated mice showed levels of WASp expression comparable to those observed in Was KO mice treated with wild-type BM. These cells contained an average of 1.17 vector copy/cells, which is consistent with physiological levels of WASp expression. In contrast, the level of WASp expression in splenocytes, B-cells, and platelets was relatively lower in mice treated with gene therapy compared to controls. While these finding may be due to lower average transduction levels (0.84 copy/cell in splenocytes), they may also reflect reduced selective advantage conveyed by WASp expression in myeloid and B-cells compared to T lineages.36,38
FV vector-treated mice showed improvement of several functional assay as compared to Was KO mice. Central consequences of WASp deficiency in T cells are defective cytokine production and cell proliferation. WASp gene transfer by FV vectors achieved restoration of these functions and rescued B-cell dysfunction as assayed by migration ability in response to CXCL12. These results are in line with prior publications that have used gammaretroviral and lentiviral vectors in the Was KO animal model5,6,7,33,39,40,41,42 and suggest that low levels of WASp expression are sufficient for functional improvement of B-cells in this model.
WASp has been implicated in αIIbβ3-mediated adhesion and the binding ability of platelets to fibrinogen, a ligand of αIIbβ3, is inhibited in WAS.27 While Was KO mice do not present profound thrombocytopenia and bleeding tendency, WASp-deficient platelets from these mice show impaired capability to spread on fibrinogen after stimulation with thrombin. Importantly, such a defect was corrected in FV vector-treated mice. As an additional effect of FV-mediated WAS gene transfer on the myeloid compartment, we observed generation of podosome-positive DC in treated mice at a frequency approaching that detected in Was KO animals transplanted with wild-type donor cells. These finding are consistent with previously published results using WASp-expressing lentiviral vectors.39,40,42 Overall, we conclude that FV vector-mediated WAS gene transfer into Lin-Sca+ cells results in multilineage WASp expression and significant functional improvement in treated mice.
One goal of our study was to compare two different internal promoting sequences for their ability for sustain WAS transgene expression and functional restoration.
The 0.5-kb endogenous WAS proximal promoter has attractive tissue-specific expression features and has been used in previous studies that showed adequate WASp expression and successful functional restoration of hematopoietic cells.7,43 The A2UCOE sequence has been known to be devoid of enhancer function21,44,45 and therefore represents an equally appealing promoting element. Our analysis of WASp expression in patients' T- and B-cell lines, short-term BM cultures and cells obtained from treated mice 3 months after transplantation did not reveal significant differences between the two promoters. Of interest, murine BM cells transduced with FV vectors carrying the UCOE promoter were found to express WASp as early 5 days after transduction, a time-point when cells transduced with that containing endogenous WAS promoter did not show WASp expression. Both promoters, however, afforded long-term WASp and EGFP expression in primary (up to 10 and 15 months, respectively) and secondary (up to 4 months) recipient mice.
In summary, our data demonstrate that short-term repeated exposure of BM stem/progenitor cells to FV vectors can results in effective WAS gene transfer in repopulating cells and restoration of expression and function of WASp in lymphoid and myeloid lineages similar to that reported in the literature for gammaretroviral and lentiviral vectors.5,6,7,33,39,40,41,42,46 Importantly, such results were obtained with the insertion of relatively low vector copy numbers, which, together with the favorable safety features of FV vectors, supports the development of such vectors for future clinical application of gene therapy for WAS.
Materials and Methods
FV vectors construction and production. FV vector plasmids, pΔΦ, pCinES, pCinGS, and pCinPS47 were kindly provided by Dr David W. Russell (University of Washington, Seattle, WA).
To isolate the WAS endogenous promoter region, the 0.5-kb 5′-flanking sequence of the human WAS gene was amplified from genomic DNA by PCR.8 The obtained fragment was subcloned into the multiple cloning sites of the pΔΦ vector plasmid together with either the WAS or EGFP cDNAs to generate the pΔΦ Wa0.5-WAS (wW) and pΔΦ Wa0.5-EGFP (wE) constructs.
A 2.6-kb fragment of the ubiquitously acting chromatin-opening element from the human HNRPA2B1-CBX3 locus (A2UCOE)21 was a kind gift of Dr Adrian J. Thrasher (University College of London, London, UK). A 631-bp, BspEI and TthIIII restriction fragment of such region was isolated and inserted into the pΔΦ vector plasmid together with the WAS or EGFP cDNA to generate pΔΦ UCOE631-WAS (uW) and pΔΦ UCOE631-EGFP (uE).
FV vector particles were produced by calcium phosphate transfection of 293T cells with the resultant gene transfer vector plasmids and the three helper plasmids (pCiGS, pCiPS, and pCiES), as previously described.47 Culture supernatant was harvested after 72 hours and concentrated by ultracentrifugation.
Titers of FV stocks were determined by exposing Lin-/Sca-1+ cells to dilutions of vector stocks. Cells (1 × 103) were then resuspended in 2 ml of Methocult methylcellulose-based medium (Stem Cell Technologies, Vancouver, British Columbia, Canada) and seeded in 35-mm plates. Colony-forming unit in culture were counted after 7 days of culture, harvested, and analyzed by real-time PCR using genomic DNA extracted from each colony with a QIAamp DNA blood mini kit (QIAGEN, Hilden, Germany). Viral titers (transduction units/ml) were calculated based on the dilution of the vector and percentage of transduced colonies.
Cell preparation and culture. Peripheral blood mononuclear cells were isolated by Ficoll–Hypaque (BioWhittaker, Walkersville, MD) gradient centrifugation from healthy controls and patients with WAS under procedures approved by the institutional review board of the National Human Genome Research Institute (ClinicalTrials.gov: NCT00006319). Peripheral blood mononuclear cells from patient WAS16 (described in ref. 48 as WAS1) was used to establish a T-cell line by transformation with the human lymphotropic virus type 1. The T-cell line was maintained as previously described.48
BM cell transduction and transplantation. Was KO mice23 were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed under specific pathogen-free conditions in individually ventilated cages and supplied with sterile food, water, and bedding. All procedures were reviewed and approved by the Animal Care and Use Committee of the National Human Genome Research Institute. Lineage maker depleted (Lin-) cells and Sca1+ cells (Sca1+) cells were purified from male Was KO mice using magnetic bead separation (Miltenyi Biotec, Aurburn, CA). Purified cells were exposed directly to FV vectors once or twice for 12–17 hours on CH-296 (Retronectin)-coated plates (Takara Shuzo, Otsu, Japan) at a multiplicity of infection of 10–20 in StemSpan medium (Stem Cell Technologies), supplemented with stem cell factor (100 ng/ml), IL-3 (20 ng/ml), Flt-3 ligand (20 ng/ml), and IL-6 (50 ng/ml) (all from Peprotech, Rocky Hill, NJ). Transduced cells (2–5 × 105) were then transplanted intravenously into irradiated (700 rad) female Was KO primary recipients of 6–8 weeks of age. Secondary transplants were performed using BM cells that were harvested from primary recipient mice 4–5 months after transplant and injected into female Was KO secondary recipients (2 × 106 cells /mouse).
The percentage of transduced cells was determined either by FACS analysis of EGFP expression or by real-time PCR on colonies generated from wE- and uE-transduced Lin-/Sca1+ cells after 7 days of culture in methylcellulose medium.
Immunofluorescence staining. For FACS analysis, splenocytes and peripheral blood cells were collected from the mice, treated with ACK lysis buffer, washed with phosphate-buffered saline, and incubated with anti-CD3 APC, B220 PE, and Gr1 APC antibodies (eBioscience, San Diego, MA) for 30 minutes at 4 °C. Stained cells were analyzed on a FACSCalibur flow cytometer (BD Bioscience Immunocytometry Systems, Mountain View, CA) using the CellQuest software (BD Bioscience Immunocytometry Systems).
Western blot. WB analysis of WASp expression was performed as previously described.48 Briefly, protein lysates were separated by sodium dodecyl sulfate-poryacrylamide gel electrophoresis, transferred onto polyvinylidene fluoride membranes and blotted with anti-WASp polyclonal antibodies B-9 (Santa Cruz Biotechnology, Santa Cruz, CA) recognizing mouse and human WASp, or D-1 (Santa Cruz Biotechnology), specific for human WASp. Detection was performed using the Western Breeze chemiluminescence WB immunodetection kit (Invitrogen, Carlsbad, CA).
The quantification of WASp and tubulin levels was carried out by densitometry analysis using the ImageJ software (National Institutes of Health, Bethesda, MD) and expressed as percentage of expression observed in samples from WT mice or mice transplanted with WT Lin-/Sca-1+ cells.
T-cell proliferation and IL-2 production. CD90+ spleen cells were selected with immunomagnetic beads (Miltenyi Biotec), and resuspended in RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum, 2 mmol/l glutamine, 50 µmol/l 2-mercaptethanol, penicillin (100 IU/ml), and streptomycin (100 IU/ml). Cells (1 × 105 cells/well) were then stimulated with anti-CD3 antibody (2 µg/ml) coated onto 96-well plates. T-cell proliferation was measured after 72 hours of stimulation by [3H]thymidine incorporation and scintillation counting. IL-2 production was assessed with OptiEIA ELISA kit (BD Biosciences, San Jose, CA) in conditioned medium after 48 hours of stimulation following manufacturer's specifications.
Cell migration assay. Transwells plates (6.5-mm diameter) with 5 µmol/l pore filter (Corning Costar, Corning, NY) were coated with fibronectin (10 µg/ml; Sigma-Aldrich, St Louis, MO) for 3 hours at 37 °C. CD45R/B220+ cells (2 × 105 cells in 100 µl medium) were seeded on the upper compartment of the Transwell and CXCL12 (50 ng/ml; Peprotech) was added to the lower compartment. After 6 hours incubation at 37 °C, cells were collected from the lower compartment and stained with PE-CD45R/B220. CountBright Absolute Counting Beads (Invitrogen) were added and the cells were counted with FACSCalibur flowcytometer. The assay was performed in duplicate and the percent migration was calculated as compared to the input cell number.
Platelet adhesion assay. Peripheral blood, collected in acid/citrate/dextrose, was centrifuged at 200g for 6 minutes to obtain platelet-rich plasma. Washed platelets were prepared by centrifugation of platelet-rich plasma at 1,000g for 10 minutes in the presence of prostacyclin (0.1 µg/ml; Santa Cruz Biotechnology). The pellet was resuspended in HEPES/Tyrodes buffer (in mmol/l: 129 NaCl, 0.34 Na2HPO4, 2.9 KCL, 12 NaHCO3, 20 HEPES, 5 glucose, 1 MgCl2; pH 7.3). 24-well plates were coated with a suspension of fibrinogen (200 µg/ml; Sigma-Aldrich) overnight at 4 °C. Wells were then washed with phosphate-buffered saline and blocked with denatured bovine serum albumin (5 mg/ml) for 1 hour at room temperature. Washed platelets were incubated on coated plates at 37 °C for 45 minutes in the presence of thrombin (1 U/ml; Abcam, Cambridge, MA) and assessed for adhesion. Surface area of spreading platelets was quantified using Image J software, as previously described.49
Detection of podosomes in BM-derived DCs. BM cells were harvested and cultured for 12–14 days in the presence of mGM-CSF (20 ng/ml) and IL-4 (25 ng/ml) to produce immature DCs. Resultant DCs were then cultured for 24 hours on cover slips coated with fibronectin (100 ng/ml for overnight at 4 °C), fixed and permeabilized as previously described.6 The combinations of Anti-Vinculin (Sigma-Aldrich) plus Alexa 488 goat anti-mouse (Molecular Probes) antibodies, and Alexa Fluor 594 phalloidin (Molecular Probes, Grand Island, NY) were used for the detection of podosomes. Nuclei were labeled with DAPI and cells were examined by fluorescence microscopy. For each experiment, at least 100 cells were counted and the percentage of cells displaying podosomes was calculated.
Real-time PCR analysis. FV provirus copy numbers were determined using a TaqMan 5′ nuclease quantitative real-time PCR assay. Genomic DNA was isolated from BM cells, CD90+ splenic T cells, and CD45/B220 splenic B cells and purified with immunomagnetic beads (Miltenyi Biotec). Genomic DNA was amplified with the following FV-specific primers/probe combination: forward primer 5′-AATCCTTTACATGGAGAAGTTATAGGTCTT-3′ and reverse primer 5′-TGGCCAAATCCATAGCCTTAGA-3′ probe, 5′ FAM-ATCTGAAATCTCTCAATTTGTCCCCACCA-3′. To adjust for equal loading of genomic DNA, β-actin was amplified using the following specific primers/probe combination: forward primer 5′-AGAGGGAAATCGTGCGTGAC-3′ and reverse primer 5′- CAATAG TGATGACCTGGCCGT-3′ probe, 5′ FAM-CACTGCCGCATCCTC TTCCTCCC-3′. A standard curve was established using dilutions of DNA extracted from cell lines carrying a single copy of the FV vector plasmid.
To determine the engraftment of donor male cells in treated mice, Y-chromosome-specific real-time PCR was performed with the following primers/probe set: forward primer, 5′-GCGCCCCATGAATGCAT-3′ and reverse primer, 5′-TCCACCTGCATCCCAGCT-3′ probe, 5′ FAM—TGAGAGGCACAAGTTGGCCCAGC-3′.
Reactions were performed using the ABI master mix (Applied Biosystems, Foster city, CA) on a ABI Prism 7900 HT system (Applied Biosystems) using the following conditions: 50 °C for 2 minutes and 95 °C for 10 minutes, followed by 40 cycles of 95 °C for 15 seconds and 60 °C for 1 minute.
Linker-mediated PCR. For detection of FV vector IS, linker-mediated PCR was performed on genomic DNA isolated from BM cells and splenocytes of treated mice. Genomic DNA was digested with MseI and PstI and the fragments were ligated to an MseI linker (5′-GTAATACGACTCATAGGGCTCCGCTTAAGGGAC-3′ and 5′ PO4- TAGTCCCTTAAGCGGAG-NH2-3′). PCR was then performed with a linker-specific primer (5′-GTAATACGACTCACTATAGGGC-3′) and a FV long-terminal repeats-specific primer (5′-GTCTATGAGGAGCAGG AGTA-3′). A nested PCR was then performed using 2% of the first PCR product as template, and the linker-specific primer 5′-AGGGTCCGCTT AAGGGAC-3′ and the FV long-terminal repeats-specific primer 5′-CC TCCTTCCCTGTAATACTC-3′. The PCR products were subcloned into pCR2.1 using the TOPO cloning kit (Invitrogen, Frederick, MD) and the FV vector/DNA junction sites were sequenced on ABI 3100 genetic Analyzer (Applied Biosystems). Recovered vector/DNA junctions were matched to the murine genome using the BLAT software (UCSC Mouse genome, February 2006 freeze, mm8 version). Genes within 30 kb from IS were compared to the list of annotated cancer in the Atlas of Genetics and Cytogenetics in Oncology and Hematology database (http://atlasgeneticsoncology.org/) and to the RTCGD database (http://variation.osu.edu/rtcgd).
SUPPLEMENTARY MATERIAL Figure S1 Distribution of frequency of FV IS and mouse genes on chromosomes. Table S1. Podosome formation in FV-treated mice and controls.
Acknowledgments
The authors are grateful to David W. Russell (University of Washington, Seattle, WA) and Adrian J. Thrasher (University College of London, London, UK) for sharing reagents, Julia Fekecs (NHGRI/NIH, Bethesda, MD) for artwork, and the NIH Building 49 Animal Care Staff for excellent and humane care of laboratory animals. This research was supported by funding from the NHGRI intramural program and the Japanese Society for Promotion of Science (JSPS). The authors declared no conflict of interest.
Supplementary Material
Distribution of frequency of FV IS and mouse genes on chromosomes.
Podosome formation in FV-treated mice and controls.
REFERENCES
- Ochs HD., and, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol. 2006;117:725–38; quiz 739. doi: 10.1016/j.jaci.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Derry JM, Ochs HD., and, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;78:635–644. doi: 10.1016/0092-8674(94)90528-2. [DOI] [PubMed] [Google Scholar]
- Thrasher AJ. WASp in immune-system organization and function. Nat Rev Immunol. 2002;2:635–646. doi: 10.1038/nri884. [DOI] [PubMed] [Google Scholar]
- Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, Schulz A, Thrasher AJ, Mazzolari E.et al. (2008Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation Blood 111439–445. [DOI] [PubMed] [Google Scholar]
- Klein C, Nguyen D, Liu CH, Mizoguchi A, Bhan AK, Miki H.et al. (2003Gene therapy for Wiskott-Aldrich syndrome: rescue of T-cell signaling and amelioration of colitis upon transplantation of retrovirally transduced hematopoietic stem cells in mice Blood 1012159–2166. [DOI] [PubMed] [Google Scholar]
- Strom TS, Turner SJ, Andreansky S, Liu H, Doherty PC, Srivastava DK.et al. (2003Defects in T-cell-mediated immunity to influenza virus in murine Wiskott-Aldrich syndrome are corrected by oncoretroviral vector-mediated gene transfer into repopulating hematopoietic cells Blood 1023108–3116. [DOI] [PubMed] [Google Scholar]
- Dupré L, Marangoni F, Scaramuzza S, Trifari S, Hernández RJ, Aiuti A.et al. (2006Efficacy of gene therapy for Wiskott-Aldrich syndrome using a WAS promoter/cDNA-containing lentiviral vector and nonlethal irradiation Hum Gene Ther 17303–313. [DOI] [PubMed] [Google Scholar]
- Charrier S, Dupré L, Scaramuzza S, Jeanson-Leh L, Blundell MP, Danos O.et al. (2007Lentiviral vectors targeting WASp expression to hematopoietic cells, efficiently transduce and correct cells from WAS patients Gene Ther 14415–428. [DOI] [PubMed] [Google Scholar]
- Bosticardo M, Marangoni F, Aiuti A, Villa A., and, Grazia Roncarolo M. Recent advances in understanding the pathophysiology of Wiskott-Aldrich syndrome. Blood. 2009;113:6288–6295. doi: 10.1182/blood-2008-12-115253. [DOI] [PubMed] [Google Scholar]
- Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA.et al. (2010Stem-cell gene therapy for the Wiskott-Aldrich syndrome N Engl J Med 3631918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneine W, Schweizer M, Sandstrom P., and, Folks T. Human infection with foamy viruses. Curr Top Microbiol Immunol. 2003;277:181–196. doi: 10.1007/978-3-642-55701-9_8. [DOI] [PubMed] [Google Scholar]
- Falcone V, Schweizer M., and, Neumann-Haefelin D. Replication of primate foamy viruses in natural and experimental hosts. Curr Top Microbiol Immunol. 2003;277:161–180. doi: 10.1007/978-3-642-55701-9_7. [DOI] [PubMed] [Google Scholar]
- Vassilopoulos G, Trobridge G, Josephson NC., and, Russell DW. Gene transfer into murine hematopoietic stem cells with helper-free foamy virus vectors. Blood. 2001;98:604–609. doi: 10.1182/blood.v98.3.604. [DOI] [PubMed] [Google Scholar]
- Josephson NC, Trobridge G., and, Russell DW. Transduction of long-term and mobilized peripheral blood-derived NOD/SCID repopulating cells by foamy virus vectors. Hum Gene Ther. 2004;15:87–92. doi: 10.1089/10430340460732481. [DOI] [PubMed] [Google Scholar]
- Josephson NC, Vassilopoulos G, Trobridge GD, Priestley GV, Wood BL, Papayannopoulou T.et al. (2002Transduction of human NOD/SCID-repopulating cells with both lymphoid and myeloid potential by foamy virus vectors Proc Natl Acad Sci USA 998295–8300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leurs C, Jansen M, Pollok KE, Heinkelein M, Schmidt M, Wissler M.et al. (2003Comparison of three retroviral vector systems for transduction of nonobese diabetic/severe combined immunodeficiency mice repopulating human CD34+ cord blood cells Hum Gene Ther 14509–519. [DOI] [PubMed] [Google Scholar]
- Bauer TR, Jr, Allen JM, Hai M, Tuschong LM, Khan IF, Olson EM.et al. (2008Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors Nat Med 1493–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiem HP, Allen J, Trobridge G, Olson E, Keyser K, Peterson L.et al. (2007Foamy-virus-mediated gene transfer to canine repopulating cells Blood 10965–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si Y, Pulliam AC, Linka Y, Ciccone S, Leurs C, Yuan J.et al. (2008Overnight transduction with foamyviral vectors restores the long-term repopulating activity of Fancc-/- stem cells Blood 1124458–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobridge GD, Miller DG, Jacobs MA, Allen JM, Kiem HP, Kaul R.et al. (2006Foamy virus vector integration sites in normal human cells Proc Natl Acad Sci USA 1031498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Thornhill SI, Howe SJ, Ulaganathan M, Schambach A, Sinclair J.et al. (2007Lentiviral vectors containing an enhancer-less ubiquitously acting chromatin opening element (UCOE) provide highly reproducible and stable transgene expression in hematopoietic cells Blood 1101448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupré L, Aiuti A, Trifari S, Martino S, Saracco P, Bordignon C.et al. (2002Wiskott-Aldrich syndrome protein regulates lipid raft dynamics during immunological synapse formation Immunity 17157–166. [DOI] [PubMed] [Google Scholar]
- Snapper SB, Rosen FS, Mizoguchi E, Cohen P, Khan W, Liu CH.et al. (1998Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation Immunity 981–91. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shehabeldin A, da Cruz LA, Butler J, Somani AK, McGavin M.et al. (1999Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott-Aldrich syndrome protein-deficient lymphocytes J Exp Med 1901329–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerberg L, Larsson M, Hardy SJ, Fernández C, Thrasher AJ., and, Severinson E. Wiskott-Aldrich syndrome protein deficiency leads to reduced B-cell adhesion, migration, and homing, and a delayed humoral immune response. Blood. 2005;105:1144–1152. doi: 10.1182/blood-2004-03-1003. [DOI] [PubMed] [Google Scholar]
- Snapper SB, Meelu P, Nguyen D, Stockton BM, Bozza P, Alt FW.et al. (2005WASP deficiency leads to global defects of directed leukocyte migration in vitro and in vivo J Leukoc Biol 77993–998. [DOI] [PubMed] [Google Scholar]
- Tsuboi S, Nonoyama S., and, Ochs HD. Wiskott-Aldrich syndrome protein is involved in αIIb β3-mediated cell adhesion. EMBO Rep. 2006;7:506–511. doi: 10.1038/sj.embor.7400665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder S, Nelson D, Weiss M., and, Aepfelbacher M. Wiskott-Aldrich syndrome protein regulates podosomes in primary human macrophages. Proc Natl Acad Sci USA. 1999;96:9648–9653. doi: 10.1073/pnas.96.17.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Li Y, Crise B., and, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- Moebes A, Enssle J, Bieniasz PD, Heinkelein M, Lindemann D, Bock M.et al. (1997Human foamy virus reverse transcription that occurs late in the viral replication cycle J Virol 717305–7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SF, Baldwin DN, Gwynn SR, Yendapalli S., and, Linial ML. Human foamy virus replication: a pathway distinct from that of retroviruses and hepadnaviruses. Science. 1996;271:1579–1582. doi: 10.1126/science.271.5255.1579. [DOI] [PubMed] [Google Scholar]
- Trobridge G., and, Russell DW. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. J Virol. 2004;78:2327–2335. doi: 10.1128/JVI.78.5.2327-2335.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani J, Charrier S, Eckenberg R, Saurin W, Danos O, Perea J.et al. (2009Diverse genomic integration of a lentiviral vector developed for the treatment of Wiskott-Aldrich syndrome J Gene Med 11645–654. [DOI] [PubMed] [Google Scholar]
- Mucenski ML, Taylor BA, Ihle JN, Hartley JW, Morse HC., 3rd, , Jenkins NA.et al. (1988Identification of a common ecotropic viral integration site, Evi-1, in the DNA of AKXD murine myeloid tumors Mol Cell Biol 8301–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustikova O, Fehse B, Modlich U, Yang M, Düllmann J, Kamino K.et al. (2005Clonal dominance of hematopoietic stem cells triggered by retroviral gene marking Science 3081171–1174. [DOI] [PubMed] [Google Scholar]
- Westerberg LS, de la Fuente MA, Wermeling F, Ochs HD, Karlsson MC, Snapper SB.et al. (2008WASP confers selective advantage for specific hematopoietic cell populations and serves a unique role in marginal zone B-cell homeostasis and function Blood 1124139–4147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Bahlburg A, Becker-Herman S, Humblet-Baron S, Khim S, Weber M, Bouma G.et al. (2008Wiskott-Aldrich syndrome protein deficiency in B cells results in impaired peripheral homeostasis Blood 1124158–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno A, Wada T, Schurman SH, Garabedian EK, Kirby M, Anderson SM.et al. (2004Differential contribution of Wiskott-Aldrich syndrome protein to selective advantage in T- and B-cell lineages Blood 103676–678. [DOI] [PubMed] [Google Scholar]
- Charrier S, Stockholm D, Seye K, Opolon P, Taveau M, Gross DA.et al. (2005A lentiviral vector encoding the human Wiskott-Aldrich syndrome protein corrects immune and cytoskeletal defects in WASP knockout mice Gene Ther 12597–606. [DOI] [PubMed] [Google Scholar]
- Blundell MP, Bouma G, Calle Y, Jones GE, Kinnon C., and, Thrasher AJ. Improvement of migratory defects in a murine model of Wiskott-Aldrich syndrome gene therapy. Mol Ther. 2008;16:836–844. doi: 10.1038/mt.2008.43. [DOI] [PubMed] [Google Scholar]
- Marangoni F, Bosticardo M, Charrier S, Draghici E, Locci M, Scaramuzza S.et al. (2009Evidence for long-term efficacy and safety of gene therapy for Wiskott-Aldrich syndrome in preclinical models Mol Ther 171073–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanta-Boussif MA, Charrier S, Brice-Ouzet A, Martin S, Opolon P, Thrasher AJ.et al. (2009Validation of a mutated PRE sequence allowing high and sustained transgene expression while abrogating WHV-X protein synthesis: application to the gene therapy of WAS Gene Ther 16605–619. [DOI] [PubMed] [Google Scholar]
- Martín F, Toscano MG, Blundell M, Frecha C, Srivastava GK, Santamaría M.et al. (2005Lentiviral vectors transcriptionally targeted to hematopoietic cells by WASP gene proximal promoter sequences Gene Ther 12715–723. [DOI] [PubMed] [Google Scholar]
- Williams S, Mustoe T, Mulcahy T, Griffiths M, Simpson D, Antoniou M.et al. (2005CpG-island fragments from the HNRPA2B1/CBX3 genomic locus reduce silencing and enhance transgene expression from the hCMV promoter/enhancer in mammalian cells BMC Biotechnol 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou M, Harland L, Mustoe T, Williams S, Holdstock J, Yague E.et al. (2003Transgenes encompassing dual-promoter CpG islands from the human TBP and HNRPA2B1 loci are resistant to heterochromatin-mediated silencing Genomics 82269–279. [DOI] [PubMed] [Google Scholar]
- Bosticardo M, Draghici E, Schena F, Sauer AV, Fontana E, Castiello MC.et al. (2011Lentiviral-mediated gene therapy leads to improvement of B-cell functionality in a murine model of Wiskott-Aldrich syndrome J Allergy Clin Immunol 1271376–84.e5. [DOI] [PubMed] [Google Scholar]
- Trobridge G, Josephson N, Vassilopoulos G, Mac J., and, Russell DW. Improved foamy virus vectors with minimal viral sequences. Mol Ther. 2002;6:321–328. doi: 10.1006/mthe.2002.0672. [DOI] [PubMed] [Google Scholar]
- Wada T, Jagadeesh GJ, Nelson DL., and, Candotti F. Retrovirus-mediated WASP gene transfer corrects Wiskott-Aldrich syndrome T-cell dysfunction. Hum Gene Ther. 2002;13:1039–1046. doi: 10.1089/104303402753812449. [DOI] [PubMed] [Google Scholar]
- McCarty OJ, Larson MK, Auger JM, Kalia N, Atkinson BT, Pearce AC.et al. (2005Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow J Biol Chem 28039474–39484. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distribution of frequency of FV IS and mouse genes on chromosomes.
Podosome formation in FV-treated mice and controls.