

Over the past two decades, incredible progress has been made using gene therapy for inherited severe immunodeficiency disorders, such as X-linked severe combined immunodeficiency disorder (SCID-X1) and adenosine deaminase deficiency–severe combined immunodeficiency disorder (ADA-SCID).1,2,3 However, for reasons that remain unclear, gene transfer for SCID-X1 has also been associated with some cases of vector-induced leukemia whereas no cases have been seen in the ADA-SCID trials despite the common use of g-retroviral vectors. The first case was reported in a French gene transfer trial for SCID-X1.4 Over the next six years, an additional three cases were reported in that trial and one in a second SCID-X1 trial that enrolled a combined total of 20 subjects.2 Unfortunately, genotoxicity would not remain confined to SCID-X1. Recent reports of insertional mutagenesis leading to myelodysplastic syndrome in a trial for chronic granulomatous disease and a case of leukemia in a trial for Wiskott-Aldrich syndrome (WAS), both of which used g-retroviral vectors, underscored that this type of toxicity can also apply to other disease settings.5,6,7 In all these cases, insertion of the g-retroviral vector near known proto-oncogenes led to enhancer-mediated expression of these proto-oncogenes.
To address this toxicity, investigators have developed new vectors that, in experimental models, can achieve long-term gene correction with less risk of insertional mutagenesis. The field is also seeking to develop new animal models and in vitro assays that can accurately predict whether these vectors will indeed reduce the risk of insertional mutagenesis.
In light of these innovations and new trials, the National Institutes of Health (NIH) Office of Biotechnology Activities, in partnership with the NIH Recombinant DNA Advisory Committee and the European Network for the Advancement of Clinical Gene Transfer and Therapy (CliniGene; http://www.clinigene.eu) hosted a conference in December 2010 to examine new developments in this field so as to inform the scientific community, regulators, and the public. The focus of the conference was not only on the scientific advances but also on the potential development of predictive assays, regulatory oversight challenges, and ethical questions regarding the design of new trials with these vectors. This article reviews the key issues raised during that conference. (The presentations and the webcasts for the conference are available at http://oba.od.nih.gov/rdna/rdna_symposia.html#CONF_003h.)
Clinical experience with retroviral vectors for long-term gene correction and insertion-site analysis
Some of the earliest trials using g-retroviral vectors for long-term gene correction focused on the severe combined immunodeficiency disorders including ADA-SCID and SCID-X1 and on the neutrophil oxidase–deficiency chronic granulomatous disease (Table 1).
Table 1. Clinical experience with vectors.
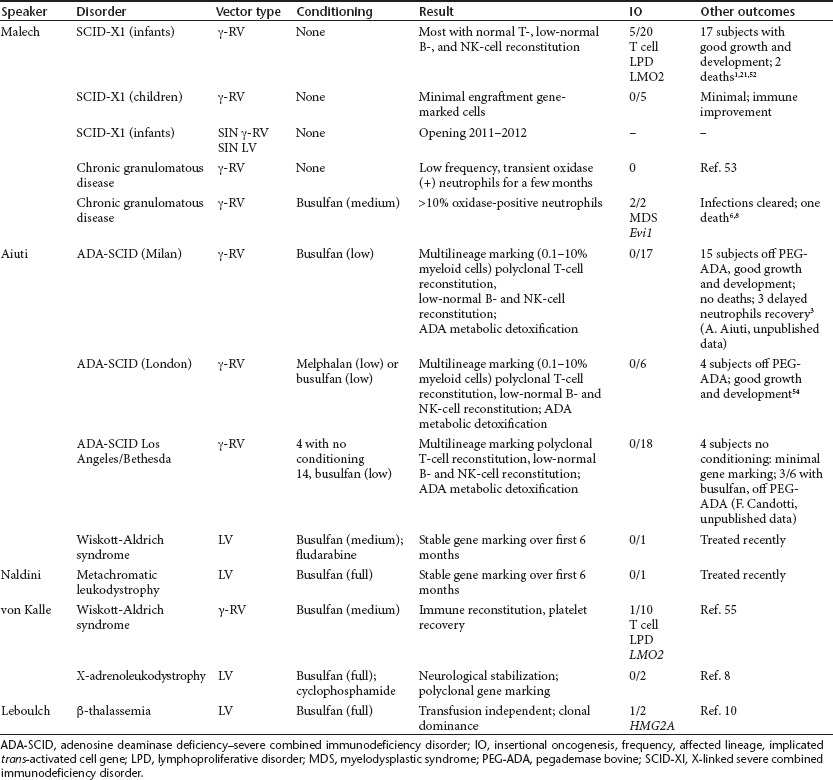
Review of these g-retroviral clinical trials demonstrated that gene transfer has the potential to provide long-term gene correction that is comparable to alternative therapies, such as bone marrow transplant (BMT), and—depending on the disease, the age of the patient at diagnosis, and the availability of alternative therapy—gene transfer may prove equally efficacious with potentially less toxicity. However, the factors leading to development of genotoxicity from integration of the vector in a particular trial or subject are not completely understood.
Early data on trials using lentiviral vectors were also discussed. Lentiviral vectors are being used in trials for leukodystrophies, including adrenoleukodystrophy (ALD), metachromatic leukodystrophy (MLD), and WAS. In MLD, a defect in the enzyme arylsulfatase A leads to impaired development of the myelin sheath that insulates nerve fibers. ALD, an X-linked disorder, is caused by a mutation in the ALD protein resulting in disruption of myelin and early death due to demyelination. In 2009, in a trial administering autologous CD34+ hematopoietic cells transduced with a lentiviral vector expressing the ALD protein, two subjects achieved polyclonal reconstitution with 9–14% of granulocytes, monocytes, and T and B lymphocytes containing the transferred ALDP gene and that, beginning at 14–16 months, progressive demyelination stopped.8 Luigi Naldini reported the results of the first treated patient in the MLD trial who received autologous CD34+ cells transduced with a self-inactivating (SIN) lentiviral vector containing arylsulfatase A complementary DNA following busulfan preconditioning. At 3- to 6-month follow-ups, there was sustained gene marking, and expression of the arylsulfatase enzyme at levels 5- to 10-fold above normal were detected in the peripheral blood cells. Eugenio Montini, via analysis of unique integration sites in that subject, demonstrated polyclonal hematopoiesis and displayed a genomic integration profile similar to that of ALD patients and in human mouse hematochimeras.9 Alessandro Aiuti presented the preclinical data and rationale supporting a novel clinical trial for WAS using a lentiviral vector encoding WAS protein under the control of its homologous promoter. A clinical trial using lentiviral vector-transduced autologous CD34+ cells combined with reduced-intensity conditioning has started at his institution, and the first subject was treated in June 2010.
Whereas both of these trials reported polyclonal hematopoiesis, another trial, using a lentiviral vector for b-thalassemia, reported the development of an oligoclonal population of vector-containing cells. An HIV-based SIN vector expressing a functional b-globin gene was introduced into hematopoietic stem cells with the goal of correcting deficiencies in hemoglobin production, the underlying defect in b-thalassemia. The first study subject engrafted with the transduced cells has shown improved hemoglobin production and displayed significant clinical improvement.10 However, a clone carrying the gene transfer vector has become dominant. The clone overexpresses the gene encoding HMGA2, a protein that regulates gene transcription. Analyses reveal that cells bearing this insertion express a truncated form of HMGA2 messenger RNA (mRNA) lacking 3ʹ binding sites for a regulatory microRNA (miRNA) that arises as a consequence of aberrant splicing to a cryptic splice acceptor site in the vector insulator sequences. In contrast to findings in the earlier g-retrovirus vector-based SCID-X1 trials, in which five study subjects developed cases of leukemia associated with vector-mediated activation of LMO2, the subject in the b-thalassemia trial has remained healthy for nearly four years. In addition, there is no evidence of other abnormalities, including karyotype changes, abnormal blood counts, or cytology. Aberrant expression of HMGA2 has been associated with lipomas and some hematological disorders including malignancies.11,12 However, integrations similar to the one discussed here have also been detected in the SCID-X1 trials in study subjects who remain healthy.13 The mechanism(s) that led to partial clonal dominance of this clone are not fully understood. A stochastic event related to low transduction of hematopoietic stem cells could be responsible. Alternatively, the integration affecting HMGA2 may confer a homeostatic advantage to cells. A recent article demonstrated that HMGA2 overexpression could result in benign hematopoietic stem and progenitor cell expansion in a murine model.14
Beyond enhancer-mediated genotoxicity
Although the clonal population of cells detected in the b-thalassemia trial has remained clinically benign, it is illustrative of how vector insertion can affect adjacent cellular gene expression through non-enhancer-mediated mechanisms. There are essentially four “alternative” mechanisms other than oncogenic activation by which retroviruses and, more recently, transposons have been shown to induce cancer in animal models: alternative splicing, gene inactivation, truncation of cellular mRNA or protein, and miRNA activation.
Drawing from in vitro and in vivo preclinical models, numerous examples were presented, in which each of the four alternative mechanisms was responsible for transformation by both replication-competent retroviruses and translocating Sleeping Beauty (SB) transposons (Table 2). In contrast to murine leukemia viruses (MLVs), which primarily cause hematopoietic lineage cancers, genetically engineered SB transposons have been used to induce tumors in many different cell types. Linda Wolff presented several murine models in which infection with replication-competent MLV causes a variety of hematopoietic cancers, including erythroleukemias, myeloid tumors, and T- and B-cell lymphomas/leukemias, by non-enhancer-mediated mechanisms. David Largaespada, using the SB system, provided evidence that alternative mechanisms of oncogenesis are not limited to hematopoietic tumors.
Table 2. Non-enhancer-mediated mechanisms by which integrating elements affect expression of adjacent cellular genes.
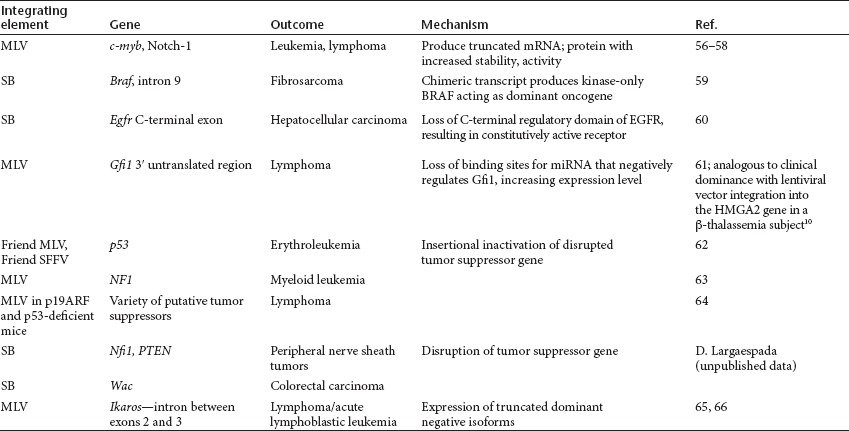
A common theme gathered from research on retroviruses, transposon systems such as SB, and oncogenesis is that it may take multiple hits to different cellular genes (genetic or epigenetic) to convert a normal cell into a tumor cell. Many mice transgenic for a single proto-oncogene have long latency periods for the development of tumors, and crossing two lines carrying “cooperating” oncogenes (e.g., c-myc and Runx2) can result in more rapid tumor development.15 Because of the requirement for multiple events, many models, including those discussed by Wolff and Largaespada, incorporate the use of mice with genetic predisposition to particular cancers (e.g., tumor suppressor gene knockouts or transgenics that express different oncogenes), chemical mutagens, or inflammatory agents (e.g., pristane) to accelerate transformation by the integrating elements (retroviruses/transposons). However, malignancies secondary to retrovirus-induced changes still occur in wild-type animals, albeit with lower incidence and longer latency.
Oncogenesis by retroviral gene therapy vectors may also be influenced by age and genetics. Marc Sitbon reviewed MLV leukemogenesis and pointed out that both age and genetic background influence the efficiency and specificity of disease. In humans, individual genetic makeup may also influence the likelihood of developing a tumor after retroviral gene therapy. MLV infection of young (neonatal) animals leads to much more efficient development of leukemias than infection with the same virus in adults. This could reflect the increased availability of target cells with the potential to become leukemic in young animals or to more robust antiviral immunity in older animals. These findings in mice suggest that human gene transfer by retroviral vectors in neonates or young children may entail a higher oncogenic risk than in older subjects. However, younger age may also underlie the better clinical responses that have been seen for gene transfer to hematopoietic stem cells (HSCs) with integrating vectors, such as the infants with SCID.
Lee Ratner discussed human T-lymphotrophic virus type 1, which has direct transforming activity and causes adult T-cell leukemia in humans,16 and the long latency between infection and oncogenesis. Interestingly, almost all infections that result in leukemia occur early in life through vertical transmission (predominantly breastfeeding),17 but the time between infection and development of leukemia is typically several decades, and only about 5% of infected individuals ultimately develop adult T-cell leukemia. By contrast, infection of adults is associated with a neurological disease that occurs with a shorter latency. Of relevance to the field of gene transfer is that even when a retrovirus carries genes with transforming potential, the lag between infection and development of a tumor can be decades, again underscoring the importance of long-term follow-up of subjects in gene correction trials. If activation or inactivation of a single cellular gene does not immediately result in development of oncogenesis, tumorigenicity may take years to develop, leading to a false presumption of safety.
Altogether, these observations of the cooperative effects of multiple integrations into different genes have implications for considering oncogenic risk from integrating vectors in gene transfer trials. On the positive side, a single genetic hit (e.g., insertional activation or mutagenesis of one cellular gene) may not be sufficient to induce a tumor cell by itself. Minimizing the numbers of vector integrants per target cell while achieving sufficient percentages of gene-modified cells should be a goal in clinical applications. On the negative side, it has long been recognized that human cancers are the result of multiple genetic changes; dysregulation of cellular gene expression by vector insertion could be an initiating event, and long-term follow-up of subjects may be required.
Approaches to improving design and safety of gene transfer vectors
Following the reports of severe adverse events directly attributable to gene transfer vectors integration, several approaches have been pursued separately or in combination, as follows: (i) genetic insulator elements (GIEs) to act as both enhancer-blockers and boundary against potential silencing; (ii) inhibition of integration by knocking down in target cells lens epithelium-derived growth factor (LEDGF), the cell partner of HIV integrase, which may also have the potential to allow integration to be redirected toward defined genetic regions; (iii) selection of genetic regions as safe harbors for integration; (iv) alternative integration systems with a more random integration profile in human cells, such as foamy virus vectors or nonviral vectors derived from transposons; and (v) inhibition of unwanted expression of the transgene in nontarget cells.
Odile Cohen-Haguenauer reviewed studies of safety modifications in MLV and lentiviral vectors to determine whether new short synthetic transcriptional insulator modules, identified in collaboration with Nic Mermod (University of Lausanne), could inhibit cis-acting transcriptional effects.18 Further studies of a particular GIE that showed sustained and robust expression were undertaken to establish how the system would perform in primary cells. Of note, in a study with David Klatzmann (Paris, France), a strong CD4-specific promoter was constrained by the insulators in human kidney 293 cells, and expression in Jurkat T-cell lines was observed. Comparative high-throughput integration-site analysis performed with Christof von Kalle in collaboration with Montini, using an in vivo genotoxicity assay on Cdkn2a–/– tumor-prone mice,19 provided evidence that this new GIE system might lead to a lower risk of genotoxicity.20 Insulated lentiviral vectors expressing the Fanconi anemia A complementary DNA are currently being developed. Additional experiments are exploring the retargeting of insulated vectors to transcriptionally inactive chromatin through use of chimeric integrases.21
Frederic Bushman gave an update on data his laboratory has accumulated from studies of integration sites using high-throughput 454 sequencing of sequences flanking HIV-1 integration sites. Integrase determines viral integration–site preference, as demonstrated by studies in which substitution of the MLV integrase into HIV-1 vectors resulted in integration profiles similar to those of MLV. Host cell factors are also involved; in particular, knockdown of a transcriptional co-activator LEDGF that binds to HIV-1 integrase drastically reduces titer, and the remaining integration events are not targeted into transcriptional units.22 In proof-of-principle experiments (with Zeger Debyser and Rik Gjisbers, Katholieke Universiteit Leuven, Belgium) using LEDGF knockdown cells with a chimeric LEDGF fusion to HP1-b that binds heterochromatic regions, integration was redirected to nontranscribing regions.
Naldini elaborated on studies using zinc-finger nucleases to direct integration into safe genomic sites, which may allow robust transgene expression without disrupting endogenous transcription.23 Initially, CCR5 and AAVS loci were selected, as zinc-finger nucleases for these targets have been developed and extensively characterized. Integration of an enhanced green fluorescent protein (EGFP) expression vector was targeted to the CCR5 locus, and when EGFP-positive cells were then sorted, 90% of cells had a site-specific integration. Upregulation of the CCR5 transcript was observed when phosphoglycerate kinase or spleen focus-forming virus promoters were used in the CCR5-targeted integrating expression cassette, and some flanking genes were also upregulated. By contrast, targeting into the AAVS1 site did not result in deregulation of flanking genes, demonstrating locus- and promoter-dependent effects that do not correlate with the strength of the promoter. Ultimately this site-specific gene modification technology may allow for the correction of a mutation at an endogenous locus.
Other vector systems with different integration profiles may also have less mutagenic properties, and Scott McIvor and Thierry VandenDriessche described advances with the transposon vector SB. This bipartite system consists of a source of transposase (typically a plasmid) and a transposon. Although the integration sites for SB are spread uniformly across the genome with no bias toward transcribed genes, some approaches are also in the pipeline to target integration of transposons. For example, the group of Zoltán Ivics (Max Delbrück Center for Molecular Medicine, Berlin) has used a fusion protein that contains a DNA-binding moiety and the SB transposase.24 In addition, VandenDriessche presented an overview of recent advances with the SB and piggyBac (PB) transposons systems for gene transfer, including the development of a hyperactive transposase, SB100 (ref. 25). Integration-site analysis of SB100 integrants confirmed no bias of integration into genes. A side-by-side comparison of the SB system with the PB system in HSCs showed that the SB100 system was slightly more efficient than the PB system. However, the PB system has been reported to carry larger transgenes more efficiently. Several disease models and target organs are being examined, including targeting the liver for hemophilia A and B, and hereditary tyrosinemia and human T cells for Fanconi anemia. Hydrodynamic delivery of a factor IX–encoding transposon and SB100 transposase resulted in prolonged and stable expression from the liver in mice. The SB system can also be used to mediate gene transfer into induced pluripotent stem cells, and the cells retain pluripotency to differentiate into neurons, glia, muscle, and hepatocytes.26
Naldini presented a novel approach that relies on incorporating an miRNA targeting sequence into the 3ʹ untranslated region of lentiviral vectors so that the vector mRNA is degraded if expressed in a cell type that expresses the cognate miRNA. Based on a bicistronic vector system to screen candidates, miRNA-126 was defined as specific for HSC expression in murine and human models. Because overexpression of galactocerebrosidase (GALC) in differentiated cells has been proposed for correction of globoid cell leukodystrophy but is limited by the toxicity of GALC in human HSCs, the target sequence for miRNA-126 was incorporated into a GALC-expressing lentiviral vector. While allowing expression in differentiated progeny, as required, GALC expression was successfully prevented in HSCs. This approach resulted in significant survival in a mouse model.27
Is it safer? Defining an optimal preclinical testing strategy
One of the key questions for the field is what preclinical testing needs to be done before bringing a new vector into the clinic, and, more specifically, how one can predict the risk of genotoxicity of a new vector. Several of the available in vitro and in vivo systems and their relative benefits and limitations were described (Table 3).
Table 3. Bioassays of vector safety.
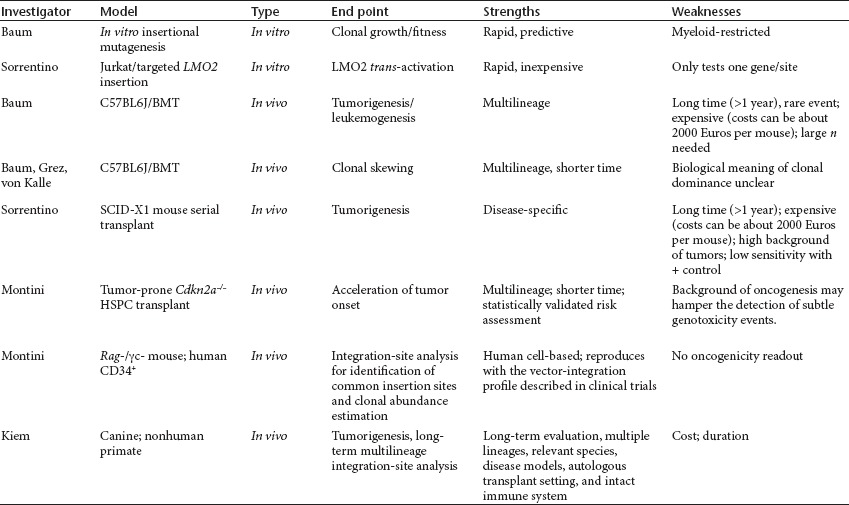
For example, in vitro assays using immortalized C57BL/6J bone marrow cells can be used to determine the incidence of mutants based on the number of cells that need to be exposed before development of a transformant.28 Evaluation of the fitness of the mutant is an important factor. In general, a weaker enhancer in the vector tends to reduce the fitness of the insertional mutants, perhaps by lowering proto-oncogene upregulation levels. This assay has also been used to test the impact of different insulators on development of a clone, and it revealed that, in comparison to the g-retroviral integration pattern, the lentiviral integration pattern reduces the risk of insertional transformation. When SIN vectors contain cellular promoters that lack strong enhancer activity, such as the intronless promoter fragment of the human elongation factor 1a gene, both g-retroviral and lentiviral vectors did not induce transformation in this assay. Of note, in this assay the culture conditions and cell density impact the results, requiring standardized operating procedures to obtain reproducible results.
Numerous models have been developed in which murine or human HSCs are transduced and transplanted into mice, allowing for in vivo observations. End points are leukemogenesis in the more severe cases or “clonal skewing,” in which integrants near certain subclasses of genes involved in cell growth, proliferation, and survival are relatively amplified, which may lead to clonal expansion even to the point of clonal dominance. Although providing the comfort of having performed an in vivo assessment, these assays may be of low sensitivity, given that they test only a small fraction of a patient dose, take a long time to perform, and are costly when performed under good laboratory practice (GLP) conditions.
An unanswered question is whether large-animal models would be better able to predict the risk of genotoxicity. Hans-Peter Kiem presented studies in large animals with follow-up of more than five years—in some cases, more than seven years. A major advantage of large-animal studies is the ability to follow gene marking or correction levels in multiple lineages for a very long time. Analysis reveals that both lentiviral and g-retroviral vectors show increased frequency of integration near proto-oncogenes and also suggested that foamy virus vector may have a more favorable integration-site profile.29,30,31 However, despite this observation with g-retroviral and lentivirus vectors, there has been long-term polyclonal hematopoiesis in the animal studies using vectors with lower-risk transgenes. The substitution of a growth-promoting gene, however, can lead to cases of leukemia in these models. Large-animal studies allow for the evaluation of engraftment kinetics of gene-modified cells using different conditioning regimens, impact of stem cell dose, and long-term follow-up of expression. In addition, the vector performance can be studied in the presence of an intact immune system in contrast to the immunodeficient mouse models. Disease-specific large-animal models are also available.
von Kalle summarized the advances in methods for vector insertion-site analyses and the associated bioinformatics. The advent of high-throughput sequencing has allowed characterization of the number of g-retroviral and lentiviral integration events in one afternoon that previously required years of sequencing. The common core technology currently used by many laboratories is 454 pyrosequencing because of the usefulness of its relatively long sequence reading, allowing up to half a million amplicons in two to three days. But improved methods such as a combination of linear amplification-mediated polymerase chain reaction (LAM-PCR) with next-generation sequencing promise a two-log yearly growth or greater of unbiased sequence capability in the immediate future. Those next advances face technical obstacles standing in the way of a truly nonrestricted approach to defining the integration events and their consequences. Many of these issues have been addressed through the development of bioinformatical integration-site sequence analyses performed by QuickMap (http://www.gtsg.org/quickmap.jsp), HISAP (von Kalle laboratory, unpublished data, or SeqMap (http://seqmap.compbio.iupui.edu). These bioinformatical data-analysis pipelines allow the characterization of integration sites, including compression of sequence-read information and quick clustering to reduce redundant reads and cross-sample clone-tracking and “read-count” assessment. Such tools permit simplified and automated tracking of clonal inventories and provide support for trends toward a read-count interpretation. The result is large-scale chromosomal integration site distribution, gene-related integration site distribution, functional integration site analysis, and ingenuity pathway analysis.
One of the important impediments to integration site retrieval is the limitation imposed by the use of restriction enzyme sites to chop DNA into fragments of more than 500 base pairs. As underscored by Bushman, the use of restriction enzymes in older studies led to bias based on the restriction enzyme–site distributions in the genome, and there is currently no method to fully sequence any integration-site population. The limits of current methods were highlighted in an article first published online in November 2008 (ref. 32) and precisely addressed in 2009 by von Kalle's group.33 It is therefore important to develop a nonrestrictive integration-site approach that combines LAM-PCR methods (nrLAM-PCR)33 to reach a more comprehensive integration-site analysis. This approach, which is under development, promises to remove DNA-processing biases due to restriction enzymes and reduce the biases to those associated with polymerase errors, as recently described.34
Another important issue regarding the use of these insertion site–analysis methods is that they require trained labs and in inexperienced hands may yield discrepant results. Christopher Baum noted the results of an interlaboratory comparison of a protocol in which DNA from a K562 clone that had a number of insertions was sent around, as well as primers for the PCR; surprisingly, all the labs produced different results. Hence, although the assays may be a powerful tool, the experience of the lab may also need to be taken into account. If such assays are to be widely used, the operating procedures will need to be standardized to ensure uniform results. Standardization would apply equally to any animal model that might become widely adopted. Currently, there is no consensus on the optimal model(s) that can definitively answer the question of whether a new vector will be safe for a particular condition. A combination of models may be used to add to the risk assessment, but even when in vitro assays are combined with in vivo animal models, the results may not be entirely predictive. Moreover, the appropriate end point for an animal model and assay is unclear. If one chooses as the end point a rare event such as the development of leukemia or other tumors, it is likely to take considerable time and resources to reach. Alternatively, one could look at the development of clonal skewing, which will allow for a more rapid readout, but clonal dominance is not always predictive of an adverse clinical outcome. As Janis Abkowitz's review of mathematical models demonstrated, even in the absence of any oncogenic event related to insertional mutagenesis, when transplanting limiting stem cell doses, clonal dominance is a very common outcome.35
Although most of the models build on data obtained in clinical trials to try to develop methods for comparing relative risks of insertional mutagenesis, a model may not always be able to recapitulate the event. For example, after the development of an HMGA2-insertion-site clone in the b-thalassemia trial, the investigators performed extensive studies in thalassemic mice, including secondary transplants with the GMP vector that was used in the subject at a very similar multiplicity of infection and dose of cells and the identical type of conditioning (adapted to a mouse), and yet there was no skewing in the population of cells as seen in the clinical trial.36
Quantitative assay systems will be essential to monitor the outgrowth of premalignant clones and to assess the practical value of preventive actions. Data obtained in murine models of BMT in combination with an in vitro immortalization assay suggest that the modification of the vector's cis-active elements (most importantly, transcriptional enhancers and splice sites) is even more important to prevent insertional oncogene activation than a modification of the integration pattern of semirandomly integrating vectors.37 For newer vectors lacking strong enhancers, it remains to be determined which of the currently established semirandomly integrating vectors (lentiviral, g-retroviral, foamy viral, a-retroviral, transposons) has the safest integration pattern. Furthermore, there is growing evidence for the importance of milieu factors, probably connected to the underlying disease, in the selection of insertional mutants.
The development of rational prevention strategies thus depends on the identification of the specific mutations forming premalignant clones and a better knowledge of the mechanisms underlying their creation, expansion, and homeostatic control.37 Finally, it was noted that, given the diversity of models and assays being developed to test new vectors, it is critical that investigators using such models to support new clinical trials also have the opportunity to publish these data, both positive and negative, because public distribution of these data will foster more rapid comparison of the preclinical models and development of the field. One potential opportunity for this exchange will be through the National Gene Vector Biorespository (NGVB), which currently has a listing of GLP pharmacology and toxicology results. NGVB leadership is proposing to expand this database to include non-GLP studies that have been submitted as part of an investigational new drug application. A European Commission–funded counterpart currently is under consideration.
Monitoring for clonality: regulatory paradigms
In the United States, the Food and Drug Administration (FDA) has codified recommendations regarding monitoring for insertional mutagenesis into their guidance on long-term follow-up of subjects in gene transfer trials.38 Peripheral blood mononuclear cells can be used for monitoring in a protocol that used transduced CD34+ cells. It is recommended that vector sequences should be tested at least every 6 months for the first 5 years and then yearly for the next 10 years or until no vector is detected.
When at least 1% of the surrogate cells have detectable vector by PCR or another sensitive method, then the pattern of vector integration should be assessed. The FDA does not prescribe one specific method, only that the method should be shown to be specific, sensitive, and reproducible and be based on data with appropriate positive and negative controls, such as a target cell with known number and sites of integrated vector versus target cells with no vector integrants. LAM and ligation-mediated PCR have typically been used.
If this integration analysis reveals the development of a predominant clone or monoclonality, the investigator is asked to identify the integration site(s) in that clone. A predominant clone is not defined by the FDA, but some investigators use a cutoff of >20% of gene-modified cells being derived from a single clone. Analysis should include determining whether the vector is integrated in the vicinity of a known proto-oncogene, monitoring for signs of malignancy, and performing additional clonality analysis within three months. The optimal methods for this analysis are not yet prescribed.
European gene transfer trials are subject to individual country regulation and approval required for early phases of clinical trials as well as to regulations of the European Medicines Agency (EMA) when applying for market authorization. For gene transfer, new regulations for advanced-therapy medicinal products, which includes gene and cell therapies, were issued in 2007 (ref. 39). Out of these regulations arose a consolidated regulatory framework that included the establishment of a Committee for Advanced Therapeutics (CAT), a multidisciplinary scientific committee of experts representing all members of the European Union and countries from the European Economic Area and the European Free Trade Association as well as patient and medical associations.39 The CAT is primarily responsible for evaluation of applications for marketing authorization of advanced-therapy medicinal products for the EMA. Several European regulatory guidance documents address the issues relevant to monitoring for development of insertional mutagenesis and malignancy in gene transfer trials. The risk of integration of the vector and the risk of oncogenicity are used to develop an appropriate monitoring plan. Although close monitoring is expected, current guidelines are not detailed in terms of the type of monitoring and time frames for particular events.40 A monitoring plan with stopping rules is part of the application to begin the clinical trial and is reviewed on a case-by-case basis. Each application dossier is expected to state the rationale for the proposed monitoring plan based on a risk-centered approach. Dedicated guidelines have been issued that are consistent with this41,42 because both the EMA-CAT and national-level regulatory bodies consider such an approach paramount when considering both first-in-human administration and later clinical trials.
Putting it all together: design of new clinical trials and ethical and scientific considerations
Perhaps the greatest challenge in any first-in-human trial is dealing with uncertainty. Despite strong preclinical models, uncertainty about toxicity in humans remains. A key ethical question is how one determines the point at which the potential benefit of proceeding into the clinic outweighs the risks of unforeseen toxicities.43,44,45 Because many diseases targeted for long-term gene correction are pediatric diseases, an additional consideration is that if the intervention presents greater than minimal risk, which a new gene transfer trial does, current regulatory standards in the United States require that there be the prospect of direct benefit to the subject (45 CFR 46 Subpart D). Finally, in the face of uncertainty about risks of insertional mutagenesis, what is the optimal consent process?
The ethical questions of when to move into the clinic, for which diseases, and in which populations, are often addressed in scientific ways by minimizing uncertainty and risks of harm through preclinical testing and by implementing plans to detect and address rare events, such as insertional mutagenesis. Notably, determining whether and when a study has sufficient value to go forward is both a scientific and an ethical determination.46 As in science, much in ethics is about anticipating issues likely to be encountered later. Thus, it is important for researchers to begin thinking early about the harm–benefit balance that would be appropriate in order to move forward at different research stages.
When patients are selected as research subjects, their selection should reflect the goals of the research. Because safety is the foremost consideration in first-in-human trials, it may not always be possible to start with the most informative patient group if the research poses undue risks for them. If it becomes necessary to choose between the most informative subjects and the subjects for whom the risks can best be minimized, the latter are often chosen; nonetheless, those two considerations should be balanced, and it is critical to develop a more systematic sense of what goes into that balance.46
For pediatric subjects, the calculus is more complex because US regulations require that, even in an initial trial involving pediatric subjects, there must be a prospect of direct benefit. What investigators hope for is often different from what they can meaningfully expect from the data.47 This distinction is difficult not only in the consent process but in the harm–benefit calculus itself. Moreover, there may be reasonable disagreements about the meaning of the available data, about what the harm–benefit balance should look like, and about how to value the risks of harm and the chance of benefit under the particular circumstances of a given trial.48 In the context of phase I trials, the prospect of direct-benefit requirement is particularly problematic because it can result in “benefit creep” that is, researchers and oversight bodies may perceive themselves as having to invent or inflate a potential direct benefit in order to do important research in children. Although there is some debate about the usefulness of this regulatory requirement,49 it must be addressed so as to help develop a clearer consensus on what “the prospect of direct benefit” means, especially in early-phase research.
There is irreducible uncertainty in predicting insertional mutagenesis in these studies. The key question is when that uncertainty is small enough that it becomes fair to ask subjects to be involved in the research. Ethical research demands a robust discussion of uncertainty with potential subjects and their families, at the levels of both science and policy.50,51 Patients with few options may have unrealistic expectations of the potential benefit and perhaps minimize the risks of harms—that is, the therapeutic misconception. It is not problematic when researchers' expectations for what is likely to happen in a study and patient-subjects' hopes for what will happen to them differ as long as the risks are minimized and clear information is shared. The key question, then, is how to share the right amount of information in the right amount of detail to facilitate informed decisions about research participation.
The investigator is the most appropriate person to facilitate the information sharing required in the consent process for a given trial, given his or her in-depth understanding of the trial; moreover, the investigator is ultimately responsible, both ethically and legally, for the adequacy of the consent form and process. However, this does not mean that the investigator should be the only participant in the process. When patients have few therapeutic options for a serious or fatal disease, the patient's desire to enroll in a trial strongly aligns with the investigator's goal of enrollment, and it may be beneficial to separate the recruitment process from the consent process, the latter being where the investigator's input is critical. Waiting periods between recruitment, screening, consent, and dosing may also provide mechanisms to promote careful consideration of the risks of harm and potential benefits of participation. Finally, including other parties who do not have a direct interest in the research—for example, clinicians involved in alternative treatments such as a BMT—and even using consent monitors to help test how effectively information has been conveyed to potential subjects may facilitate an effective informed-consent process.48
Because long-term follow-up is critical for gene transfer trials that use integrating vectors for long-term gene correction, the rationale for follow-up, the length of follow-up, and the testing that will be undertaken need to be understood at the outset. Although subjects can always opt out of long-term follow-up, enrolling subjects who are not willing to participate in long-term follow-up at the outset would be a matter for concern. Long-term follow-up is an ethical obligation as well. As a result, it is important to determine how long-term follow-up will be implemented and, in particular, who will undertake long-term follow-up if a commercial or other sponsor is no longer able to support the trial or if an investigator leaves the institution. For NIH-supported research or research carried out at institutions that receive NIH support for recombinant DNA research, the responsibility for long-term follow-up falls on the principal investigator. This does not preclude an institution from placing the responsibility on the department and not just the principal investigator or from devising other arrangements that provide mechanisms to share this responsibility.
There was agreement that the NGVB (previously National Gene Vector Laboratories) plays a critical role in allowing investigators to carry out their responsibilities in long-term follow-up. Without the banking of specimens that the NGVB offers and the LAM-PCR analysis, only very affluent institutions would have the resources to engage in this type of research.
Conclusion
Trials in several blood cell diseases have now demonstrated that long-term gene correction is feasible and in some patients may provide equal clinical benefit with less risk as compared with standard treatments (Table 4). With these therapeutic benefits has also come recognition of real clinical risks, such as leukemogenesis. The increasingly sophisticated understanding of the mechanisms by which these vectors caused leukemia provides new avenues to develop vectors that maintain or even increase the efficacy seen in the first-generation retroviral vectors but that potentially offer a safer alternative. These vectors must undergo preclinical testing before moving into the clinic, but despite great strides made in developing animal models and in vitro assays to predict the safety of new vectors, there are limits to our ability to accurately predict the risk of genotoxicity with a new vector. Ideally, standard assays or platforms might be developed to use across trials. Verification across multiple labs will be key for establishing validity. It is important to understand and be vigilant for alternative mechanisms that may have already been detected in other studies, in which these viruses are used to study oncogenesis. Further ongoing research should address the impact of the target cell biology and disease background on the risk of oncogenic complications induced by integrating gene vectors. Given the latencies between dosing and the cases of leukemia seen in these trials, it may take several years of clinical experience to appreciate whether a new vector truly offers a “safer” alternative. Until a greater understanding is in hand, risk will need to be taken into account in determining whether integrating vector-based gene therapy is appropriate for the disease and patient population being targeted.
Table 4. Summary of key observations.

Acknowledgments
This article reflects the individual views of the authors and is not a consensus paper, nor does it reflect the views of the NIH, the FDA, the European Commission, or the EMA. CLINIGENE is funded by the EC-DG research as the FP6-Network of Excellence contract LSHB-CT-2006-018933.
References
- Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC.et al. (2010Efficacy of gene therapy for X-linked severe combined immunodeficiency N Engl J Med 363355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth C, Gaspar HB., and, Thrasher AJ. Gene therapy for primary immunodeficiency. Curr Opin Pediatr. 2011;23:659–666. doi: 10.1097/MOP.0b013e32834cd67a. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L.et al. (2009Gene therapy for immunodeficiency due to adenosine deaminase deficiency N Engl J Med 360447–458. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P.et al. (2003LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1 Science 302415–419. [DOI] [PubMed] [Google Scholar]
- Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U.et al. (2006Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1 Nat Med 12401–409. [DOI] [PubMed] [Google Scholar]
- Persons DA., and, Baum C. Solving the problem of g-retroviral vectors containing long terminal repeats. Mol Ther. 2011;19:229–231. doi: 10.1038/mt.2010.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A.et al. (2010Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease Nat Med 16198–204. [DOI] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I.et al. (2009Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy Science 326818–823. [DOI] [PubMed] [Google Scholar]
- Biffi A, Bartolomae C, Cesana D, Cartier N, Aubourg P, Ranzani M.et al. (2011Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection Blood 1175332–5339. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F.et al. (2010Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia Nature 467318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco A., and, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer. 2007;7:899–910. doi: 10.1038/nrc2271. [DOI] [PubMed] [Google Scholar]
- Inoue N, Izui-Sarumaru T, Murakami Y, Endo Y, Nishimura J, Kurokawa K.et al. (2006Molecular basis of clonal expansion of hematopoiesis in 2 patients with paroxysmal nocturnal hemoglobinuria (PNH) Blood 1084232–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GP, Berry CC, Malani N, Leboulch P, Fischer A, Hacein-Bey-Abina S.et al. (2010Dynamics of gene-modified progenitor cells analyzed by tracking retroviral integration sites in a human SCID-X1 gene therapy trial Blood 1154356–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Mason P., and, Bessler M. 3′UTR-truncated Hmga2 cDNA causes MPN-like hematopoiesis by conferring a clonal growth advantage at the level of HSC in mice. Blood. 2011;117:5860–5869. doi: 10.1182/blood-2011-02-334425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaillant F, Blyth K, Terry A, Bell M, Cameron ER, Neil J.et al. (1999A full-length Cbfa1 gene product perturbs T-cell development and promotes lymphomagenesis in synergy with MYC Oncogene 187124–7134. [DOI] [PubMed] [Google Scholar]
- Kannian P., and, Green PL. Human T lymphotropic virus type 1 (HTLV-1): molecular biology and oncogenesis. Viruses. 2010;2:2037–2077. doi: 10.3390/v2092037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Take H, Umemoto M, Kusuhara K., and, Kuraya K. Transmission routes of HTLV-I: an analysis of 66 families. Jpn J Cancer Res. 1993;84:1265–1267. doi: 10.1111/j.1349-7006.1993.tb02832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaussin A, Modlich U, Bauche C, Niederländer NJ, Schambach A, Duros C.et al. (2012CTF/NF1 transcription factors act as potent genetic insulators for integrating gene transfer vectors Gene Ther 1915–24. [DOI] [PubMed] [Google Scholar]
- Montini E, Cesana D, Schmidt M, Sanvito F, Bartholomae CC, Ranzani M.et al. (2009The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy J Clin Invest 119964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duros C, Artus A, Gaussin A, Scholtz S, Cesana D, Montini E.et al. (2011Insulated lentiviral vectors towards safer gene transfer to stem cells (abstract 383) Presented 20 May 2011 at the Annual Meeting of the Society of Gene and Cell Therapy, Seattle, WA; < http://www.nature.com/mt/journal/ v19/n1s/pdf/mt201185a.pdf > ( 2011 [Google Scholar]
- Artus A, Duros C, Botbol Y, Scholtz S, Schmidt M, von Kalle C.et al. (2011Directed integration of insulated lentiviral vectors to the heterochromatin towards safer gene transfer to stem cell (abstract 389) Presented 20 May 2011 at the Annual Meeting of the Society of Gene and Cell Therapy, Seattle, WA; < http://www.nature.com/mt/journal/ v19/n1s/pdf/mt201185a.pdf > ( 2011 [Google Scholar]
- Marshall HM, Ronen K, Berry C, Llano M, Sutherland H, Saenz D.et al. (2007Role of PSIP1/LEDGF/p75 in lentiviral infectivity and integration targeting PLoS ONE 2e1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A, Cesana D, Genovese P, Di Stefano B, Provasi E, Colombo DF.et al. (2011Site-specific integration and tailoring of cassette design for sustainable gene transfer Nat Methods 8861–869. [DOI] [PubMed] [Google Scholar]
- Ivics Z, Katzer A, Stüwe EE, Fiedler D, Knespel S., and, Izsvák Z. Targeted Sleeping Beauty transposition in human cells. Mol Ther. 2007;15:1137–1144. doi: 10.1038/sj.mt.6300169. [DOI] [PubMed] [Google Scholar]
- Mátés L, Chuah MKL, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A.et al. (2009Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates Nat Genet 41753–761. [DOI] [PubMed] [Google Scholar]
- Belay E, Dastidar S, VandenDriessche T., and, Chuah MK. Transposon-mediated gene transfer into adult and induced pluripotent stem cells. Curr Gene Ther. 2011;11:406–413. doi: 10.2174/156652311797415836. [DOI] [PubMed] [Google Scholar]
- Gentner B, Visigalli I, Hiramatsu H, Lechman E, Ungari S, Giustacchini A.et al. (2010Identification of hematopoietic stem cell–specific miRNAs enables gene therapy of globoid cell leukodystrophy Sci Transl Med 258ra84. [DOI] [PubMed] [Google Scholar]
- Modlich U, Bohne J, Schmidt M, von Kalle C, Knöss S, Schambach A.et al. (2006Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity Blood 1082545–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hematti P, Hong B-K, Ferguson C, Adler R, Hanawa H, Sellers S.et al. (2004Distinct genomic integration of MLV and SIV vectors in primate hematopoietic stem and progenitor cells PLoS Biol 2e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiem H-P, Sellers S, Thomasson B, Morris JC, Tisdale JF, Horn PA.et al. (2004Long-term clinical and molecular follow-up of large animals receiving retrovirally transduced stem and progenitor cells: no progression to clonal hematopoiesis or leukemia Mol Ther 9389–395. [DOI] [PubMed] [Google Scholar]
- Beard BC, Dickerson D, Beebe K, Gooch C, Fletcher J, Okbinoglu T.et al. (2007Comparison of HIV-derived lentiviral and MLV-based gammaretroviral vector integration sites in primate repopulating cells Mol Ther 151356–1365. [DOI] [PubMed] [Google Scholar]
- Ciuffi A, Ronen K, Brady T, Malani N, Wang G, Berry CC.et al. (2009Methods for integration site distribution analyses in animal cell genomes Methods 47261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel R, Eckenberg R, Paruzynski A, Bartholomae CC, Nowrouzi A, Arens A.et al. (2009Comprehensive genomic access to vector integration in clinical gene therapy Nat Med 151431–1436. [DOI] [PubMed] [Google Scholar]
- Paruzynski A, Arens A, Gabriel R, Bartholomae CC, Scholz S, Wang W.et al. (2010Genome-wide high-throughput integrome analyses by nrLAM-PCR and next-generation sequencing Nat Protoc 51379–1395. [DOI] [PubMed] [Google Scholar]
- Abkowitz JL, Catlin SN., and, Guttorp P. Evidence that hematopoiesis may be a stochastic process in vivo. Nat Med. 1996;2:190–197. doi: 10.1038/nm0296-190. [DOI] [PubMed] [Google Scholar]
- Ronen K, Negre O, Roth S, Colomb C, Malani N, Denaro M.et al. (2011Distribution of lentiviral vector integration sites in mice following therapeutic gene transfer to treat b-thalassemia Mol Ther 191273–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum C, Modlich U, Göhring G., and, Schlegelberger B. Concise review: managing genotoxicity in the therapeutic modification of stem cells. Stem Cells. 2011;29:1479–1484. doi: 10.1002/stem.716. [DOI] [PubMed] [Google Scholar]
- US Food and Drug Administration, Center for Biologics Evaluation and Research (2006). Guidance for industry: gene therapy clinical trials—observing subjects for delayed adverse events < http://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/ Guidances/CellularandGeneTherapy/ucm072957.htm > ( November 2006).
- Committee for Advanced Therapies (CAT); CAT Scientific Secretariat, Schneider, CK, Salmikangas , P, Jilma, B, Flamion, B, Todorova, LR, Paphitou, A et al. (2010Challenges with advanced therapy medicinal products and how to meet them Nat Rev Drug Discov 9195–201. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP) (2008). Guideline on follow-up of patients administered with gene therapy medicinal products < http://www.ema.europa.eu/docs/en_GB/document_library/ Scientific_guideline/2009/11/WC500013424.pdf > ( 22 October 2009).
- European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP) (2009). Concept paper on the development of a guideline on the risk-based approach according to Annex I, Part IV of Directive 2001/83/EC applied to advanced therapy medicinal products < http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/ general/general_content_000405.jsp&mid=WC0b01ac058002958a&jsenabled=true > ( January 2012).
- European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP) (2008). Guideline on safety and efficacy follow-up—risk management of advanced therapy medicinal products < http://www.emea.europa.eu/docs/en_GB/document_library/ Regulatory_and_procedural_guideline/2009/10/WC500006326.pdf > ( 20 November 2008).
- Dresser R. First-in-human trial participants: not a vulnerable population, but vulnerable nonetheless. J Law Med Ethics. 2009;37:38–50. doi: 10.1111/j.1748-720X.2009.00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmelman J. Cambridge University Press: New York; 2010. Gene Transfer and the Ethics of First-in-Human Trials: Lost in Translation. [Google Scholar]
- King NMP., and, Cohen-Haguenauer O. En route to ethical recommendations for gene transfer clinical trials. Mol Ther. 2008;16:432–438. doi: 10.1038/mt.2008.13. [DOI] [PubMed] [Google Scholar]
- Emanuel EJ, Wendler D., and, Grady C. What makes clinical research ethical. JAMA. 2000;283:2701–2711. doi: 10.1001/jama.283.20.2701. [DOI] [PubMed] [Google Scholar]
- King NMP. Defining and describing benefit appropriately in clinical trials. J Law Med Ethics. 2000;28:332–343. doi: 10.1111/j.1748-720x.2000.tb00685.x. [DOI] [PubMed] [Google Scholar]
- Kent A, King NMP., and, Cohen-Haguenauer O. Toward a proportionate regulatory framework for gene transfer: a patient group–led initiative. Hum Gene Ther. 2011;22:126–134. doi: 10.1089/hum.2010.163. [DOI] [PubMed] [Google Scholar]
- Ross LF.2006Phase I research and the meaning of direct benefit J Pediatr 149Suppl 1): S20–S24. [DOI] [PubMed] [Google Scholar]
- King NMP, Henderson GE, Churchill LR, Davis AM, Hull SC, Nelson DK.et al. (2005Consent forms and the therapeutic misconception: the example of gene transfer research IRB 271–8. [PubMed] [Google Scholar]
- National Institutes of Health. NIH guidance on informed consent for gene transfer research < http://oba.od.nih.gov/oba/rac/ic/index.html > ( December 2003).
- Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Adams S, Howe SJ.et al. (2011Long-term persistence of a polyclonal T cell repertoire after gene therapy for X-linked severe combined immunodeficiency Sci Transl Med 397ra79. [DOI] [PubMed] [Google Scholar]
- Kang E, Choi U, Theobald N, Linton G, Long Priel D, Kuhns D.et al. (2010Retrovirus gene therapy for X-linked chronic granulomatous disease can achieve stable long-term correction of oxidase activity in peripheral blood neutrophils Blood 115783–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Zhang F., and, Adams S. Hematopoietic stem cell gene therapy for adenosine deaminase–deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med. 2011;3:97ra80. doi: 10.1126/scitranslmed.3002716. [DOI] [PubMed] [Google Scholar]
- Boztug K, Schmidt M, Schwarzer A, Banerjee PP, Díez IA, Dewey RA.et al. (2010Stem-cell gene therapy for the Wiskott-Aldrich syndrome N Engl J Med 3631918–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bies J, Feikova S, Bottaro D., and, Wolff L. Hyperphosphorylation and increased proteolytic breakdown of c-Myb induced by the inhibition of Ser/Thr protein phosphatases. Oncogene. 2000;19:2846–2854. doi: 10.1038/sj.onc.1203613. [DOI] [PubMed] [Google Scholar]
- Bies J., and, Wolff L. Oncogenic activation of c-Myb by carboxyl-terminal truncation leads to decreased proteolysis by the ubiquitin-26S proteasome pathway. Oncogene. 1997;14:203–212. doi: 10.1038/sj.onc.1200828. [DOI] [PubMed] [Google Scholar]
- Hoemann C, Beaulieu N, Girard L, Rebai N., and, Jolicoeur P. Two distinct Notch1 mutant alleles are involved in the induction of T-cell leukemia in c-myc transgenic mice. Mol Cell Biol. 2000;20:3831–3842. doi: 10.1128/mcb.20.11.3831-3842.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier LS, Carlson CM, Ravimohan S, Dupuy AJ., and, Largaespada DA. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature. 2005;436:272–276. doi: 10.1038/nature03681. [DOI] [PubMed] [Google Scholar]
- Keng V, Villanueva A, Chiang DY, Dupuy AJ, Ryan BJ, Matise I.et al. (2009A conditional transposon-based insertional mutagenesis screen for genes associated with mouse hepatocellular carcinoma Nat Biotechnol 27264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowska MJ, Dybkaer K, Johnsen HE, Wang B, Wabl M., and, Pedersen FS. Loss of microRNA targets in the 3′ untranslated region as a mechanism of retroviral insertional activation of growth factor independence 1. J Virol. 2009;83:8051–8061. doi: 10.1128/JVI.00427-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben David Y, Prideaux VR, Chow V, Benchimol S., and, Bernstein A. Inactivation of the p53 oncogene by internal deletion or retroviral integration in erythroleukemic cell lines induced by Friend leukemia virus. Oncogene. 1988;3:179–185. [PubMed] [Google Scholar]
- Cho BC, Shaughnessy JD, Jr, Largaespada DA, Bedigian HG, Buchberg AM, Jenkins NA.et al. (1995Frequent disruption of the Nf1 gene by a novel murine AIDS virus-related provirus in BXH-2 murine myeloid lymphomas J Virol 697138–7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren AG, Kool J, Matentzoglu K, de Ridder J, Mattison J, van Uitert M.et al. (2008Large-scale mutagenesis in p19ARF- and p53-deficient mice identifies cancer genes and their collaborative networks Cell 133727–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beverly LJ., and, Capobianco AJ. Perturbation of Ikaros isoform selection by MLV integration is a cooperative event in NotchIC-induced T cell leukemogenesis. Cancer Cell. 2003;3:551–564. doi: 10.1016/s1535-6108(03)00137-5. [DOI] [PubMed] [Google Scholar]
- Dail M, Li Q, McDaniel A, Wong J, Akagi K, Huang B.et al. (2010Mutant Ikzf1, KrasG12D, and Notch1 cooperate in T lineage leukemogenesis and modulate responses to targeted agents Proc Natl Acad Sci USA 1075106–5111. [DOI] [PMC free article] [PubMed] [Google Scholar]