

Abstract
Foamy viruses (FVs) (spumaretroviruses) are good alternative to retroviruses as gene therapy vector. Despite four decades since the discovery of FV, its receptor molecule is still unknown. FV vector transduction of human CD34+ cells was inhibited by culture with fibronectin. Because fibronectin contains heparin-binding domain, the interactions of fibronectin with heparan sulfate (HS) on cells might be inhibitory to FV transduction. These observations led us to investigate whether HS is a receptor for FV. Two mutant CHO cell lines (but not parental wild type) lacking cell surface HS but not chondroitin sulfate (CS) were largely resistant to FV attachment and transduction. Inhibition of HS expression using enzymes or chemicals greatly reduced FV transduction in human, monkey, and rodent cells. Raji cells, which lack HS and were largely resistant to FV, were rendered more permissive through ectopic expression of syndecan-1, which contains HS. In contrast, mutant syndecan-1-expressing cells were largely resistant to FV. Our findings indicate that cellular HS is a receptor for FV. Identifying FV receptor will enable better understanding of its entry process and optimal use as gene therapy vector to treat inherited and pathogenic diseases.
Introduction
Spumaretroviruses, commonly known as foamy viruses (FVs), belong to the retrovirus subfamily, Spumaretrovirinae. Their name is derived from the formation of highly vacuolating, foamy-like cytoplasm in productively infected cells and the presence of multinucleated syncytia formed by cell fusion. The replication pattern of FV is similar to that of Hepadnaviridae, another family of reverse-transcribing viruses.1,2,3
FV vectors have several advantages over other retroviral vectors for gene transfer: wild-type viruses lack pathogenicity, and are the largest of all retroviruses, with a packaging capacity of more than 9 kb.1,3 FV vectors have broad host and tissue tropism with a favorable integration profile.4 In contrast, other retroviral vectors can have toxicity as well as gene regulation and targeting issues. The oncoretroviral vector used in the X-SCID gene therapy trial activated the LMO2 proto-oncogene, which caused leukemia in treated patients.5,6 FV vector-mediated gene transfer of hematopoietic stem cells has been used to successfully treat genetic diseases in preclinical animal models such as CD18 deficiency in a canine model,4 and Fanconi's anemia in a murine model.7 The success of these preclinical studies may stimulate the use of FV vectors in future human gene therapy clinical trials.
The FV envelope glycoprotein (gp130) is synthesized as a precursor protein that is cleaved by cellular proteases into surface, transmembrane, and leader peptide subunits.8 The FV receptor–binding domain of the SU at amino acid (aa) 225 to 396 and aa 484 to 555, with N-glycosylation at aa 391 plays a crucial role in cellular receptor binding.9
Virus particles bind to cellular receptors through an envelope or a capsid to enter cells. Receptors are the primary determinants in the early step of virus infection. Although the exact mechanism of uptake of FV into target cells remains unknown, it is thought that FV particles bind to a ubiquitous, yet unidentified cellular receptor. After attachment and endocytosis, the FV capsid can remain in the cytoplasm until uncoating. The viral genome migrates toward the cellular nucleus by yet-unknown cellular signaling pathways.10
Identifying FV receptors and understanding the FV–host cell interactions are important to elucidate the entry process as well as effectively using FV as a gene therapy vector. Interestingly, we have found that FV vector transduction of human CD34+ cells was inhibited when cells were cultured overnight on fibronectin-coated plates. Because fibronectin has a heparin-binding domain,11 it is possible that the interaction of fibronectin with the cell surface heparan sulfate (HS) on target cells might inhibit FV transduction. Moreover, FV can infect a wide variety of human, murine, and nonhuman primates cells,1,12 suggesting that it uses a ubiquitous cell surface molecule for transduction. As HS is present on numerous cell types and is ubiquitously expressed throughout the animal kingdom, we investigated the possibility that FV uses cell surface HS to mediate transduction. It has been reported that proteoglycans (PGs) are not absolutely essential for FV susceptibility but seemed to contribute significantly to FV infection.13
HSPG consists of a PG core such as syndecan-1 to which HS chains are attached.14,15 HS interacts with growth factors and their receptors, extracellular matrix proteins, and cell–cell adhesion molecules14,15,16 and acts as a receptor for viruses, such as adeno-associated virus 2,17 herpes simplex virus 1 (HSV-1),18,19 and dengue virus.20
Here, we have provided evidences that HS serves as a receptor for FV attachment and transduction of human, monkey, and rodent cells.
Results
Fibronectin-inhibited FV transduction through downregulation of cell surface HS
If human CD34+ cells and FV- green fluorescent protein (GFP) vector containing a native envelope were added simultaneously onto a fibronectin (CH-296)-coated plate, cells were successfully transduced (Figure 1a, middle panel, black bar). In contrast, when cells were cultured on a fibronectin-coated plate overnight, FV transduction was reduced about 93% (Figure 1a, middle panel, gray bar). Vesicular stomatitis virus (VSV)-G envelope pseudotyped HIV-based lentiviral GFP vector (LV) is routinely used in our laboratory to transduce various types of cells. It was interesting to investigate whether LV transduction could be inhibited by fibronectin when used in conditions similar to those used for FV. After LV transduction of CD34+ cells under both conditions, it was found that fibronectin had little but statistically significant effect on LV transduction (Figure 1a, right panels, black versus gray bars). Because fibronectin has a heparin-binding domain,11 it raised the possibility that interactions of fibronectin with cell surface HS on target cells might inhibit FV transduction. Cell surface expression of HS was found in CD34+ cells after staining with anti-HS antibody (Figure 1b, upper graph). However, HS expression was abolished if cell were grown overnight on fibronectin-coated plate (Figure 1b, lower graph).
Figure 1.

Fibronectin-inhibited FV transduction through downregulation of cell surface HS. (a) Human mobilized peripheral blood CD34+ cells were transduced with FV-GFP or VSV-G pseudotyped LV-GFP. Black bars (FN−) showed CD34+ cells that were transduced with FV or LV vector overnight immediately after seeding on fibronectin-coated plates. Gray bars (FN+) showed cells were cultured overnight on fibronectin-coated plates and transduced with FV or LV vector overnight. Cells were analyzed for GFP expression by flow cytometry 3–7 days after transduction. For each FV and LV groups, the transduction of “FN−” cells was converted into 100% and the transduction of “FN+” cells was calculated according to their proportional percentages. Data represented mean and standard error of the mean (SEM) of three independent experiments. * P = 1.6 × 10−5 and **P = 0.03. (b) CD34+ cells were stained with anti-HS antibody. Upper graph, HS expression in the absence of fibronectin; lower graph, inhibition of cell surface HS expression when cells were grown overnight on fibronectin. Light lines, isotype control; dark solid area, HS expression detected with anti-HS antibody staining. FITC, fluorescein isothiocyanate; FN, fibronectin; FV, foamy virus; GFP, green fluorescent protein; HS, heparan sulfate; LV, lentivirial vector; VSV, vesicular stomatitis virus.
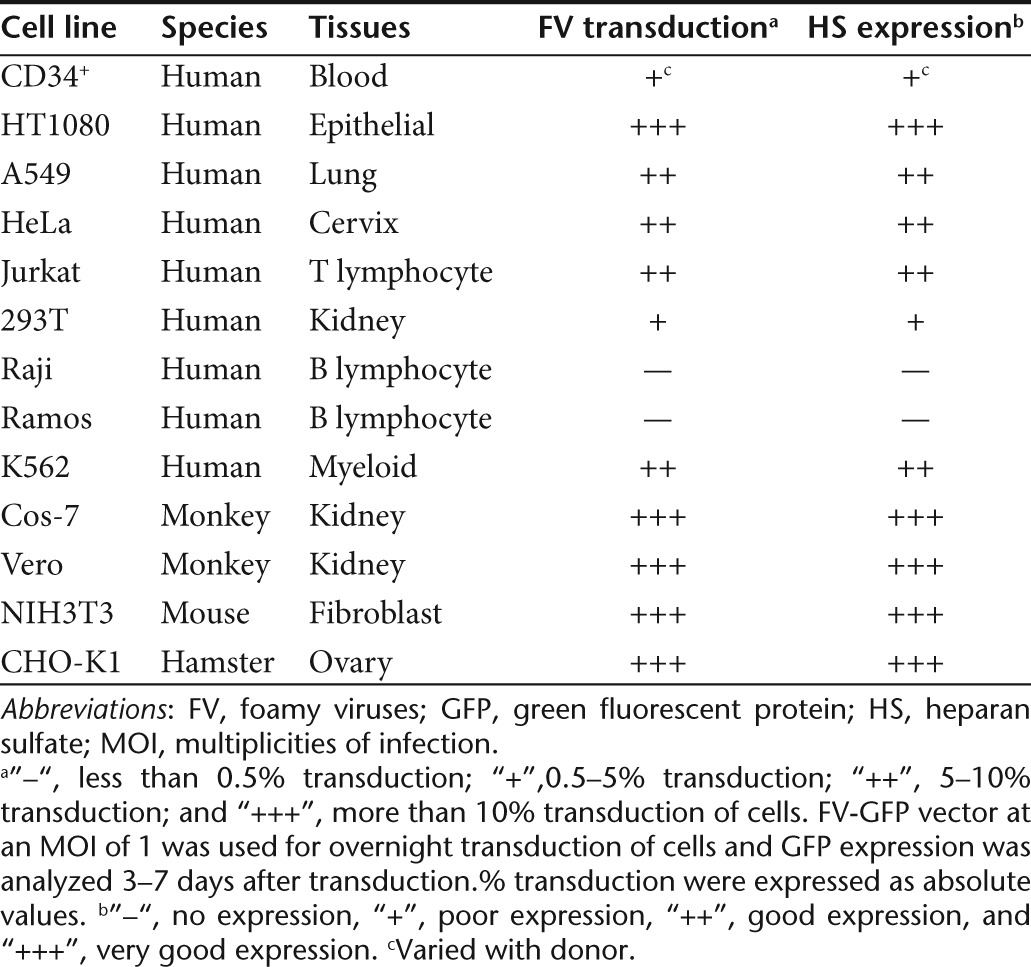
In addition, there is a relationship between FV transduction and HS expression in several human, monkey, hamster, and murine cell lines (Table 1). These findings indicate that cell surface HS is essential for FV transduction.
Table 1. Cell lines tested for FV transduction and HS expression.
FV transduction of cells deficient in HS synthesis pathways is strongly reduced
CHO-K1 and its mutant cell lines defective in glycosaminoglycan (GAG) synthesis were used to define the requirement of HS for FV attachment and transduction. The mutant cell line, pgsA-745 is deficient in xylosyltransferase, an enzyme necessary for GAG synthesis, and does not produce detectable levels of HS or chondroitin sulfate (CS).21 The pgsD-677 cells have a single mutation affecting both N-acetyl-glucosaminyltransferase and glucuronosyltransferase,which are necessary for the polymerization of HS chains, and do not synthesize HS but does produce three times more CS than wild-type CHO-K1 cells.22,23 We confirmed that the mutant cells were deficient in HS expression whereas the wild-type CHO-K1 expressed HS, by staining cells with anti-HS antibody (Figure 2a). FV vector was used to evaluate the transduction efficiency of CHO-K1 and its mutants. The lack of HS expression severely impaired the ability of FV to transduce mutant cells. Compared with wild-type CHO-K1 cells, there was a 21 to 36 times reduction in FV transduction in pgsA-745 cells and 6 to 13 times reduction in FV transduction in pgsD-677 cells (Figure 2b). Despite overproduction of CS in pgsD-677 cells, the poor transduction in mutant cells further demonstrated the specificity of FV for HS and not for CS. A few mutant cells express residual HS (because of residual xylotransferase activity)21,22 which allowed FV transduction to a lesser extent. However, much higher multiplicities of infections (MOIs) of FV were required to obtain a level of transduction similar to that of wild-type CHO-K1cells. This low HS expression could not be detected by flow cytometric analysis. In contrast, wild-type CHO-K1 cells as well as HS-deficient cells were susceptible to LV transduction at similar efficiencies (Figure 2c).
Figure 2.

HS-deficient CHO-K1 cells were largely resistant to FV transduction. FV and VSV-G LV transduction of wild-type CHO-K1 and mutant cells (pgsA-745 and pgsD-677) defective in HS synthesis were assessed. (a) Flow cytometric analyses of wild-type and mutant CHO cells were done to check for HS expression. Cells were stained with a monoclonal antibody (F58-10E4) specific for HS or its isotype control. (b) FV transduction of wild-type CHO-K1 and mutant cells was done at MOIs of 0, 1, 2, 5, 10, and 20 for 1 hour at 37 °C. Cells were harvested 3–7 days after transduction, and GFP analyses were done by flow cytometry. (c) As a control, LV-GFP vector transduction and analyses were done as for the FV group. (d) Binding of 35S-labeled FV to wild-type CHO-K1 and mutant lines, pgsA-745 and pgsD-677, was done at MOIs of 0, 1, 2, 5, 10, and 20 for 1.5 hours at 4 °C. After unbound virus particles were removed by washing, cells were lysed and radioactivity was counted. Counts of wild-type CHO-K1 cells were converted to 100% and counts of pgsA-745 and pgsD-677 cells were assessed according to their proportional percentages. Data represented the mean and standard error of the mean (SEM) of three independent experiments. FITC, fluorescein isothiocyanate; FV, foamy virus; GFP, green fluorescent protein; HS, heparan sulfate; LV, lentivirial vector; MOI, multiplicities of infection; VSV, vesicular stomatitis virus.
To investigate FV binding to CHO-K1 and its mutants, cells were incubated with 35S-labeled FV. At 4 °C, FV could bind to but did not enter cells. Compared with wild-type CHO-K1 cells, 17 to 20 times and 7 to 9 times reduction in FV binding was found in pgsA-745 and pgsD-677cells, respectively (Figure 2d). The poor binding and transduction of FV to HS-mutants support that HS is a receptor for FV binding and transduction of cells.
Heparinase III digestion of HS Reduced FV Transduction
We examined the ability of FV to transduce cells after treatment with heparinase III which specifically digests HS from cells. FV transduction was reduced in heparinase III-treated cells than in untreated cells. For NIH3T3 cells, a heparinase III concentration of 0.01 mIU/ml reduced FV transduction by more than 95% (Figure 3b, left panel, black bars). For HT1080 and Cos-7 cells, an enzyme concentration of 0.1 mIU/ml reduced FV transduction by more than 80% (Figure 3a,c, left panels, black bars). LV transduction levels were similar for buffer-treated and heparinase III-treated cells (Figure 3, left panels, gray bars). Anti-HS antibody (F58-10E4) specifically recognized intact HS in the untreated control cells (Figure 3a–c, middle panels). Heparinase III specifically digested the HS from PG moieties and thus eliminated the HS epitope recognized by the antibody. Therefore, heparinase III-treated cells had lower HS expression than untreated cells (Figure 3a–c, right panels). The reduced susceptibility of HT1080, NIH3T3, and Cos-7 cells after enzymatic removal of HS indicates that HS plays an important role in FV transduction in human, murine, and monkey cells.
Figure 3.

Heparinase III digestion of cell surface HS severely reduced FV transduction. Cells were incubated for 1 hour at 37 °C with the indicated concentrations of heparinase III. After washing, one group of cells was transduced with FV (black bars) or LV (gray bars) at MOI of 2 for 1 hour at 37 °C. Unbound virus was removed by extensive washing with PBS. Cells were analyzed for GFP expression 3–7 days after transduction. (a) HT1080, (b) NIH3T3, and (c) Cos-7. For each FV and LV groups, the transduction of untreated control cells was converted to 100% and the transduction of heparinase III-treated cells was calculated according to their proportional percentages. Data represented the mean and standard error of the mean (SEM) of three independent experiments. Another group of cells was fixed with paraformaldehyde and stained with anti-HS antibody to check HS expression. Middle and right panels showed the flow cytometric analysis of HS expression of buffer-treated control cells and heparinase III-treated cells, respectively. FITC, fluorescein isothiocyanate; FV, foamy virus; GFP, green fluorescent protein; HS, heparan sulfate; LV, lentivirial vector; MOI, multiplicities of infection; PBS, phosphate-buffered saline.
Sulfation of cellular HS is essential for efficient FV transduction
To test whether sulfated HS is required for FV transduction, cellular adenosine triphosphate sulfurylase (which causes sulfation of cellular GAGs or other molecules) was inhibited by adding sodium chlorate to cells. Sodium chlorate at 25 mmol/l inhibited FV transduction by 95% in A549 (Figure 4a, black bars) and NIH3T3 (Figure 4b, black bars) cells and by 82% in Vero cells (Figure 4c, black bars). In contrast, the same concentration of sodium chlorate inhibited LV transduction by only 37% in A549 cells (Figure 4a, gray bars); 61% in NIH3T3 cells (Figure 4b, gray bars); and 36% in Vero cells (Figure 4c, gray bars). Compared with untreated control cells, there was no significant toxicity in sodium chlorate-treated cells. Anti-HS antibody specifically recognized sulfated HS in untreated control cells (Figure 4a–c, middle panels). Sodium chlorate inhibited the sulfation of HS and thus eliminated the epitope recognized by the antibody (Figure 4a–c, right panels). Therefore, sodium chlorate-treated cells showed reduction in HS expression compared with untreated control cells. The reduced susceptibility of A549, NIH3T3, and Vero cells after inhibition of sulfation indicates that the sulfate group of HS plays an important role in FV transduction in human, murine, and monkey cells.
Figure 4.

FV transduction was strongly dependent on cellular sulfation of HS. Cells were cultured in a low-sulfate F12 medium in the presence of increasing concentrations of sodium chlorate for 36 hours. One group of cells was transduced with FV (black bars) or LV (gray bars) at MOI of 2 for 1 hour at 37 °C, and another group of cells was fixed and stained with anti-HS antibody. (a) A549, (b) NIH3T3, and (c) Vero. Left graphs showed transduction efficiency. The transduction of untreated control cells was converted to 100% and the transduction of sodium chlorate-treated cells was assessed according to their proportional percentages. Data represented the mean and standard error of the mean (SEM) of three independent experiments. Middle and right graphs showed the flow cytometric analysis of HS expression of untreated control cells and sodium chlorate-treated cells, respectively. FITC, fluorescein isothiocyanate; FV, foamy virus; HS, heparan sulfate; LV, lentivirial vector; MOI, multiplicities of infection.
Ectopic expression of syndecan-1 and HS permits efficient FV transduction
Raji cells do not express HS (Figure 5a, first row, left) and are less permissive to FV. Raji cell susceptibility to FV transduction was strongly increased through ectopic expression of HS. Raji cells transfected with syndecan-1 cDNA (a PG-expressing molecule in which HS binds through serine residues) expressed both cell surface HS (Figure 5a, first row) and PG (Figure 5a, second row) in bulk culture and clones. Compared with mock cells, FV transduction was 20 to 30 times higher in both bulk culture and individual clones those expressed HS (Figure 5b, bars 3–8). We also investigated whether FV uses either HS or PG to transduce cells. Syndecan-1 contains three HS chains that originate from evolutionarily conserved serine residues.24 We obtained a triple deletion mutant (TDM) of syndecan-1 that lacks HS chains because of replacement of serines with alanines and therefore HS cannot bind to PG (Supplementary Figure S1). Two CS-binding serines of the TDM syndecan-1 were not mutated.24 TDM syndecan-1-transfected cells expressed PG (Figure 5a, fourth row) but not HS (Figure 5a, third row). Indeed, cells transfected with wild-type syndecan-1 had high levels of HS, whereas cells transfected with TDM syndecan-1 expressed HS but not attached to TDM syndecan-1 (Figure 5a, first versus third row). We also found that FV transduction of cells expressing HS-deficient mutant (TDM) was 20–30 times lower than that of cells expressing wild-type syndecan-1 both in bulk culture and cell clones (Figure 5b, bars 9 to 13 versus bars 3 to 8). These findings indicate that FV transduces cells through the HS chain and not the PG (which acts as binding molecule for HS) and that HS but not CS is needed for FV transduction.
Figure 5.

Ectopic expression of HS in Raji cells made them more permissive to FV transduction. (a) Raji cells transfected with syndecan-1 (first and second rows), and TDM-syndecan-1(third and fourth rows) expression plasmids were investigated for the cell surface expression of HS (first and third rows) and proteoglycan (second and fourth rows). (b) Mock, syndecan-1-, and TDM syndecan-1-transfected bulk cultures and cell clones were evaluated for FV transduction. Cells were transduced with FV at MOI of 2 overnight and checked for GFP expression 3–7 days after transduction. Data represented the absolute values of mean and standard error of the mean (SEM) of three independent experiments. (c) Southern blot analysis was done to check FV provirus DNA entry into the mock (untransfected), syndecan-1-, and TDM syndecan-1-transfected cells of both bulk cultures and cell clones. Lane 1, 3, and 7 were untransduced and lane 2, 4, 5, 6, 8, 9, and 10 were transduced with FV-GFP. Genomic DNA was extracted from cells and resolved on agarose gel. 32P-labeled FV pol sequence was used to probe the membranes. FITC, fluorescein isothiocyanate; FV, foamy virus; GFP, green fluorescent protein; HS, heparan sulfate; MOI, multiplicities of infection; TDM, triple deletion mutant.
Although the same MOIs of FV were used in both wild-type syndecan-1 and TDM syndecan-1-transfected cells, the number of GFP-positive cell was 20–30 times lower in TDM syndecan-1-expressing cells than in wild-type syndecan-1-expressing cells. The reason for the low percentages of GFP expression in TDM syndecan-1-positive cells was clarified by investigating the FV provirus DNA in cells. As the wild-type syndecan-1-positive Raji cells were vigorously transduced by FV because of expression of HS, the amount of FV provirus DNA in these cells were 20–30 times higher than in TDM syndecan-1-positive cells (Figure 5c, lanes 4–6 versus lanes 8–10). These data provide genetic evidence that HS is required for FV entry to cells.
Discussion
In this study, we have found that cell membrane-associated HS serves as a receptor for FV based on the following evidence: (i) HS-deficient cells are less permissive to FV whereas wild-type cells are permissive, (ii) enzymatic removal of HS or chemical removal of the sulfate group from cells greatly reduces FV permissiveness, and (iii) induction of HS expression makes cells more permissive to FV.
It has been reported that PGs seemed to contribute significantly to FV infection,13 but detailed information were not given whether these molecules are receptor for FV. The expression of PGs or HS in SOG9 cell and its parental cell was not compared in this study. In fact, this mutant cell was still susceptible to FV infection.
Overnight culture of hCD34+ cells in the presence of fibronectin-inhibited HS expression and FV transduction. But FV needs fibronectin to transduce hCD34+ cells. Therefore, we seeded hCD34+ cells on fibronectin-coated plate during FV transduction and immediately added FV so that HS could mediate FV transduction by interacting with fibronectin and no inhibition of HS occurred by fibronectin.
Nuclear localization of HS occurs in primary corneal fibroblast cultured in the presence of fibronectin. Matrices containing the heparin-binding domain of fibronectin but not the integrin-activating domain show increased nuclear localization of HS mediated by protein kinase C signaling pathways.25
Genetic defects in the cellular pathways for HS synthesis can make cells less permissive to FV. The requirement of FV for HS cannot be fulfilled by other GAGs such as CS commonly found on the cell membrane. Mutant CHO cells, pgsD-677, which express three times more CS than the wild-type but do not express HS, were less permissive to FV attachment and transduction. These findings indicate that FV envelope glycoprotein recognizes structural features of HS that are not present in other GAG molecules such as CS. HS composed of repeating disaccharide units of alternating glucosamine and hexuronic acid residues,26 whereas CS composed of repeating disaccharide units of alternating galactosamine and hexuronic acid residues.27 Therefore, it is possible that FV may have a specific affinity for the glucosamine residues of HS.
We found that enzymatic removal of HS molecules from cells reduced FV transduction. HS is expressed on the cell surface rapidly if heparinase III is removed from cells. To analyze HS expression after heparinase III treatment, the cells need to be fixed immediately. If the cells are stained without fixation, significant amount of HS was found in heparinase III-treated cells (Md. Nasimuzzaman and D.A. Persons, unpublished observations).
We showed that ectopic expression of HS by transfection with syndecan-1 made cells more permissive to FV. Three HS-binding serines at positions 37, 45, and 47 in the N-terminal region as well as two CS-binding serines at 210 and 220 near the C-terminal region has been reported in syndecan-1.24 The syndecan-1 mutant protein, TDM lacks all three HS chains, and FV does not transduce cells that express this mutant. Despite the presence of two CS chains, the syndecan-1 mutant cells are less well transduced by FV than wild-type syndecan-1 cells, indicating the requirement of HS for FV transduction.
HS is present almost ubiquitously on cell surfaces but is extensively heterogeneous with respect to their compositions and quantities among different species, cell types, tissues, and developmental stages. HS chains are repeating disaccharide units of N-acetylglucoseamine and glucuronic/iduronic acid, covalently linked to a membrane protein, such as syndecans, β-glycans, and glypicans.28 It would be interesting to analyze the affinity of FV to the HS of PGs other than syndecan-1. HS moieties derived from a variety of sources have high heterogeneity and vary in their sequences, and the level of sulfation. Also, there are subtle differences in the HS structure for binding to different ligands, for example, HSV-1 requires specific sulfation to infect host cells.29 It is therefore necessary to analyze the type of HS structure that interacts with FV.
We found that sodium chlorate inhibited cellular sulfation of HS and FV transduction as well as LV transduction. The LV particles may use an unknown sulfated receptor molecule for transduction. It is reported that dengue virus infection in Vero cell is inhibited with sodium chlorate through inhibition of sulfation of HS.20 Despite the same concentration of sodium chlorate was used under the same cell culture conditions, HS inhibition was variable in different cell lines, likely due to heterogeneity in the structure of HS chains.14 As low-sulfate F12 medium and sodium chlorate are toxic to HT1080 and Cos-7 cells, we used A549 and Vero cells.
The basic amino acids of a viral envelope or capsid protein interact with the acidic sulfate group of cell surface HS. For example, the feline immunodeficiency virus needs two highly conserved arginine residues, R379 and R389 at the N-terminal side of the V3 region, for contact between feline immunodeficiency virus envelope glycoprotein and HS;30 adeno-associated virus 2 needs two arginine residues, 585 and 588 for binding of the capsid protein to HS.31 It would be interesting to analyze the amino acid sequences needed for FV envelope binding to HS.
HS and fibroblast growth factor receptor 1 (FGFR1) act in a coordinated manner in cellular signaling pathways.32,33 HS acts as a primary receptor for HSV-118 and AAV-217; and FGFR1 as a co-receptor for HSV-134 and adeno-associated virus 2.35 There is a controversy regarding using FGFR1 as a co-receptor for HSV- 1.36,37 FGFR1 may not be a co-receptor for FV. Raji cells do not express FGFR1.35 We have shown that transfecting Raji cells with only syndecan-1 made them more permissive to FV transduction. Moreover, siRNA-mediated downregulation of FGFR1 in cells did not inhibit FV transduction (Md. Nasimuzzaman and D.A. Persons, unpublished observations). It would be interesting to investigate whether FV uses any co-receptor as an entry portal.
We have provided biochemical and genetic evidences supporting that HS is a receptor for FV. Although HS is expressed in almost all host species and tissues, the transduction of FV to human CD34+ hematopoietic cells varies by donor. The induction of HS expression in hematopoietic stem cells may increase their transduction with FV. Identification of the FV receptor molecule will provide information on the entry mechanism of FV and promote further development of the FV vector to treat inherited and pathogenic diseases in clinical human gene therapy trial.
Materials and Methods
Cell culture. Chinese hamster ovary cell lines, CHO-K1, pgsA-745, and pgsD-67721,22 were purchased from American Type Culture Collection (ATCC) (Rockville, MD) and cultured in F12K medium (Invitrogen, San Diego, CA) supplemented with 10% fetal bovine serum (FBS; Invitrogen). Raji,38 Jurkat,39 and Ramos cells (a kind gift from Dr Mary Ellen Conley) were cultured in RPMI 1640 medium (Invitrogen) supplemented with 15% FBS. HT1080,40 Vero,41 Cos-7,42 A549,43 NIH3T3,44 and 293T45 cells were grown in Dulbecco's Modified Eagle Medium (Invitrogen) supplemented with 10% FBS. Human CD34+ cells were either cultured in Ex Vivo 10 (Lonza, Walkersville, MD) with cytokine mixtures (SCF, TPO, and Flt-3 ligand, each 100 µg/ml) overnight on a fibronectin-coated plate (Retronectin, Takara, Shiga, Japan) or seeded on a fibronectin-coated plate immediately before vector transduction.
FV vector production. Prototype FV vector plasmid DNA based on pHSRV13 (infectious clone of SFVcpz(hu)) was described previously.46 FV particles were produced by using the four plasmid system47 by transient transfection of 293T cells with polyethyleneimine (PEI). In brief, 293T cells were transfected with FV plasmid DNA consisting of 12 µg of pCIGS (gag), 1.6 µg of pCIPS (pol), 0.75 µg of pCIES (envelope), and 12 µg of gene transfer vector plasmid, pPFV-GFP per 10-cm dish. The PEI-DNA mixture was added to cells and incubated for 60–72 hours. For 35S-labeled FV production, transfected cells were washed with phosphate-buffered saline (PBS) 24 hours after transfection and cysteine-methionine-free Dulbecco's Modified Eagle Medium (Invitrogen) with 5% FBS was added. Trans-35S- labeled cysteine-methionine (MP Biomedicals, Solana, OH) was added to a final concentration of 100 µCi/ml.48 Both unlabeled and 35S-labeled FV particles were concentrated by ultracentrifugation at 50,000g for 2 hours at 20 °C. Virus supernatants were stored at –80 °C in 5% DMSO. Titers of the vector preparations were determined after transduction of the HT1080 cell.47 The unconcentrated vector titer was ~107 transduction units (TU)/ ml and was concentrated upto 100 times.
VSV-G pseudotyped LV production. 293T cells were transfected with HIV-1-based plasmid DNA consisting of 6 µg of pCAGkGP1R (Gag/Pol), 2 µg of pCAG4-RTR2 (Rev/Tat), and 10 µg of gene transfer vector expressing GFP; and 2 µg of pCAG-VSV-G (VSV-G envelope) plasmid per 10-cm dish using the calcium phosphate method.49 The next day, cells were washed with PBS and then grown in Dulbecco's Modified Eagle Medium with 10% FBS. After 24 hours, LV supernatants were harvested, cleared using low-speed centrifugation, and filtered through a 0.2-µm filter. LV supernatants were concentrated by ultracentrifugation for 1.5 hours at 50,000g at 4 °C (Beckman SW28 rotor), aliquoted, and kept at −80 °C until use. Titers of vector preparations were determined by transduction of HeLa cells with 6 µg/ml of polybrene (Sigma, St. Louis, MO). The unconcentrated vector titer was ~5 × 107 transduction units (TU)/ml and was concentrated upto 100 times.
Development of syndecan-1/HS-positive stable raji cells. Raji cell is less permissive to FV transduction and does not express HS. Raji cell was separately transfected with syndecan-1 expression plasmid and TDM syndecan-1 (a kind gift from Dr Sanderson, University of Alabama, Birmingham, AL).24 These plasmids also contain a neomycin-resistant gene. After selecting cells with 500 µg/ml G418, expression of HS and syndecan-1 (PG) was checked after staining cells with anti-HS and anti-syndecan-1 antibodies, respectively. Raji cells transfected with syndecan-1 expressed both cell surface HS and PG moieties whereas TDM did not showed HS. Both bulk culture and cell clones were expanded. Expression of HS and PG was checked after staining cells with their corresponding antibodies. Both bulk culture and several cell clones expressed high level of both HS and PG for wild-type syndecan-1 whereas mutant cells did not showed HS. Both bulk culture and cell clones were also checked for FV transduction.
FV and VSV-G pseudotyped LV vector transduction of target cells. Cells were seeded and cultured at 37 °C in 5% CO2 for at least 18 hours before vector transduction. Cells were transduced with FV or VSV-G pseudotyped LV-GFP at multiplicities of infection (MOIs) of 1–20, where applicable. Cells were washed extensively with PBS to remove unbound vector particles, and GFP expression was analyzed 3–7 days after transduction by using a BD FACScan cytometer (BD Pharmingen, San Diego, CA). In individual FACS samples, 10,000 events (viable cells) were acquired and the absolute values of negative control were 0.1 to 0.5%. (GFP expression levels of FV-GFP and LV-GFP vector-transduced cells were 200–1,000 times above the detection limit.)
FV binding assay. Binding of FV vector to the cellular receptor was done using 35S-labeled FV particles. Cells were detached, and 3 × 105 cells were resuspended in ice-cold virus binding buffer (0.5 mmol/l MgCl2, 0.5 mmol/l CaCl2, and 0.5 % bovine serum albumin in PBS). FV vector was added to the cells at MOIs of 0, 1, 2, 5, 10, and 20. Cells were mixed with FV vector and incubated at 4 °C for 1.5 hours with mild shaking. Cells were washed extensively with ice-cold PBS to remove unbound virus, lysed in 1% SDS, and cell-associated radioactivity was counted by a liquid scintillation counter (Beckman Instruments, Fullerton, CA).
Heparinase III treatment of cells. Heparinase III, also known as heparitinase (E.C. 4.2.2.8), an enzyme that specifically digests cell surface HS,50 was purchased from Sigma (St. Louis, MO). Heparinase III concentrations were used in milli international units (mIU) per milliliter (1 IU is equivalent to 600 Sigma units). Cells were seeded 18 hours before enzyme treatment and FV transduction. Cells were incubated with various concentrations of heparinase III in 20 mmol/l Tris-HCl (pH 7.5), 4 mmol/l CaCl2, and 0.01% bovine serum albumin at 37 °C, in 5% CO2 for 1 hour. Cells were washed with PBS and immediately transduced with the FV or VSV-G pseudotyped LV vectors at an MOI of 2 at 37 °C for 1 hour. Cells were washed extensively with PBS to remove unbound vectors. The transduction efficiency of both heparinase III-treated and untreated control cells were determined after 3–7 days. For FACS analysis of HS expression, HT1080, NIH3T3, and Cos-7 cells were untreated or treated with 1.0, 4.0, and 2.0 mIU/ml heparinase III, respectively, for 1 hour at 37 °C. Cells were detached from plate, washed with PBS, and fixed with 2% paraformaldehyde for 30 minutes.
Sodium chlorate treatment of cells to inhibit sulfation of HS. To prevent sulfation of cell surface HS, cells were cultured for 36 hours in the presence of various concentrations of sodium chlorate (Sigma, MO) in F12 medium (reduced sulfate) with 5% dialyzed FBS. Cells were washed and transduced with FV or LV vector at MOI of 2 at 37 °C for 1 hour. For FACS analysis of HS expression, A549, NIH3T3, and Vero cells were untreated or treated with 25 mmol/l sodium chlorate. Cells were detached from plates, washed with PBS, and fixed with 2% paraformaldehyde for 30 minutes.
Flow cytometric analysis of HS expression in cells. To investigate cell surface expression of HS or PG, 2 × 105 cells were fixed with 2% paraformaldehyde for 30 minutes, washed with PBS, and incubated in Fc receptor blocking buffer (100 mg/ml human γ globulin, 5% FBS, and 2 mmol/l sodium azide) for 10 minutes to prevent nonspecific binding of antibodies. Cells were washed and resuspended in primary antibodies; mouse anti-HS (F58-10E4; Seikagaku, Japan), mouse anti-syndecan-1 (BD Biosciences, San Diego, CA) or isotype control antibody at a dilution of 1:200 for 30 minutes and washed to remove the primary antibody. Fluorescein isothiocyanate-conjugated secondary anti-mouse antibody (Bio Legend, San Diego, CA) was added at a dilution of 1:200 for 30 minutes. Finally, cells were washed extensively with PBS and analyzed by a flow cytometer, BD FACScan (BD Pharmingen, San Diego, CA).
Southern blot analysis. Southern blot analysis was done by a modified protocol.49 Genomic DNA was prepared from transduced cells by using an Archivepure DNA purification kit (5Prime, Gaithersburg, MD). Five micrograms of genomic DNA was digested with EcoNI restriction enzyme (which cut at both ends of the provirus, liberating near unit length FV provirus). DNA was resolved on a 0.8% agarose gel and transferred onto a nylon membrane. A 32P-labeled FV pol sequence (a 551-bp EcoRI-EcoRV fragment) was used to probe the membranes in Hibrisol I blocking buffer (Millipore, Temecula, CA) overnight at 42 °C on a rotator. The membrane was washed at 65 °C for 1 hour in 2× saline-sodium citrate/0.05% SDS, and 1 hour in 0.2× saline-sodium citrate/0.05% SDS wash buffer. The membrane was kept in a Phospho-Image cassette aligned to the grid. The cover was placed on the cassette and was scanned after 24, 48, and 72 hours of exposure using a Molecular Dynamic Storm 860 Phosphorimager (Sunnyvale, CA). Equivalent loading of each lane was confirmed by ethidium bromide staining of the gel.
Statistical analyses. Statistical analyses were done using the Student's two-tailed t-test to determine statistically significant differences between mean values of data sets using Microsoft excel and GraphPad Prism software (San Diego, CA). A P value of less than 0.05 was considered statistically significant. Mean and standard error of the mean were calculated by using Microsoft Excel.
SUPPLEMENTARY MATERIAL Figure S1. Schematic maps of syndecan-1 and its mutant, TDM-syndecan-1.
Acknowledgments
We would like to thank Ralph D. Sanderson, University of Alabama, Birmingham, AL for generously providing syndecan-1 and its mutant plasmids. We would like to thank Arthur W. Nienhuis and Brian P. Sorrentino of St. Jude Children's Research Hospital, Memphis, TN for critical review of the manuscript. This work was supported by the National Heart, Lung, and Blood Institute PO1HL053749, Basic and Translational Research Program Sickle Cell Disease Grant U54HL070590, the Cooley's Anemia Foundation, and the American Lebanese Syrian Associated Charities. We declare that there is nofinancial conflict of interest of our work.
Supplementary Material
Schematic maps of syndecan-1 and its mutant, TDM-syndecan-1.
REFERENCES
- Linial M.2007Foamy viruses Fields BN, Knipe DM., and, Howley PM.eds). Fields Virology5th edn, vol. 1Lippincott Williams & Wilkins: Philadelphia; 2245–2262. [Google Scholar]
- Rethwilm A. Foamy virus vectors: an awaited alternative to gammaretro- and lentiviral vectors. Curr Gene Ther. 2007;7:261–271. doi: 10.2174/156652307781369092. [DOI] [PubMed] [Google Scholar]
- Rethwilm A. Molecular biology of foamy viruses. Med Microbiol Immunol. 2010;199:197–207. doi: 10.1007/s00430-010-0158-x. [DOI] [PubMed] [Google Scholar]
- Bauer TR, Jr, Allen JM, Hai M, Tuschong LM, Khan IF, Olson EM.et al. (2008Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors Nat Med 1493–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P.et al. (2000Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease Science 288669–672. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P.et al. (2003LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1 Science 302415–419. [DOI] [PubMed] [Google Scholar]
- Si Y, Pulliam AC, Linka Y, Ciccone S, Leurs C, Yuan J.et al. (2008Overnight transduction with foamyviral vectors restores the long-term repopulating activity of Fancc−/− stem cells Blood 1124458–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke N, Stange A, Lüftenegger D, Zentgraf H., and, Lindemann D. Ubiquitination of the prototype foamy virus envelope glycoprotein leader peptide regulates subviral particle release. J Virol. 2005;79:15074–15083. doi: 10.1128/JVI.79.24.15074-15083.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda A, Lüftenegger D, Pietschmann T., and, Lindemann D. Characterization of the prototype foamy virus envelope glycoprotein receptor-binding domain. J Virol. 2006;80:8158–8167. doi: 10.1128/JVI.00460-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann-Che J, Renault N, Giron ML, Roingeard P, Clave E, Tobaly-Tapiero J.et al. (2007Centrosomal latency of incoming foamy viruses in resting cells PLoS Pathog 3e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruoslahti E. Fibronectin and its receptors. Annu Rev Biochem. 1988;57:375–413. doi: 10.1146/annurev.bi.57.070188.002111. [DOI] [PubMed] [Google Scholar]
- Mergia A, Leung NJ., and, Blackwell J. Cell tropism of the simian foamy virus type 1 (SFV-1) J Med Primatol. 1996;25:2–7. doi: 10.1111/j.1600-0684.1996.tb00185.x. [DOI] [PubMed] [Google Scholar]
- Stirnnagel K, Lüftenegger D, Stange A, Swiersy A, Müllers E, Reh J.et al. (2010Analysis of prototype foamy virus particle-host cell interaction with autofluorescent retroviral particles Retrovirology 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JR, Schuksz M., and, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- Häcker U, Nybakken K., and, Perrimon N. Heparan sulphate proteoglycans: the sweet side of development. Nat Rev Mol Cell Biol. 2005;6:530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick CA., and, Selleck SB. Heparan sulfate proteoglycans at a glance. J Cell Sci. 2007;120 Pt 11:1829–1832. doi: 10.1242/jcs.03432. [DOI] [PubMed] [Google Scholar]
- Summerford C., and, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 1998;72:1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh MT, WuDunn D, Montgomery RI, Esko JD., and, Spear PG. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J Cell Biol. 1992;116:1273–1281. doi: 10.1083/jcb.116.5.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WuDunn D., and, Spear PG. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J Virol. 1989;63:52–58. doi: 10.1128/jvi.63.1.52-58.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Maguire T, Hileman RE, Fromm JR, Esko JD, Linhardt RJ.et al. (1997Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate Nat Med 3866–871. [DOI] [PubMed] [Google Scholar]
- Esko JD, Stewart TE., and, Taylor WH. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc Natl Acad Sci USA. 1985;82:3197–3201. doi: 10.1073/pnas.82.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko JD, Rostand KS., and, Weinke JL. Tumor formation dependent on proteoglycan biosynthesis. Science. 1988;241:1092–1096. doi: 10.1126/science.3137658. [DOI] [PubMed] [Google Scholar]
- Lidholt K, Weinke JL, Kiser CS, Lugemwa FN, Bame KJ, Cheifetz S.et al. (1992A single mutation affects both N-acetylglucosaminyltransferase and glucuronosyltransferase activities in a Chinese hamster ovary cell mutant defective in heparan sulfate biosynthesis Proc Natl Acad Sci USA 892267–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langford JK, Stanley MJ, Cao D., and, Sanderson RD. Multiple heparan sulfate chains are required for optimal syndecan-1 function. J Biol Chem. 1998;273:29965–29971. doi: 10.1074/jbc.273.45.29965. [DOI] [PubMed] [Google Scholar]
- Richardson TP, Trinkaus-Randall V., and, Nugent MA. Regulation of heparan sulfate proteoglycan nuclear localization by fibronectin. J Cell Sci. 2001;114 Pt 9:1613–1623. doi: 10.1242/jcs.114.9.1613. [DOI] [PubMed] [Google Scholar]
- Esko JD., and, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001;108:169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamari FN., and, Karamanos NK. Structure of chondroitin sulfate. Adv Pharmacol. 2006;53:33–48. doi: 10.1016/S1054-3589(05)53003-5. [DOI] [PubMed] [Google Scholar]
- David G. Integral membrane heparan sulfate proteoglycans. FASEB J. 1993;7:1023–1030. doi: 10.1096/fasebj.7.11.8370471. [DOI] [PubMed] [Google Scholar]
- Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD.et al. (1999A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry Cell 9913–22. [DOI] [PubMed] [Google Scholar]
- Hu QY, Fink E, Happer M., and, Elder JH. Identification of amino acid residues important for heparan sulfate proteoglycan interaction within variable region 3 of the feline immunodeficiency virus surface glycoprotein. J Virol. 2011;85:7108–7117. doi: 10.1128/JVI.00573-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opie SR, Warrington KH, Jr, Agbandje-McKenna M, Zolotukhin S., and, Muzyczka N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J Virol. 2003;77:6995–7006. doi: 10.1128/JVI.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapraeger AC, Krufka A., and, Olwin BB. Requirement of heparan sulfate for bFGF-mediated fibroblast growth and myoblast differentiation. Science. 1991;252:1705–1708. doi: 10.1126/science.1646484. [DOI] [PubMed] [Google Scholar]
- Roghani M., and, Moscatelli D. Basic fibroblast growth factor is internalized through both receptor-mediated and heparan sulfate-mediated mechanisms. J Biol Chem. 1992;267:22156–22162. [PubMed] [Google Scholar]
- Kaner RJ, Baird A, Mansukhani A, Basilico C, Summers BD, Florkiewicz RZ.et al. (1990Fibroblast growth factor receptor is a portal of cellular entry for herpes simplex virus type 1 Science 2481410–1413. [DOI] [PubMed] [Google Scholar]
- Qing K, Mah C, Hansen J, Zhou S, Dwarki V., and, Srivastava A. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat Med. 1999;5:71–77. doi: 10.1038/4758. [DOI] [PubMed] [Google Scholar]
- Mirda DP, Navarro D, Paz P, Lee PL, Pereira L., and, Williams LT. The fibroblast growth factor receptor is not required for herpes simplex virus type 1 infection. J Virol. 1992;66:448–457. doi: 10.1128/jvi.66.1.448-457.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh MT., and, Spear PG. Fibroblast growth factor receptor: does it have a role in the binding of herpes simplex virus. Science. 1991;253:208–210. doi: 10.1126/science.1649495. [DOI] [PubMed] [Google Scholar]
- Epstein MA, Achong BG, Barr YM, Zajac B, Henle G., and, Henle W. Morphological and virological investigations on cultured Burkitt tumor lymphoblasts (strain Raji) J Natl Cancer Inst. 1966;37:547–559. [PubMed] [Google Scholar]
- Schneider, U, Schwenk H-U., and, Bornkamm G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int J Cancer. 1977;19:621–626. doi: 10.1002/ijc.2910190505. [DOI] [PubMed] [Google Scholar]
- Rasheed S, Nelson-Rees WA, Toth EM, Arnstein P., and, Gardner MB. Characterization of a newly derived human sarcoma cell line (HT-1080) Cancer. 1974;33:1027–1033. doi: 10.1002/1097-0142(197404)33:4<1027::aid-cncr2820330419>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Desmyter J, Rawls WE., and, Melnick JL. A human interferon that crosses the species line. Proc Natl Acad Sci USA. 1968;59:69–76. doi: 10.1073/pnas.59.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen FC, Girardi AJ, Gilden RV., and, Koprowski H. INFECTION OF HUMAN AND SIMIAN TISSUE CULTURES WITH ROUS SARCOMA VIRUS. Proc Natl Acad Sci USA. 1964;52:53–59. doi: 10.1073/pnas.52.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H.et al. (1973In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors J Natl Cancer Inst 511417–1423. [DOI] [PubMed] [Google Scholar]
- Jainchill JL, Aaronson SA., and, Todaro GJ. Murine sarcoma and leukemia viruses: assay using clonal lines of contact-inhibited mouse cells. J Virol. 1969;4:549–553. doi: 10.1128/jvi.4.5.549-553.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML., and, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löchelt M, Zentgraf H., and, Flügel RM. Construction of an infectious DNA clone of the full-length human spumaretrovirus genome and mutagenesis of the bel 1 gene. Virology. 1991;184:43–54. doi: 10.1016/0042-6822(91)90820-2. [DOI] [PubMed] [Google Scholar]
- Trobridge G, Josephson N, Vassilopoulos G, Mac J., and, Russell DW. Improved foamy virus vectors with minimal viral sequences. Mol Ther. 2002;6:321–328. doi: 10.1006/mthe.2002.0672. [DOI] [PubMed] [Google Scholar]
- Sharma S, Murai F, Miyanohara A., and, Friedmann T. Noninfectious virus-like particles produced by Moloney murine leukemia virus-based retrovirus packaging cells deficient in viral envelope become infectious in the presence of lipofection reagents. Proc Natl Acad Sci USA. 1997;94:10803–10808. doi: 10.1073/pnas.94.20.10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Pestina TI, Nasimuzzaman M, Mehta P, Hargrove PW., and, Persons DA. Amelioration of murine beta-thalassemia through drug selection of hematopoietic stem cells transduced with a lentiviral vector encoding both gamma-globin and the MGMT drug-resistance gene. Blood. 2009;113:5747–5756. doi: 10.1182/blood-2008-10-186684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhardt RJ.2001Analysis of glycosaminoglycans with polysaccharide lyases Current Protocols in Molecular BiologyAusubel, FM, Brent, R, Kingston, RE, Moore, DD, Seidman, JG and Struhl, K (eds). Greene Pub. Associates, J. Wiley, order fulfillment: Brooklyn, N.Y. Media; Pa., 1994. 17.13.17–17.13.32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic maps of syndecan-1 and its mutant, TDM-syndecan-1.