

Abstract
The role of the C8 gem-dimethyl group in the A-ring of bryostatin 1 has been examined through chemical synthesis and biological evaluation of a new analogue. Assays for biological function using U937, K562 and MV4-11 cells as well as the profiles for downregulation of PKC isozymes revealed that the presence of this group is not a critical determinant for the unique pattern of biological activity of bryostatin.
Keywords: Bryostatin, Phorbol ester, Protein kinase C, Cancer, Alzheimer’s disease
Bryostatin 1 is the most thoroughly studied member of a family of over 20 complex macrolide natural products originally isolated by Pettit and coworkers from the marine bryozoans Bugula neritina.1 It was later discovered that the bacterial endosymbiont Candidatus endobugula sertula was the true source of the bryostatins.2 The bryostatins are composed of three highly functionalized pyran rings embedded in a macrocycle and differ primarily in substitution at the C7 and C20 positions.3 Bryostatin 1 has found to display a remarkable wide range of biological activity. Due to its initially promising anticancer activity, bryostatin 1 has been used in numerous phase I and II clinical trials for cancer chemotherapy. Moreover, bryostatin 1 has been shown to reverse multidrug resistance4 as well as to synergize5 with other well known oncolytic drugs such as paclitaxel, vincristine and cisplatin. However, unlike most anticancer drugs, bryostatin 1 stimulates the immune system, a property that has recently been utilized to overcome HIV latency in lymphocytes.6 Additionally, bryostatin 1 has displayed promising neurologic effects. Bryostatin 1 has also been shown to enhance memory and learning in animal models7 and has shown activity against Alzheimer’s disease in transgenic mice. Moreover, it has recently been reported that bryostatin 1 is able to stimulate repair of the neural damage and reestablishment of synapses for up to 24 hours after stroke in rats.9
The mechanism by which bryostatin 1 evokes these diverse biological effects is believed to arise largely from the modulation of protein kinase C isozymes (PKCs) and other C1 domain containing proteins upon their binding of bryostatin 1.10 Due to their important role in signaling, PKCs have emerged as an attractive target in drug discovery.11 Several natural products such as phorbol esters, bryostatins, indolactams and aplysiatoxin are known high affinity exogenous ligands for PKCs. An important distinction among various PKC ligands is that, although they bind to the same C1 domain of PKCs, the biological responses subsequent to bindings can be very different. For example phorbol esters such as phorbol 12-myristate-13-acetate (PMA) and bryostatin 1 bind to the same C1 domain of PKC with high affinity; however, PMA is tumor promoting whereas bryostatin 1 is not.12 In addition, bryostatin 1 antagonizes many of the PMA induced response which it does not induce itself.
Our group is endeavoring to understand the structural features of bryostatin 1 responsible for its unique biological activity. To this end, we have synthesized and analyzed various bryopyran structures13 using the pyran annulations methodology14 developed in our laboratories that has proven critical in recently reported total syntheses of bryostatins.15 Representative examples of our bryopyran analogues are shown in Figure 1. Examination of analogue Merle 23 revealed that substitution on A- and B-rings is critical for obtaining antagonism of phorbol ester induced biological responses. The roles of individual substituents in this region including the C30 carbomethoxy group (Merle 28), C9 hydroxy group (Merle 30) and C7 acetate group (Merle 27) have been addressed, revealing that none of these substituents alone acts as a functional switch for PMA versus bryostatin like activity. Herein we examine the role of the final such substituent, the C8 gem-dimethyl group, through chemical synthesis and biological evaluation of the new bryopyran analogue Merle 32.16
Figure 1.
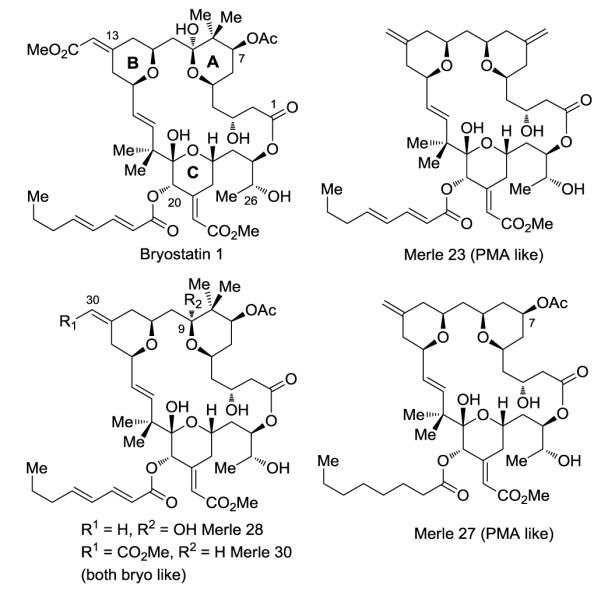
Structure of bryostatin 1 and bryopyrans
The synthesis of Merle 32 commenced from intermediate 1 which had been prepared previously enroute to analogues Merle 28 and Merle 30 (Scheme 1). The C25 alcohol was first freed by removal of the PMB group using DDQ. Subjection of the resulting hydroxy-thioester to oxidative hydrolysis using m-CPBA in aqueous THF selectively cleaved the thioester in the presence of the C7 ester and provided the corresponding seco acid. Although both the C16-C17 olefin and C20 ketone proved unreactive towards m-CPBA, it was necessary to stop the reaction after three and half hours in order to prevent the epoxidation of the C13-C30 olefin. Yamaguchi macrolactonization of the seco acid then furnished the macrolactone 2 in excellent yield. To preclude competing enolization of the C7 acetate in the ensuing aldol reaction,13c,13d the C7 acetate was removed using K2CO3/MeOH (without interference from the macrolactone ester functionality) and the resulting C7 alcohol was protected as the TES ether. The aldol reaction of the C-ring ketone 3 with freshly distilled methyl glyoxylate using LDA then provided the corresponding aldol adduct as a mixture of diastereomers which were subjected to elimination by stirring with acetic anhydride. It proved necessary to keep the reaction at room temperature in order to avoid deprotection of the C7 TES group. This reaction provided the desired α,β- unsaturated ester 4 as a single isomer.
Scheme 1.
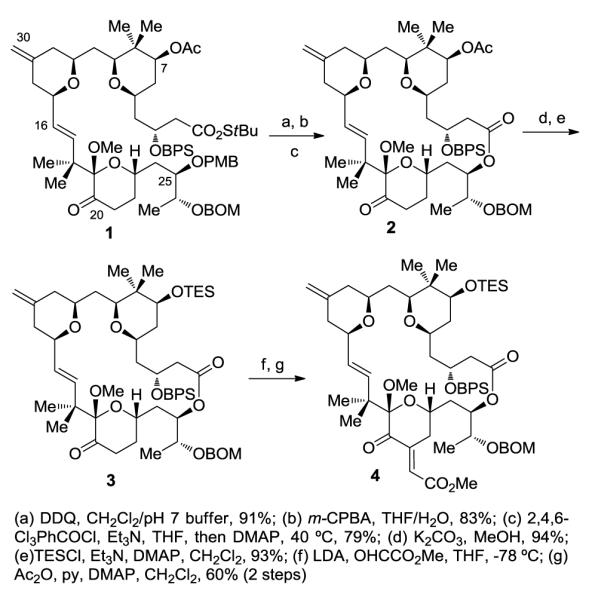
Preparation of macrolactone and further functionalization
Luche reduction of the C20 ketone provided the alcohol which was found to be unstable for isolation and purification purposes (Scheme 2). Thus, this intermediate was immediately subjected to esterification by reaction of the crude alcohol with 2,4-octadienoic anhydride. The C7 alcohol was then revealed by removal of the TES group by reaction with PPTS in MeOH.
Scheme 2.

Completion of the C8 gem-dimethyl analogue Merle 32
Oxidation of the alcohol to the corresponding ketone proved uneventful using the Dess-Martin oxidation.18 However, subsequent conversion of the ketone 6 to the desired alkene proved to be a daunting task. Attempted olefination of the ketone using Petasis reaction conditions provided a complex mixture of products.19 On the other hand, we were initially reluctant to use the Wittig reaction20 on such a complex substrate due to the following concerns: (i) low reactivity of the sterically hindered C7 ketone towards the Wittig reagent, leading to enolization, (ii) potential β-elimination of the C3-OBPS group, and (iii) migration of the C-ring olefin to an internal position. The latter two processes had been observed in our laboratory during the synthesis of related structures. However, when the ketone 7 was subjected to Wittig reaction conditions, we were pleasantly surprised to find that the reaction provided the desired product in excellent yield in just fifteen minutes. Removal of the BPS group followed by global deprotection using LiBF4 then completed the synthesis of Merle 32.21
The biological evaluation of Merle 32 began by determining its binding affinity (Ki) towards PKC in vitro.22 Its Ki of 1.08 ± 0.16 nM proved similar to that of bryostatin 1 (Ki = 1.35 nM) and to other bryopyran analogues prepared previously. The biological profile of Merle 32 was initially addressed in the U937 human lymphoma cell line using proliferation and attachment assays (both measures of differentiation in this cell line). The U937 cells display differential response towards tumor-promoting PMA and bryostatin 1.23 Specifically, PMA induces attachment and inhibits the proliferation of U937 cells whereas bryostatin 1 has little effect. On the other hand, bryostatin 1 blocks both responses to PMA in a dose dependent manner when the two agents are administered together. It can be seen (Figure 2) that Merle 32 induced the attachment of U937 cells in a manner largely similar to PMA, although the maximal attachment was not quite as great. Similar observations were made in the proliferation assay, but with even closer resemblance to the PMA response.
Figure 2.

Effect of Merle 32 on proliferation and attachment of U937 cells. Cells were treated 24 hrs after seeding with the indicated concentrations of the different compounds. The floating and attached cells were counted 60 hr after treatment as described earlier (13a). Values represent the mean ± SEM of five independent experiments.
Merle 32 was also evaluated using the human leukemia cell lines K56224 and MV4-11,25 which both show distinctive dose dependent patterns for inhibition of proliferation in the presence of PMA or bryostatin 1. As can be seen in Figure 3, Merle 32 was strongly antiproliferative, as was PMA but not bryostatin 1. Moreover Merle 32 did not block the response to PMA, while bryostatin 1 did. These results indicate that the behavior shown by Merle 32 is not limited to just the U937 cell line and indeed was even more PMA-like in these other two cell lines. Here Merle 32 showed no ability to block the effect of 10nM PMA, while bryostatin 1 was effective in that regard.
Figure 3.

Effect of Merle 32 on proliferation of K562 and MV4-11 cells. Cells were seeded and treated as described for the U937 cells (13a). Total cell numbers were determined 60 hr after treatment. Values represent the mean ± SEM of three independent experiments.
Downregulation of PKC isoforms and other C1 domain containing proteins subsequent to ligand binding is another characteristic phenomenon displayed by PKCs and is also important in determining their functional response to different ligands.26 Dose dependent patterns of downregulation were determined for PMA, bryostatin 1, and Merle 32 in the K562 cells (Figure 4). Here bryostatin 1 and PMA showed distinctly different patterns. Bryostatin 1 was more effective in downregulating PKC-α and β and showed biphasic downregulation of PKC-δ (Figure 4). Moreover, it did not cause the prominent induction of PKC-ε and RasGRP3 observed in these cells with PMA. Merle 32 showed a pattern largely similar to that of PMA but with approximately 10-fold weaker potency.
Figure 4.
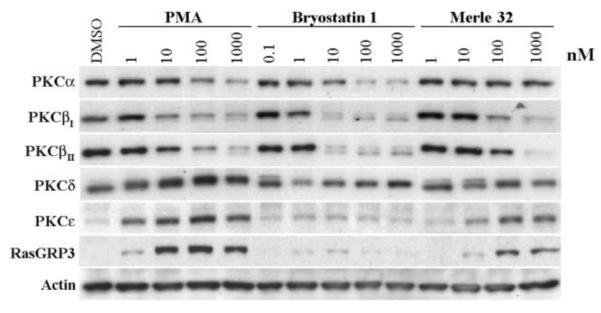
Modulation of C1 domain containing proteins upon 24 hr treatment of K562 cells. K562 cells (4 ml of 150,000 cells/ml) were treated 24 hrs after plating with the indicated concentrations of the compounds for 24 hrs. Western blot analysis was performed on the total cell lysates as described earlier.27 Actin provides a loading control. A representative image from three independently performed experiment is shown.
The results from the proliferation assays in the various cell lines together with these downregulation assays indicate that Merle 32 induces biological responses largely similar to those of PMA rather than bryostatin 1. If the C8 gem-dimethyl group was to serve as a functional switch, the biological results displayed by Merle 32 in these systems would be closer to those of bryostatin 1 than to those of Merle 23, which differs from Merle 32 only at C8, whereas they very closely resemble those of Merle 23.
To date, we have studied the role of four individual groups in the A-B region of bryostatin 1 that have been implicated as responsible for its unique biological activity. None of these groups was found to function singly as a biological switch, thus it appears that the unique activity of bryostatin 1 relies on a more subtle interplay between some combinations of these groups. It is interesting to note that those analogues with two polar substituents in the A-B ring region (Merle 28 and 30) give biological results similar to those for bryostatin 1, while those with fewer or no polar groups (Merle 32, 27 and 23) are PMA-like or largely so. Thus, it appears that a proper combination of polar substituents in the A-B ring region may be needed for mimicking the activity of bryostatin 1. Efforts to identify suitable combinations of groups in this region are in progress and will be reported in due course.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH through grant GM28961 and through the Intramural Research Program, CCR, NCI, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material Supplementary data associated with this article can be found, in the online version, at
References and notes
- 1.Pettit GR, Herald CL, Douobek DL, Herald DL. J. Am. Chem. Soc. 1982;104:6846. [Google Scholar]
- 2.Lopanik N, Gustafson KR, Lindquist N. J. Nat. Prod. 2004;67:1412. doi: 10.1021/np040007k. [DOI] [PubMed] [Google Scholar]
- 3.For a recent review of the bryostatins, see: Hale KJ, Manaviazar S. Chem. Asian J. 2010;5:704. doi: 10.1002/asia.200900634.
- 4.Spitaler M, Utz I, Hilbe W, Hofmann J, Grunicke HH. Biochem. Pharmacol. 1998;56:861. doi: 10.1016/s0006-2952(98)00107-5. [DOI] [PubMed] [Google Scholar]
- 5 a).Koutcher JA, Motwani M, Zakian KL, Li X–K, Matei C, Dyke JP, Ballon D, Yoo H–H, Schwartz GK. Clin. Cancer Res. 2000;6:1498. [PubMed] [Google Scholar]; (b) Mohammad RM, Wall NR, Dutcher Julie A., Al-Katib AM. Clin. Cancer Res. 2000;6:4950. [PubMed] [Google Scholar]; (c) Basu A, Lazo JS. Cancer Res. 1992;52:3119. [PubMed] [Google Scholar]
- 6.Mehla R, Bivalkar-Mehla S, Zhang R, Handy I, Albrecht H, Giri S, Nagarkatti P, Nagarkatti M, Chauhan A. PLoS One. 2010;5:e11160. doi: 10.1371/journal.pone.0011160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7 (a).Alkon DL, Epstein H, Kuzirian A, Bennett MC, Nelson T. J Proc. Natl. Acad. Sci., U.S.A. 2005;102:16432. doi: 10.1073/pnas.0508001102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sun M-K, Alkon DL. Eur. J. Pharmacol. 2005;512:43. doi: 10.1016/j.ejphar.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 8.Etcheberrigaray R, Tan M, Dewachter I, Kuiperi C, van der Auwera I, Wera S, Qiao L, Bank B, Nelson TJ, Kozikowski AP, van Leuven F, Alkon DL. Proc. Natl. Acad. Sci. U. S. A. 2004;101:11141. doi: 10.1073/pnas.0403921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun M-K, Hongpaisan J, Nelson TJ, Alkon DL. Proc. Natl. Acad. Sci. U. S. A. 2008;105:13620. doi: 10.1073/pnas.0805952105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reyland ME, Insel PA, Messing RO, Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP. Am. J. Physiol. Lung Cell Mol. Physiol. 2000;279:429. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 11.Kinase C Protein, Parker PJ, Dekker LV., editors. Molecular Biology Intellegence Unit. R. G. Landes; Austin: 1997. [Google Scholar]
- 12.Blumberg PM, Pettit GR. In: New Leads and Targets in Drug Research. Krosgaard–Larsen P, Christensen CB, Kodof H, editors. Munksgaard; Copenhagen: 1992. p. 273. [Google Scholar]
- 13 (a).Keck GE, Kraft MB, Truong AP, Li W, Sanchez CC, Kedei N, Lewin N, Blumberg PM. J. Am. Chem. Soc. 2008;130:6660. doi: 10.1021/ja8022169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Keck GE, Li W, Kraft MB, Kedei N, Lewin NE, Blumberg PM. Org. Lett. 2009;11:2277. doi: 10.1021/ol900585t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Keck GE, Poudel YB, Welch DS, Kraft MB, Truong AP, Stephens JC, Kedei N, Lewin NE, Blumberg PM. Org. Lett. 2009;11:593. doi: 10.1021/ol8027253. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Keck GE, Poudel YB, Rudra A, Stephens JC, Kedei N, Lewin NE, Peach ML, Blumberg PM. Angew. Chem. Int. Ed. 2010;49:4580. doi: 10.1002/anie.201001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14 (a).Keck GE, Covel JA, Schiff T, Yu T. Org. Lett. 2002;4:1189. doi: 10.1021/ol025645d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Keck GE, Truong AP. Org. Lett. 2005;7:2149. doi: 10.1021/ol050511w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Keck GE, Truong AP. Org. Lett. 2005;7:2153. doi: 10.1021/ol050512o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keck GE, Poudel YB, Cummins TJ, Rudra A, Covel JA. J. Am. Chem. Soc. 2011;133:744. doi: 10.1021/ja110198y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wender PA, Schrier AJ. J. Am. Chem. Soc. 2011;133:9228. doi: 10.1021/ja203034k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lu Y, Woo SK, Krische MJ. J. Am. Chem. Soc. 2011;133:13876. doi: 10.1021/ja205673e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For gem-dimethyl containing analogues with a B-ring acetal moiety, see: Wender PA, Baryza JL, Brenner SE, DeChristopher BA, Loy BA, Schrier AJ, Verma VA. Proc .Natl. Acad. Sci. USA. 2011;108:6721. doi: 10.1073/pnas.1015270108.
- 17.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989. [Google Scholar]
- 18.Dess DB, Martin JC. J. Org. Chem. 1983;48:4155. [Google Scholar]
- 19.Petasis N,A, Bzowej E,I. J. Am. Chem. Soc. 1990;112:6392. [Google Scholar]
- 20.Wittig G, Schollkof U. Chem. Ber. 1954;87:1318. [Google Scholar]
- 21.Lipschutz BH, Harvey DF. Synth. Commun. 1982;14:267. [Google Scholar]
- 22.Lewin NE, Blumberg PM. Methods Mol. Biol. 2003;233:129. doi: 10.1385/1-59259-397-6:129. [DOI] [PubMed] [Google Scholar]
- 23.Vrana JA, Saunders AM, Srikumar PC, Grant S. Differentiation. 1998;63:33. doi: 10.1046/j.1432-0436.1998.6310033.x. [DOI] [PubMed] [Google Scholar]
- 24.Hocevar BA, Morrow DM, Tykocinski ML, Fields AP. J. Cell Sci. 1992;101:671. doi: 10.1242/jcs.101.3.671. [DOI] [PubMed] [Google Scholar]
- 25.Lange B, Valtieri M, Santoli D, Caracciolo D, Mavilio F, Gemperlein I, Griffin C, Emanuel B, Finan J, Nowell P. Blood. 1987;70:192. [PubMed] [Google Scholar]
- 26.Leontieva OV, Black JD. J. Biol. Chem. 2004;279:5788. doi: 10.1074/jbc.M308375200. [DOI] [PubMed] [Google Scholar]
- 27.Kedei N, Telek A, Czap A, Lubart ES, Czifra G, Yang D, Chen J, Morrison T, Goldsmith PK, Lim L, Mannan P, Garfield SH, Kraft MB, Li W, Keck GE, Blumberg PM. Biochem. Pharmacol. 2011;81:1296. doi: 10.1016/j.bcp.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.