

Abstract
Liver ischemia/reperfusion (IR) injury is caused by a heavily toothed network of interactions of cells of the immune system, cytokine production, and reduced microcirculatory blood flow in the liver. These complex networks are further elaborated by multiple intracellular pathways activated by cytokines, chemokines, and danger-associated molecular patterns. Furthermore, intracellular ionic disturbances and especially mitochondrial disorders play an important role leading to apoptosis and necrosis of hepatocytes in IR injury. Overall, enhanced production of reactive oxygen species, found very early in IR injury, plays an important role in liver tissue damage at several points within these complex networks. Many contributors to IR injury are only incompletely understood so far. This paper tempts to give an overview of the different mechanisms involved in the formation of IR injury. Only by further elucidation of these complex mechanisms IR injury can be understood and possible therapeutic strategies can be improved or be developed.
1. Introduction
Ischemia/reperfusion (IR) injury of the liver results from a loss of blood supply reducing oxygen supply to the organ. Upon revascularisation the liver undergoes reperfusion injury. Together these factors lead to affection of oxygen-dependent cells within the liver causing impairment of organ function. Affected are all cells requiring mitochondrial oxidative phosphorylation for their metabolism [1]. Warm IR injury can be separated from cold IR injury. Warm IR injury occurs during prolonged surgical liver resection using clamping of the perfusion [2]. Other aetiologies are reduced liver perfusion due to shock, heart failure, respiratory failure, hemorrhage, trauma, and sepsis [3–5]. In contrast, cold IR injury follows liver transplantation with the necessity of cold preservation of the donor organ, followed by reperfusion after implantation [6, 7]. Furthermore, it has been demonstrated that tissue damage occurs in two phases, an early and a late phase [8, 9]. The early phase which occurs within the first 6 hours following reperfusion is thought to be the consequence of the fast change in the redox state of the liver tissue [9, 10]. Most likely, this change is caused by hepatocytes, Kupffer cells (KCs), and sinusoidal endothelial cells (SECs) [8, 9, 11]. In contrast, the late phase of IR injury is caused by the production of cytokines and chemokines followed by the infiltration of leukocytes into the liver tissue [8, 9, 12].
Of clinical relevance is that liver IR injury results in elevated liver enzymes, biliary strictures, clinical dysfunction, or even liver failure [13]. Furthermore, other organs can develop dysfunction secondary to the liver damage. Possible affected organs are lungs, heart, kidneys, and blood vessels [14–17]. Risk factors for IR injury include age of the liver, sex and others [18–21].
A complex network and cross talk of multiple molecular mechanisms and cellular interactions lead to liver IR injury [22, 23]. The result of these processes is cell death by apoptosis and necrosis via different pathways. Redox status, cellular ionic disturbances, cytokines, chemokines, other mediators and molecular mechanisms as well as many different cells like KC, SEC, dendritic cells, leukocytes, and lymphocytes are involved in this process and are closely interlocked. Therefore, there are still many open questions regarding this inflammatory response. This paper tempts to give a systematic overview of the different components and signalling pathways leading to IR injury.
2. Altered Redox Status and Reduced Microcirculatory Blood Flow
IR injury starts with reduced blood flow and a lack of oxygen supply [24, 25]. This ischemia leads to a lack of adenosine triphosphate (ATP) production in hepatocytes, KC, and SEC [26]. As a result the function of the ATP-dependent sodium/potassium plasma membrane pump (Na+/K+ ATPase) is impaired. This results in an increase of intracellular Na+, which is followed by a swelling of the hepatocytes, KC and SEC. Narrowing of the sinusoidals is the consequence. Within minutes after reperfusion, enhanced levels of reactive oxygen species (ROS), such as superoxide (O2 −), hydrogen peroxide (H2O2) and hydroxyl radical (OH•), can be measured [8, 9, 11, 27]. Cellular sources for these ROS are mitochondrial metabolism, hepatocyte-derived xanthine oxidase, and KC- and SEC-associated NADPH oxidase [8, 9, 11, 28]. This increase of ROS is accompanied by reduced nitric oxide (NO), most likely due to a decreased function of the NO synthase (NOS) in SEC [29, 30]. Since NO is a vasodilator [31], reduced NO aggravates the sinusoidal narrowing. However, these mechanisms are still controversially discussed [32, 33]. Especially the role of the different NOS isoforms is still unclear [34]. The effects of NO are well known. It increases the sinusoidal diameter and increases intrahepatic ATP levels via better oxygen supply. Thereby mitochondrial damage and leukocyte infiltration are reduced. Since NO is synthesized by NOS, NOS should be protective in IR injury. While this is well observed for the endothelial NOS (eNOS), the role of the inducible NOS (iNOS) is less clear [34]. In the late phase of IR injury it probably is also protective, while in the early phase, there may be a harmful role of iNOS.
Nevertheless, the changes occurring in IR injury lead to a more oxidative environment with the ROS leading to both, apoptotic and necrotic cell death of hepatocytes and SEC [35, 36]. ROS causes damage to membrane lipids resulting in cell swelling and death [35]. The damage is not limited to the plasma membrane but includes cell organelles and extracellular matrix. Besides swelling of cells and reduced NO levels, the described damage leads to an increase of vasoconstrictors like endothelin and thromboxane A2 [37]. In addition, adhesion and aggregation of platelets and leucocytes is increased (see below). This leads to further narrowing of the sinusoidals with significant reduction of microcirculatory blood flow including areas with complete absence of blood flow [38], enhancing the lack of oxygen supply. Increased ROS and decreased NO levels play further roles which will be discussed below.
3. Ionic and Mitochondrial Disturbances
In IR injury significant changes of intracellular Ca2+ concentration in the hepatocytes can be found [39]. Ca2+ is mainly found in three cellular compartments, in the cytosol, the mitochondria, and the endoplasmic reticulum (ER). The homeostatic concentration is regulated by different Ca2+ channels. In IR injury cytosolic Ca2+ concentration is increased as a result of increased entry across the plasma membrane and release from the ER. Reason for this cytoplasmic Ca2+ overload is the activation of the ryanodine receptor in the ER membrane and the so-called transient receptor potential (TRP) channels in the plasma membrane. There is evidence that ROS can activate these channels [40, 41]. Secondary to decreased Ca2+ concentration in the endoplasmic reticulum so-called store operated calcium (SOC) channels in the plasma membrane further increase Ca2+ influx [42, 43]. In addition, in IR injury the Ca2+ ATPase in ER and plasma membrane is inhibited, potentially because of ATP depletion. Normally this Ca2+ ATPase discharges cytosolic Ca2+ into extracellular space and into the ER counteracting the above mentioned Ca2+ channels [44].
Increased cytosolic Ca2+ leads to stimulation of the Ca2+ uniporter in the mitochondrial membrane [45]. As a result the mitochondrial Ca2+ concentration increases as well. The mechanism how the mitochondrial Ca2+ uniporter is activated has not been fully resolved, so far. It is believed that the mitochondrial P2Y-like receptor 1 (mP2Y1) is activated by adenosine diphosphate (ADP) and adenosine monophosphate (AMP). The mP2Y1 stimulates the PLC-dependent mP2Y-like receptor resulting in activation of the Ca2+ uniporter [46, 47]. In contrast, mP2Y2 activated by ATP leads to inhibition of the uniporter. During IR injury ATP is depleted, as discussed before, possibly leading to activation of the uniporter. As a consequence of this increased mitochondrial Ca2+ concentration the mitochondrial transmembrane potential is reduced. To maintain the mitochondrial membrane the mitochondrial ATP-synthase reverses its activity and hydrolyzes ATP to provide energy for different ionic pumps in the mitochondrial membrane [39]. However, this further increases Ca2+ influx resulting in ATP consumption instead of production in the mitochondria. This is enhanced by the fact that ROS causes oxidative damage to the enzymes of the respiratory chain in the mitochondria leading to failure of ATP production [48]. Cytosolic and mitochondrial Ca2+ and other ionic disturbance lead to damage of plasma and mitochondrial membranes including the formation and opening of mitochondrial permeability transition (MPT) pores [49]. MPT pores are formed from integral not fully identified mitochondrial membrane proteins [50, 51]. Hepatic mitochondria afflicted by MPT pores are permanently damaged due to depolarization of the mitochondria [37]. When only a few mitochondria are afflicted, they are removed from the hepatocyte by lysosomal mitophagy [52]. Such damaged mitochondria are a source for further ROS production and ATP consumption [53]. However, ROS themselves induce MPT pore opening. With the number of damaged mitochondria increasing, cytochrome C is released from the mitochondria into the cytosol triggering apoptosis [48]. When the majority of the mitochondria within the hepatocyte are afflicted by MPT pores ATP levels drop too fast resulting in cell death by necrosis [49, 54].
Other important ionic disturbances in IR injury include intracellular Na+ and hydrogen (H+) concentrations. Lack of oxygen supply leads to anaerobic respiration of the hepatocytes resulting in intracellular acidosis [55]. To stabilize intracellular pH within normal range the Na+/H+ exchanger is activated by the hepatocytes, resulting in reduced cytosolic H+ and further increased Na+ levels. In addition, the Na+/K+ exchanger is ATP dependent, so ATP depletion, as in IR injury, subsequently blocks this exchange leading to further increase of intracellular Na+ concentration resulting in cell death [56].
Furthermore, this counteracts the protective effect of an acidic pH during reperfusion [57], for example, the maintenance of an acidic pH prevents the formation of MPT pores [49]. However, these regulations are still based on experimental observations and need to be studied further to understand the relevance in IR injury (Figure 1).
Figure 1.
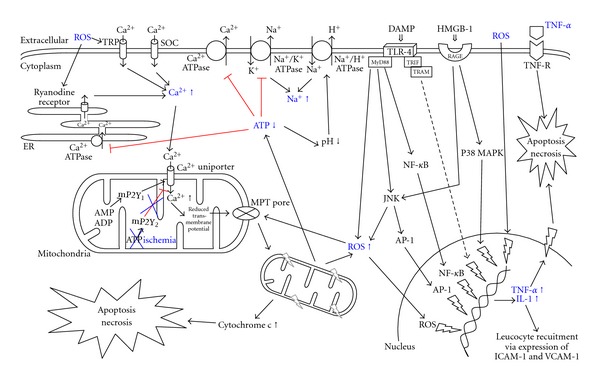
Intracellular signalling pathways and ionic disturbances engaged during IR injury, resulting in cellular swelling, apoptosis, and necrosis. ADP: adenosine diphosphate; AMP: adenosine monophosphate; AP-1: activator protein-1; ATP: adenosine triphosphate; DAMP: danger-associated molecular pattern; HMGB-1: high mobility group box-1; ICAM-1: intercellular adhesion molecule-1; IL-1: interleukin-1; JNK: c-Jun N-terminal kinase; MAPK: mitogen-activated protein kinase; MPT pore: mitochondrial permeability transition pore; MyD88: myeloid differentiation factor 88; NF-κB: nuclear factor kappa B; RAGE: receptor for advanced gylcation end product; ROS: reactive oxygen species; SOC: store operated calcium channel; TLR: toll-like receptor; TNF: tumor necrosis factor; TRAM: TRIF-related adaptor molecule; TRIF: TIR domain-containing adaptor inducing interferon; TRP: transient receptor protein; VCAM-1: vascular adhesion molecule-1.
4. Cellular Cascade in IR Injury
Many different cell types are involved in the process of hepatic damage and cell death in IR injury. The key cells initiating IR injury are the KCs [58–60]. Besides their direct damage by ROS, as discussed above, they are also activated by ROS leading to production of more ROS and thereby entering a cycle of self-activation and -destruction. In addition, KC are activated by the systemic complement system [61] which may also be liberated by damaged hepatocytes. In addition, complement leads to further liver damage by formation of a membrane attack complex in the plasma membrane, lysing liver cells [62].
Activated KCs also produce proinflammatory cytokines including interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) [63]. These cytokines lead to activation and migration of neutrophils and CD4+ T lymphocytes into the liver [64]. Furthermore, these cytokines stimulate SEC and hepatocytes to produce ROS and to express adhesion molecules on the cell surface [65]. As described above, this leads to adhesion and aggregation of leucocytes and platelets [66], influencing the microcirculatory blood flow in the liver.
The recruitment of neutrophils and CD4+ T lymphocytes is further enhanced by the matrix metalloproteinase 9 [67] after ischemic damage of the liver. Via production of interferon-γ (IFN-γ) and IL-17 by activated CD4+ T lymphocytes additional activation of KC and hepatocytes is achieved [68, 69]. Thus CD4+ T lymphocytes and KC reciprocally activate each other [64]. These chemokines furthermore activate natural killer T (NKT) cells. Activated NKT cells directly damage liver tissue and also produce IFN-γ with further activation of KC and hepatocytes [69, 70]. The net result of this circular activation and stimulation of different cell sub types is destruction of hepatocytes and SEC [71, 72].
The expressed cell-surface adhesion molecules on hepatocytes and SEC include intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1) [28, 73]. Neutrophils bind to ICAM-1 and VCAM-1 and by doing so migrate across the endothelium into the liver parenchyma enhancing ROS production and degranulation of cytoplasmic vesicles containing enzymes capable to degrade extracellular matrix and hepatocytes [28] (Figure 2).
Figure 2.
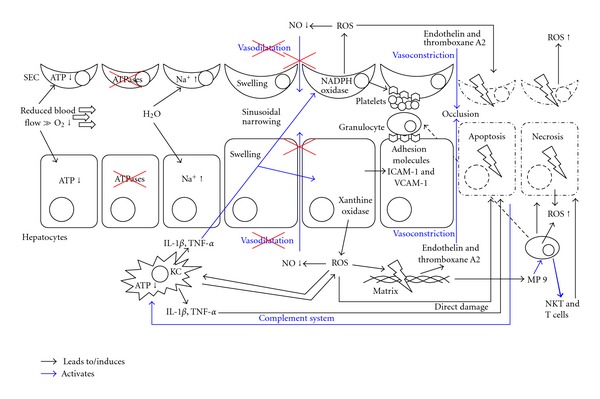
Cellular interaction involved in IR injury, resulting in cellular swelling, apoptosis, and necrosis. ATP: adenosine triphosphate; ICAM-1: intercellular adhesion molecule-1; KC: Kupffer cell; IL-1, interleukin-1; NKT: natural killer T cell; NO: nitric oxide; ROS: reactive oxygen species; SEC: sinusoidal endothelial cells; T cell: CD4+ T lymphocyte; TNF: tumor necrosis factor; VCAM-1: vascular adhesion molecule-1.
5. Death Signalling Pathways
Besides direct damage of hepatocytes by neutrophils, NKT cells, the complement system and ROS, the main destruction of cells is mediated by endogeneous pathways leading to apoptosis or necrosis of hepatic cells during IR injury.
This paper is not capable to focus on all cytokine cascades with pro- and antiinflammatory effects [74] as well as their effect during IR injury, but will concentrate on some important signalling pathways. The most important component in IR injury seems to be TNF-α [75, 76]. The pathways leading to up regulation of TNF-α have been described above. TNF-α binds to specific TNF-receptors, as for example TNF-R1 and TNF-R2, on the hepatocyte surface which leads to increased production of cytokines and ROS. In addition, activation of CD95 leads to apoptosis [77–79]. Furthermore, CD95 also binds NKT cells leading to direct destruction of hepatocytes [80].
Furthermore, downstream of the receptor the nuclear factor kappa B (NF-κB), the mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) are activated [81–83]. The various cytokines and the mentioned molecules lead to alteration of various factors further downstream like transcription factors, activator protein-1 (AP-1), heat shock factor, signal transducer and activators of transcription (STATs), antioxidants, inflammation-stimulated inducible enzymes (COX-2), intracellular signalling molecules, antiapoptotic proteins (Bcl-2, Bcl-x), and many more [37]. The damage in IR injury therefore spreads throughout the entire cell. NF-κB furthermore upregulates the expression of cytokines, like TNF-α [84], and of ICAM-1 and VCAM-1 [75, 82], enhancing the recruitment of neutrophils. AP-1 promotes apoptosis of liver cells by activation of caspase-3 and release of cytochrome C [82].
ROS furthermore inherit a direct oxidative damage of DNA within the nucleus resulting in further failure of protein transcription and translation. In addition, ROS cause posttranslational protein modification [85]. These alterations and pathways lead to apoptosis of the damaged cells.
The intracellular damage and alterations as well as the damage of extracellular matrix are followed by the release of danger-associated molecular patterns (DAMPs). Examples of DAMPs released during IR injury are the nuclear transcription factor high mobility group box-1 (HMGB-1), the cytoplasmic Ca2+ regulator S100, ATP, DNA, and hyaluronic acid [86–90]. DAMPs bind to a group of so-called pattern recognition receptors (PRRs) on the cell surface as well as in the cytoplasm [37]. In IR injury mainly two PRRs are involved, the toll-like receptors (TLRs), specifically TLR-4, and the receptor for advanced glycation end products (RAGE). To present knowledge TLR-4 provides an important link between liver damage and activation of the immune system. Activation of TLR-4 triggers intracellular signalling cascades in IR injury [91]. The Toll-IL-1 receptor domain (TIR) of TLR-4 interacts with intracellular adaptors. One may be the myeloid differentiation factor 88 (MyD88), others are TIR domain-containing adaptor inducing interferon-β (TRIF), and TRIF-related adaptor molecule (TRAM) [92]. Via production of proinflammatory cytokines the inflammatory response is mediated leading to liver IR injury [91–93]. Included in the downstream process of these activation are further transcription factors like NF-κB, AP-1, STAT, the MAP kinase JNK, and ROS [86, 94–96].
The best characterized DAMP is HMGB-1 which is expressed by all nucleated cells within the liver and is released upon necrosis and apoptosis [86, 97]. HMGB-1 binding to RAGE in IR injury leads to a signalling cascade involving activation of JNK and other kinases, increasing expression and activation of the inducible transcription factor early growth response-1. As a consequence the upregulation of several gene families is found, recruiting immune cells into the post ischemic liver [98]. RAGE is mainly expressed on dendritic cells and to lesser extent on KC [98]. Furthermore, dendritic cells and KC also express TLR-4 [99]. This hints to an important, but at present unclear, function of dendritic cells during IR injury of the liver.
This complex communication of the described networks is responsible to initiate and propagate IR injury.
6. Conclusions
The understanding of the molecular mechanisms underlying cell death in hepatic IR injury will provide the basis for the development of new strategies for inhibition of liver injury and improvement of survival of the graft. The initial phase of IR injury involves the release of ROS and proinflammatory mediators by KC. ROS lead to oxidative damage, induction of p53, apoptosis and necrosis of hepatocytes and endothelial cells. The late phase (6–48 hours after reperfusion) is characterized by neutrophil-mediated inflammatory responses. Thus, proteins regulating the cellular redox equilibrium, p53-dependent apoptosis and cellular death receptors represent potential targets for novel pharmaceutical interventions to protect hepatocytes from IR injury-induced cell death.
Author's Contribution
Peter Schemmer and Martina Müller equally contributed to this work.
References
- 1.de Groot H, Rauen U. Ischemia-reperfusion injury: processes in pathogenetic networks: a review. Transplantation Proceedings. 2007;39(2):481–484. doi: 10.1016/j.transproceed.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Kim YI. Ischemia-reperfusion injury of the human liver during hepatic resection. Journal of Hepato-Biliary-Pancreatic Surgery. 2003;10(3):195–199. doi: 10.1007/s00534-002-0730-x. [DOI] [PubMed] [Google Scholar]
- 3.Rushing GD, Britt LD. Reperfusion injury after hemorrhage: a collective review. Annals of Surgery. 2008;247(6):929–937. doi: 10.1097/SLA.0b013e31816757f7. [DOI] [PubMed] [Google Scholar]
- 4.Birrer R, Takuda Y, Takara T. Hypoxic hepatopathy: pathophysiology and prognosis. Internal Medicine. 2007;46(14):1063–1070. doi: 10.2169/internalmedicine.46.0059. [DOI] [PubMed] [Google Scholar]
- 5.Nickkholgh A, Barro-Bejarano M, Liang R, et al. Signs of reperfusion injury following CO2 pneumoperitoneum: an in vivo microscopy study. Surgical Endoscopy and Other Interventional Techniques. 2008;22(1):122–128. doi: 10.1007/s00464-007-9386-6. [DOI] [PubMed] [Google Scholar]
- 6.Kupiec-Weglinski JW, Busuttil RW. Ischemia and reperfusion injury in liver transplantation. Transplantation Proceedings. 2005;37(4):1653–1656. doi: 10.1016/j.transproceed.2005.03.134. [DOI] [PubMed] [Google Scholar]
- 7.Liang R, Bruns H, Kincius M, et al. Danshen protects liver grafts from ischemia/reperfusion injury in experimental liver transplantation in rats. Transplant International. 2009;22(11):1100–1109. doi: 10.1111/j.1432-2277.2009.00925.x. [DOI] [PubMed] [Google Scholar]
- 8.Fan C, Zwacka RM, Engelhardt JF. Therapeutic approaches for ischemia/reperfusion injury in the liver. Journal of Molecular Medicine. 1999;77(8):577–592. doi: 10.1007/s001099900029. [DOI] [PubMed] [Google Scholar]
- 9.Zwacka RM, Zhou W, Zhang Y, et al. Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-κB activation. Nature Medicine. 1998;4(6):698–704. doi: 10.1038/nm0698-698. [DOI] [PubMed] [Google Scholar]
- 10.Caldwell CC, Okaya T, Martignoni A, Husted T, Schuster R, Lentsch AB. Divergent functions of CD4+ T lymphocytes in acute liver inflammation and injury after ischemia-reperfusion. American Journal of Physiology. 2005;289(5):G969–G976. doi: 10.1152/ajpgi.00223.2005. [DOI] [PubMed] [Google Scholar]
- 11.Hines IN, Hoffman JM, Scheerens H, et al. Regulation of postischemic liver injury following different durations of ischemia. American Journal of Physiology. 2003;284(3):G536–G545. doi: 10.1152/ajpgi.00400.2002. [DOI] [PubMed] [Google Scholar]
- 12.Husted TL, Lentsch AB. The role of cytokines in pharmacological modulation of hepatic ischemia/reperfusion injury. Current Pharmaceutical Design. 2006;12(23):2867–2873. doi: 10.2174/138161206777947597. [DOI] [PubMed] [Google Scholar]
- 13.Pine JK, Aldouri A, Young AL, et al. Liver transplantation following donation after cardiac death: an analysis using matched pairs. Liver Transplantation. 2009;15(9):1072–1082. doi: 10.1002/lt.21853. [DOI] [PubMed] [Google Scholar]
- 14.Takeuchi D, Yoshidome H, Kurosawa H, et al. Interleukin-18 exacerbates pulmonary injury after hepatic ischemia/reperfusion in mice. Journal of Surgical Research. 2010;158(1):87–93. doi: 10.1016/j.jss.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 15.Weinbroum AA, Kidron A, Hochhauser E, Hochman A, Rudick V, Vidne BA. Liver glutathione level influences myocardial reperfusion injury following liver ischemia-reperfusion. Medical Science Monitor. 2001;7(6):1137–1144. [PubMed] [Google Scholar]
- 16.Behrends M, Hirose R, Park YH, et al. Remote renal injury following partial hepatic ischemia/reperfusion injury in rats. Journal of Gastrointestinal Surgery. 2008;12(3):490–495. doi: 10.1007/s11605-007-0252-6. [DOI] [PubMed] [Google Scholar]
- 17.Weinbroum AA. N-acetyl-L-cysteine mitigates aortic tone injury following liver ischemia-reperfusion. Journal of Cardiovascular Pharmacology. 2005;45(6):509–515. doi: 10.1097/01.fjc.0000159640.36900.5d. [DOI] [PubMed] [Google Scholar]
- 18.Okaya T, Blanchard J, Schuster R, et al. Age-dependent responses to hepatic ischemia/reperfusion injury. Shock. 2005;24(5):421–427. doi: 10.1097/01.shk.0000181282.14050.11. [DOI] [PubMed] [Google Scholar]
- 19.Yokoyama Y, Nagino M, Nimura Y. Which gender is better positioned in the process of liver surgery? Male or female? Surgery Today. 2007;37(10):823–830. doi: 10.1007/s00595-007-3508-6. [DOI] [PubMed] [Google Scholar]
- 20.Manekeller S, Sioutis M, Hirner A, Minor T. Influence of neoadjuvant chemotherapy on liver integrity and ischemic tolerance. Zeitschrift fur Gastroenterologie. 2008;46(1):17–21. doi: 10.1055/s-2007-963440. [DOI] [PubMed] [Google Scholar]
- 21.Schemmer P, Nickkholgh A, Hinz U, et al. Extended donor criteria have no negative impact on early outcome after liver transplantation: a single-center multivariate analysis. Transplantation Proceedings. 2007;39(2):529–534. doi: 10.1016/j.transproceed.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Vollmar B, Menger MD. Intestinal ischemia/reperfusion: microcirculatory pathology and functional consequences. Langenbeck’s Archives of Surgery. 2011;396(1):13–29. doi: 10.1007/s00423-010-0727-x. [DOI] [PubMed] [Google Scholar]
- 23.Schindler G, Kincius M, Liang R, et al. Fundamental efforts toward the development of a therapeutic cocktail with a manifold ameliorative effect on hepatic ischemia/reperfusion injury. Microcirculation. 2009;16(7):593–602. doi: 10.1080/10739680903110779. [DOI] [PubMed] [Google Scholar]
- 24.Zhong Z, Connor HD, Froh M, et al. Free radical-dependent dysfunction of small-for-size rat liver grafts: prevention by plant polyphenols. Gastroenterology. 2005;129(2):652–664. doi: 10.1016/j.gastro.2005.05.060. [DOI] [PubMed] [Google Scholar]
- 25.Eipel C, Abshagen K, Vollmar B. Regulation of hepatic blood flow: the hepatic arterial buffer response revisited. World Journal of Gastroenterology. 2010;16(48):6046–6057. doi: 10.3748/wjg.v16.i48.6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seizner M, Seizmer N, Jochum W, Graf R, Clavien PA. Increased ischemic injury in old mouse liver: an ATP-dependent mechanism. Liver Transplantation. 2007;13(3):382–390. doi: 10.1002/lt.21100. [DOI] [PubMed] [Google Scholar]
- 27.Froh M, Wheeler MD, Smutney O, Zhong Z, Bradford BU, Thurman RG. New method of delivering gene-altered Kupffer cells to rat liver: studies in an ischemia-reperfusion model. Gastroenterology. 2003;124(1):172–183. doi: 10.1053/gast.2003.50002. [DOI] [PubMed] [Google Scholar]
- 28.Jaeschke H. Mechanisms of liver injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. American Journal of Physiology. 2006;290(6):G1083–G1088. doi: 10.1152/ajpgi.00568.2005. [DOI] [PubMed] [Google Scholar]
- 29.Lefer AM, Lefer DJ. II. Nitric oxide protects in intestinal inflammation. American Journal of Physiology. 1999;276(3):G572–G575. doi: 10.1152/ajpgi.1999.276.3.G572. [DOI] [PubMed] [Google Scholar]
- 30.Grisham MB, Granger DN, Lefer DJ. Modulation of leukocyte-endothelial interactions by reactive metabolites of oxygen and nitrogen: relevance to ischemic heart disease. Free Radical Biology and Medicine. 1998;25(4-5):404–433. doi: 10.1016/s0891-5849(98)00094-x. [DOI] [PubMed] [Google Scholar]
- 31.Phillips L, Toledo AH, Lopez-Neblina F, Anaya-Prado R, Toledo-Pereyra LH. Nitric oxide mechanism of protection in ischemia and reperfusion injury. Journal of Investigative Surgery. 2009;22(1):46–55. doi: 10.1080/08941930802709470. [DOI] [PubMed] [Google Scholar]
- 32.Abe Y, Hines I, Zibari G, Grisham MB. Hepatocellular protection by nitric oxide or nitrite in ischemia and reperfusion injury. Archives of Biochemistry and Biophysics. 2009;484(2):232–237. doi: 10.1016/j.abb.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Contaldo C, Elsherbiny A, Lindenblatt N, et al. Erythropoietin enhances oxygenation in critically perfused tissue through modulation of nitric oxide synthase. Shock. 2009;31(6):599–606. doi: 10.1097/SHK.0b013e31818b9cc4. [DOI] [PubMed] [Google Scholar]
- 34.Abu-Amara M, Yang SY, Seifalian A, Davidson B, Fuller B. The nitric oxide pathway—evidence and mechanisms for protection against liver ischaemia reperfusion injury. Liver International. 2012;32:531–543. doi: 10.1111/j.1478-3231.2012.02755.x. [DOI] [PubMed] [Google Scholar]
- 35.Jaeschke H. Role of reactive oxygen species in hepatic ischemia-reperfusion injury and preconditioning. Journal of Investigative Surgery. 2003;16(3):127–140. [PubMed] [Google Scholar]
- 36.Urakami H, Abe Y, Grisham MB. Role of reactive metabolites of oxygen and nitrogen in partial liver transplantation: lessons learned from reduced-size liver ischaemia and reperfusion injury. Clinical and Experimental Pharmacology and Physiology. 2007;34(9):912–919. doi: 10.1111/j.1440-1681.2007.04640.x. [DOI] [PubMed] [Google Scholar]
- 37.Abu-Amara M, Yang SY, Tapuria N, Fuller B, Davidson B, Seifalian A. Liver ischemia/reperfusion injury: processes in inflammatory networks—a review. Liver Transplantation. 2010;16(9):1016–1032. doi: 10.1002/lt.22117. [DOI] [PubMed] [Google Scholar]
- 38.Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Piña E, Geller DA. Factors in the pathophysiology of the liver ischemia-reperfusion injury. Journal of Surgical Research. 2008;147(1):153–159. doi: 10.1016/j.jss.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Belous A, Knox C, Nicoud IB, et al. Reversed activity of mitochondrial adenine nucleotide translocator in ischemia-reperfusion. Transplantation. 2003;75(10):1717–1723. doi: 10.1097/01.TP.0000063829.35871.CE. [DOI] [PubMed] [Google Scholar]
- 40.López-Neblina F, Toledo-Pereyra LH, Toledo AH, Walsh J. Ryanodine receptor antagonism protects the ischemic liver and modulates TNF-α and IL-10. Journal of Surgical Research. 2007;140(1):121–128. doi: 10.1016/j.jss.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 41.Miller BA. The role of TRP channels in oxidative stress-induced cell death. Journal of Membrane Biology. 2006;209(1):31–41. doi: 10.1007/s00232-005-0839-3. [DOI] [PubMed] [Google Scholar]
- 42.Jiang N, Zhang ZM, Liu L, Zhang C, Zhang YL, Zhang ZC. Effects of Ca2+ channel blockers on store-operated Ca2+ channel currents of Kupffer cells after hepatic ischemia /reperfusion injury in rats. World Journal of Gastroenterology. 2006;12(29):4694–4698. doi: 10.3748/wjg.v12.i29.4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barritt GJ, Chen J, Rychkov GY. Ca2+-permeable channels in the hepatocyte plasma membrane and their roles in hepatocyte physiology. Biochimica et Biophysica Acta. 2008;1783(5):651–672. doi: 10.1016/j.bbamcr.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 44.Janicki PK, Wise PE, Belous AE, Pinson CW. Interspecies differences in hepatic Ca2+ -ATPase activity and the effect of cold preservation on porcine liver Ca2+ -ATPase function. Liver Transplantation. 2001;7(2):132–139. doi: 10.1053/jlts.2001.21459. [DOI] [PubMed] [Google Scholar]
- 45.Anderson CD, Pierce J, Nicoud I, Belous A, Knox CD, Chari RS. Modulation of mitochondrial calcium management attenuates hepatic warm ischemia-reperfusion injury. Liver Transplantation. 2005;11(6):663–668. doi: 10.1002/lt.20407. [DOI] [PubMed] [Google Scholar]
- 46.Knox CD, Pierce JM, Nicoud IB, et al. Inhibition of phospholipase C attenuates liver mitochondrial calcium overload following cold ischemia. Transplantation. 2006;81(4):567–572. doi: 10.1097/01.tp.0000199267.98971.77. [DOI] [PubMed] [Google Scholar]
- 47.Belous AE, Jones CM, Wakata A, et al. Mitochondrial calcium transport is regulated by P2Y1- and P2Y2-like mitochondrial receptors. Journal of Cellular Biochemistry. 2006;99(4):1165–1174. doi: 10.1002/jcb.20985. [DOI] [PubMed] [Google Scholar]
- 48.Zhao K, Zhao GM, Wu D, et al. Cell-permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. Journal of Biological Chemistry. 2004;279(33):34682–34690. doi: 10.1074/jbc.M402999200. [DOI] [PubMed] [Google Scholar]
- 49.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125(4):1246–1257. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- 50.Zorov DB, Juhaszova M, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Regulation and pharmacology of the mitochondrial permeability transition pore. Cardiovascular Research. 2009;83(2):213–225. doi: 10.1093/cvr/cvp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochimica et Biophysica Acta. 2009;1787(11):1395–1401. doi: 10.1016/j.bbabio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. The FASEB Journal. 2001;15(12):2286–2287. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 53.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Archives of Biochemistry and Biophysics. 2007;462(2):245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim JS, Qian T, Lemasters JJ. Mitochondrial permeability transition in the switch from necrotic to apoptotic cell death in ischemic rat hepatocytes. Gastroenterology. 2003;124(2):494–503. doi: 10.1053/gast.2003.50059. [DOI] [PubMed] [Google Scholar]
- 55.Wang D, Dou K, Song Z, Liu Z. The Na(+)/H(+) exchange inhibitor: a new therapeutic approach for hepatic ischemia injury in rats. Transplantation Proceedings. 2003;35(8):3134–3135. doi: 10.1016/j.transproceed.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 56.Carini R, De Cesaris MG, Splendore R, Bagnati M, Bellomo G, Albano E. Alterations of Na+ homeostasis in hepatocyte reoxygenation injury. Biochimica et Biophysica Acta. 2000;1500(3):297–305. doi: 10.1016/s0925-4439(99)00114-3. [DOI] [PubMed] [Google Scholar]
- 57.Vairetti M, Richelmi P, Bertè F, Currin RT, Lemasters JJ, Imberti R. Role of pH in protection by low sodium against hypoxic injury in isolated perfused rat livers. Journal of Hepatology. 2006;44(5):894–901. doi: 10.1016/j.jhep.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 58.Caban A, Oczkowicz G, Abdel-Samad O, Cierpka L. Influence of Kupffer cells on the early phase of liver reperfusion. Transplantation Proceedings. 2002;34(2):694–697. doi: 10.1016/s0041-1345(01)02891-3. [DOI] [PubMed] [Google Scholar]
- 59.Bruns H, Watanpour I, Gebhard MM, et al. Glycine and taurine equally prevent fatty livers from Kupffer cell-dependent injury: an in Vivo microscopy study. Microcirculation. 2011;18(3):205–213. doi: 10.1111/j.1549-8719.2010.00078.x. [DOI] [PubMed] [Google Scholar]
- 60.Schemmer P, Schoonhoven R, Swenberg JA, Bunzendahl H, Thurman RG. Gentle in situ liver manipulation during organ harvest decreases survival after rat liver transplantation: role of Kupffer cells. Transplantation. 1998;65(8):1015–1020. doi: 10.1097/00007890-199804270-00001. [DOI] [PubMed] [Google Scholar]
- 61.Brock RW, Nie RG, Harris KA, Potter RF. Kupffer cell-initiated remote hepatic injury following bilateral hindlimb ischemia is complement dependent. American Journal of Physiology. 2001;280(2):G279–G284. doi: 10.1152/ajpgi.2001.280.2.G279. [DOI] [PubMed] [Google Scholar]
- 62.Fondevila C, Shen XD, Tsuchihashi S, et al. The membrane attack complex (C5b-9) in liver cold ischemia and reperfusion injury. Liver Transplantation. 2008;14(8):1133–1141. doi: 10.1002/lt.21496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Llacuna L, Marí M, Lluis JM, García-Ruiz C, Fernández-Checa JC, Morales A. Reactive oxygen species mediate liver injury through parenchymal nuclear factor-κB inactivation in prolonged ischemia/reperfusion. American Journal of Pathology. 2009;174(5):1776–1785. doi: 10.2353/ajpath.2009.080857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hanschen M, Zahler S, Krombach F, Khandoga A. Reciprocal activation between CD4+ T cells and Kupffer cells during hepatic ischemia-reperfusion. Transplantation. 2008;86(5):710–718. doi: 10.1097/TP.0b013e3181821aa7. [DOI] [PubMed] [Google Scholar]
- 65.Taniai H, Hines IN, Bharwani S, et al. Susceptibility of murine periportal hepatocytes to hypoxia-reoxygenation: role for NO and Kupffer cell-derived oxidants. Hepatology. 2004;39(6):1544–1552. doi: 10.1002/hep.20217. [DOI] [PubMed] [Google Scholar]
- 66.Nakano Y, Kondo T, Matsuo R, et al. Platelet dynamics in the early phase of postischemic liver in vivo. Journal of Surgical Research. 2008;149(2):192–198. doi: 10.1016/j.jss.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 67.Khandoga A, Kessler JS, Hanschen M, et al. Matrix metalloproteinase-9 promotes neutrophil and T cell recruitment and migration in the postischemic liver. Journal of Leukocyte Biology. 2006;79(6):1295–1305. doi: 10.1189/jlb.0805468. [DOI] [PubMed] [Google Scholar]
- 68.Caldwell CC, Tschoep J, Lentsch AB. Lymphocyte function during hepatic ischemia/reperfusion injury. Journal of Leukocyte Biology. 2007;82(3):457–464. doi: 10.1189/jlb.0107062. [DOI] [PubMed] [Google Scholar]
- 69.Kuboki S, Sakai N, Tschöp J, Edwards MJ, Lentsch AB, Caldwell CC. Distinct contributions of CD4+ T cell subsets in hepatic ischemia/reperfusion injury. American Journal of Physiology. 2009;296(5):G1054–G1059. doi: 10.1152/ajpgi.90464.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. Journal of Experimental Medicine. 2006;203(12):2639–2648. doi: 10.1084/jem.20061097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khandoga A, Hanschen M, Kessler JS, Krombach F. CD4+ T cells contribute to postischemic liver injury in mice by interacting with sinusoidal endothelium and platelets. Hepatology. 2006;43(2):306–315. doi: 10.1002/hep.21017. [DOI] [PubMed] [Google Scholar]
- 72.Froh M, Zhong Z, Walbrun P, et al. Dietary glycine blunts liver injury after bile duct ligation in rats. World Journal of Gastroenterology. 2008;14(39):5996–6003. doi: 10.3748/wjg.14.5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Selzner N, Selzner M, Odermatt B, Tian Y, Van Rooijen N, Clavien PA. ICAM-1 triggers liver regeneration through leukocyte recruitment and Kupffer cell-dependent release of TNF-α/IL-6 in mice. Gastroenterology. 2003;124(3):692–700. doi: 10.1053/gast.2003.50098. [DOI] [PubMed] [Google Scholar]
- 74.Gressner O, Schilling T, Lorenz K, et al. TAp63α induces apoptosis by activating signaling via death receptors and mitochondria. EMBO Journal. 2005;24(13):2458–2471. doi: 10.1038/sj.emboj.7600708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peralta C, Fernández L, Panés J, et al. Preconditioning protects against systemic disorders associated with hepatic ischemia-reperfusion through blockade of tumor necrosis factor-induced P-selectin up-regulation in the rat. Hepatology. 2001;33(1):100–113. doi: 10.1053/jhep.2001.20529. [DOI] [PubMed] [Google Scholar]
- 76.Rüdiger HA, Clavien P. Tumor necrosis factor α, but not Fas, mediates hepatocellular apoptosis in the murine ischemic liver. Gastroenterology. 2002;122(1):202–210. doi: 10.1053/gast.2002.30304. [DOI] [PubMed] [Google Scholar]
- 77.Müller M, Strand S, Hug H, et al. Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO- 1/Fas) receptor/ligand system and involves activation of wild-type p53. Journal of Clinical Investigation. 1997;99(3):403–413. doi: 10.1172/JCI119174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Müller M, Wilder S, Bannasch D, et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. Journal of Experimental Medicine. 1998;188(11):2033–2045. doi: 10.1084/jem.188.11.2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eichhorst ST, Muller M, Li-Weber M, Schulze-Bergkamen H, Angel P, Krammer PH. A novel AP-1 element in the CD95 ligand promoter is required for induction of apoptosis in hepatocellular carcinoma cells upon treatment with anticancer drugs. Molecular and Cellular Biology. 2000;20(20):7826–7837. doi: 10.1128/mcb.20.20.7826-7837.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li B, Chen B, Zhang G, Wang K, Zhou L, Hu S. Cell apoptosis and fas gene expression in liver and renal tissues after ischemia-reperfusion injury in liver transplantation. Transplantation Proceedings. 2010;42(5):1550–1556. doi: 10.1016/j.transproceed.2010.01.055. [DOI] [PubMed] [Google Scholar]
- 81.Bradham CA, Schemmer P, Stachlewitz RF, Thurman RG, Brenner DA. Activation of nuclear factor-κB during orthotopic liver transplantation in rats is protective and does not require Kupffer cells. Liver Transplantation and Surgery. 1999;5(4):282–293. doi: 10.1002/lt.500050401. [DOI] [PubMed] [Google Scholar]
- 82.Schwabe RF, Brenner DA. Mechanisms of liver injury. I. TNF-α-induced liver injury: role of IKK, JNK, and ROS pathways. American Journal of Physiology. 2006;290(4):G583–G589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 83.Liang R, Nickkholgh A, Hoffmann K, et al. Melatonin protects from hepatic reperfusion injury through inhibition of IKK and JNK pathways and modification of cell proliferation. Journal of Pineal Research. 2009;46(1):8–14. doi: 10.1111/j.1600-079X.2008.00596.x. [DOI] [PubMed] [Google Scholar]
- 84.Sanlioglu S, Williams CM, Samavati L, et al. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-α secretion through IKK regulation of NF-κB. Journal of Biological Chemistry. 2001;276(32):30188–30198. doi: 10.1074/jbc.M102061200. [DOI] [PubMed] [Google Scholar]
- 85.Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nature Reviews Drug Discovery. 2007;6(8):662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 86.Tsung A, Sahai R, Tanaka H, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. Journal of Experimental Medicine. 2005;201(7):1135–1143. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pelinka LE, Harada N, Szalay L, Jafarmadar M, Redl H, Bahrami S. Release of S100B differs during ischemia and reperfusion of the liver, the gut, and the kidney in rats. Shock. 2004;21(1):72–76. doi: 10.1097/01.shk.0000101672.49265.14. [DOI] [PubMed] [Google Scholar]
- 88.Zhang J, Wang H, Xiao Q, et al. Hyaluronic acid fragments evoke Kupffer cells via TLR4 signaling pathway. Science in China C. 2009;52(2):147–154. doi: 10.1007/s11427-009-0002-y. [DOI] [PubMed] [Google Scholar]
- 89.Pardo M, Budick-Harmelin N, Tirosh B, Tirosh O. Antioxidant defense in hepatic ischemia-reperfusion injury is regulated by damage-associated molecular pattern signal molecules. Free Radical Biology and Medicine. 2008;45(8):1073–1083. doi: 10.1016/j.freeradbiomed.2008.06.029. [DOI] [PubMed] [Google Scholar]
- 90.Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology. 2010;51(2):621–632. doi: 10.1002/hep.23365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Katsargyris A, Klonaris C, Alexandrou A, Giakoustidis AE, Vasileiou I, Theocharis S. Toll like receptors in liver ischemia reperfusion injury: a novel target for therapeutic modulation? Expert Opinion on Therapeutic Targets. 2009;13(4):427–442. doi: 10.1517/14728220902794939. [DOI] [PubMed] [Google Scholar]
- 92.Arumugam TV, Okun E, Tang SC, Thundyil J, Taylor SM, Woodruff TM. Toll-like receptors in ischemia-reperfusion injury. Shock. 2009;32(1):4–16. doi: 10.1097/SHK.0b013e318193e333. [DOI] [PubMed] [Google Scholar]
- 93.Szabo G, Dolganiuc A, Mandrekar P. Pattern recognition receptors: a contemporary view on liver diseases. Hepatology. 2006;44(2):287–298. doi: 10.1002/hep.21308. [DOI] [PubMed] [Google Scholar]
- 94.Tsuchihashi SI, Zhai Y, Bo Q, Busuttil RW, Kupiec-Weglinski JW. Heme oxygenase-1 mediated cytoprotection against liver ischemia and reperfusion injury: inhibition of type-1 interferon signaling. Transplantation. 2007;83(12):1628–1634. doi: 10.1097/01.tp.0000266917.39958.47. [DOI] [PubMed] [Google Scholar]
- 95.Wang H, Li ZY, Wu HS, et al. Endogenous danger signals trigger hepatic ischemia/reperfusion injury through toll-like receptor 4/nuclear factor-kappa B pathway. Chinese Medical Journal. 2007;120(6):509–514. [PubMed] [Google Scholar]
- 96.Zhai Y, Shen XD, O’Connell R, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. Journal of Immunology. 2004;173(12):7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 97.Watanabe T, Kubota S, Nagaya M, et al. The role of HMGB-1 on the development of necrosis during hepatic ischemia and hepatic ischemia/reperfusion injury in mice. Journal of Surgical Research. 2005;124(1):59–66. doi: 10.1016/j.jss.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 98.Zeng S, Dun H, Ippagunta N, et al. Receptor for advanced glycation end product (RAGE)-dependent modulation of early growth response-1 in hepatic ischemia/reperfusion injury. Journal of Hepatology. 2009;50(5):929–936. doi: 10.1016/j.jhep.2008.11.022. [DOI] [PubMed] [Google Scholar]
- 99.Tsung A, Zheng N, Jeyabalan G, et al. Increasing numbers of hepatic dendritic cells promote HMGB1-mediated ischemia-reperfusion injury. Journal of Leukocyte Biology. 2007;81(1):119–128. doi: 10.1189/jlb.0706468. [DOI] [PubMed] [Google Scholar]