

Abstract
The past few years have provided substantial evidence for the vital role of the local tumor microenvironment for various aspects of tumor progression. With obesity and its pathophysiological sequelae still on the rise, the adipocyte is increasingly moving center stage in the context of tumor stroma-related studies. To date, we have limited insight into how the systemic metabolic changes associated with obesity and the concomitant modification of the paracrine and endocrine panel of stromal adipocyte-derived secretory products (“adipokines”) influence the incidence and progression of obesity-related cancers. Here, we discuss the role of adipocyte dysfunction associated with obesity and its potential impact on cancer biology.
Introduction
-
Evidence for the Role of the Adipocyte in Cancer
Dysfunction of adipose tissues in obesity
Adipocyte and cancer biology in obesity
-
How Do Adipocytes Influence Cancer Biology?
Paracrine effects of dysregulated adipocytes
Endocrine effects of dysregulated adipocytes
-
Targeting Adipose Tissues in Cancer Prevention and Treatment
Targeting adipose tissues
Targeting systemic metabolic effects
Conclusions
I. Introduction
Adipocytes are major constituents of adipose tissue. They are key players in the homeostatic control of whole body metabolism. Not only do adipocytes fulfill an important function to buffer excess triglycerides and free fatty acids (FFA); they also secrete numerous lipid and protein factors. Therefore, they are considered major endocrine cells with a profound impact on the metabolism of other tissues, including liver, muscle, brain, and pancreatic β-cells (1). During times of caloric excess, adipocytes become hypertrophic and face various physiological insults, such as hypoxia, oxidative stress, and disruptions in the protein secretory pathway, the latter originating from stress related to folding and assembly of protein complexes in the endoplasmic reticulum (ER) (2) (Fig. 1). As a result, adipocytes become dysfunctional and thus less efficient at fulfilling their primary function—the storage and neutralization of potentially harmful lipids. Adipocyte dysfunction has been widely appreciated as a major cause underlying obesity-related metabolic disorders, including insulin resistance, type 2 diabetes, cardiovascular disease, chronic inflammation, and cancer, due to inappropriate release of mitogenic and proinflammatory factors (3, 4).
Fig. 1.

Paracrine and endocrine interactions of adipocytes with cancer cells. A, Paracrine signals (tumor microenvironment). Both cell-autonomous signals and paracrine signals derived from stromal cells control tumor cell physiology. The tumor microenvironment contains a number of cell types, including adipocytes, fibroblasts, immune cells, tumor-associated macrophages (TAMs), and myoendothelial cells, all of which mediate paracrine signaling in the microenvironment. Adipocytes constitute a key stromal cell type and interact with cancer cells by secreting a variety of signaling molecules that include ECM constituents, adipokines, inflammatory cytokines, and angiogenic factors. HGF, Hepatocyte growth factor. B, Endocrine signals (systemic effects). During the process of adipocyte hypertrophy and hyperplasia associated with the obese state, adipose tissues become dysfunctional due to metabolically challenging conditions, such as hypoxia and oxidative and ER stress. The metabolic consequences of adipose tissue dysfunction, such as adipokine dysregulation, chronic inflammation, dyslipidemia, and hormonal dysregulation, result in insulin resistance. As a result, adipose tissue not only affects proximal tumor cells but also influences distal tumor cell behavior through altered expression of cytokines, insulin, and adipokines.
Currently, several epidemiological studies demonstrate positive associations between the prevalence of obesity as judged by increased body mass index (BMI), cancer incidence, and cancer-related mortality (5–8). More specifically, one of the most rigorous meta-analysis studies to date revealed strong associations of overweight (BMI, 25–29.9 kg/m2) or obesity (BMI ≥30 kg/m2) with an increased risk of a subset of cancers, such as thyroid, colon, and renal cancers, and esophageal adenocarcinoma, leukemia, and non-Hodgkin lymphoma in both sexes; rectal and malignant melanoma in men; as well as endometrial, gallbladder, pancreas, ovarian, and breast cancers in women (7). Obesity-associated colon, breast, and endometrial cancer incidence in women is critically dependent on menopausal status (6) (Table 1). Although obesity affects tumor incidence, the correlation of elevated body fat mass with cancer-related mortalities has also been described for many cancers, such as cancers deriving from tissues such as esophagus, colon, liver, gallbladder, pancreas, and kidney, as well as non-Hodgkin lymphoma and multiple myeloma in both sexes; stomach and prostate in men; and breast, uterus, cervix, and ovary in women (8). Obesity accounted for a 52% higher (in males) and an 88% higher (in females) mortality rate across all cancers (8) (Table 1). Although the epidemiological evidence connecting obesity with cancer incidence is strong, the underlying mechanistic basis linking obesity per se to tumor-initiating events remains largely elusive. Three main mechanistic connections have been suggested that may causally invoke obesity (i.e., increased fat mass) with cancer progression. These include: 1) sex hormones; 2) alterations in signaling events along the insulin and IGF-I axis; and 3) changes in the levels of adipocyte-derived factors (5) (Fig. 2). Therefore, as adipose tissue becomes increasingly dysfunctional upon weight gain, the altered physiological state of the expanding fat pads is likely to hold the key to the enhanced mitogenic effects observed on tumor development and progression, at least in part through altered paracrine and endocrine signals as well as immune cell infiltration (9).
Table 1.
Relative risk of cancer incidence and mortality in association to obesity
| Incidence (risk ratio, per 5 kg/m2 increasea) |
Mortality (risk ratio, normal vs. obeseb) |
Related adipokines and effects |
|||||
|---|---|---|---|---|---|---|---|
| Type of cancer | Men | Women | Type of cancer | Men | Women | Promotion | Inhibition |
| Liver | 1.24 | 1.07 | Liver (BMI ≥35) | 4.52 | 1.68 | IL-6, TNFα (26); leptin (167) | Adiponectin (168) |
| Pancreas | 1.07 | 1.12 | Pancreas (BMI ≥35) | 2.61 | 2.76 | Leptin (169) | |
| Esophagus (adenocarcinoma) | 1.52 | 1.51 | Esophagus (BMI ≥30) | 1.91 | 2.64 | Leptin (169, 170) | Adiponectin (171, 172) |
| Colorectal | 1.24 | 1.09 | Colon and rectum (BMI ≥35) | 1.84 | 1.46 | Leptin (173–175); TNFα (176) | Adiponectin (79, 177, 178) |
| Gallbladder | 1.09 | 1.59 | Gallbladder (BMI ≥30) | 1.76 | 2.13 | ||
| Kidney | 1.24 | 1.34 | Kidney (BMI ≥35) | 1.70 | 4.75 | Leptin (179, 180) | Adiponectin (181) |
| Prostate | 1.03 | Prostate (BMI ≥35) | 1.34 | Leptin (182, 183) | Adiponectin (184) | ||
| Breast (postmenopausal) | 1.12 | Breast (BMI ≥40) | 2.12 | Leptin (94, 186, 187); adiponectin (77, 81, 82) | Adiponectin (188–190) | ||
| Endometrium | 1.59 | Leptin (186, 191, 192) | Adiponectin (193) | ||||
| Uterus (BMI ≥40) | 6.25 | ||||||
| Cervix (BMI ≥35) | 3.20 | ||||||
| All other cancers (BMI ≥35) | 1.68 | 2.51 | |||||
| All cancers (BMI ≥40) | 1.52 | 1.88 | |||||
Summary of epidemiological data on the relative risk of cancer incidence and mortality in relation to BMI (in kilograms per square meter). Cancer incidence data are from the meta-analysis of published epidemiological studies from 1966 to 2007 (7).
Risk ratio represents increase of incidence rate per 5 kg/m2 increase in BMI. Cancer-related mortality data are from the Cancer Prevention Study II with more than 900,000 cancer patients (1982–1998) (8).
Risk ratio represents relative mortality rate of high BMI (35–39.9 kg/m2) patients compared to normal BMI (18.5–24.9 kg/m2).
Fig. 2.

Crosstalk among the signaling pathways linking obesity and breast cancer progression. The EGFR (epidermal growth factor receptor) family, such as EGFR (ErbB1), and ErbB2/HER2/Neu, plays an important role in tumorigenesis by inducing cells to proliferate and to survive. Upon ligand binding to EGFR, downstream pathways including kinases such as PI3K, ERK, and JAK/STAT are activated in a coordinated manner to promote cell proliferation and survival. The estrogen receptor ERα triggers EGFR pathways, which are also directly regulated by adipokines, such as adiponectin and leptin through their receptors AdipoR1, AdipoR2, and LEPR-B, respectively, which are expressed in mammary tumor cells. Importantly, adiponectin and leptin modulate estrogen (ER) production through regulation of aromatase activity in adipose tissue, resulting in altering ERα and EGFR pathways in cancer cells. LEPR-B mediated downstream signaling, such as PI3K, ERK and JAK/STAT3 is well-known to be involved in protumorigenic pathways. Adiponectin actions are multifaceted. Activation of ceramidase activity through AdipoR1 and AdipoR2 causes proangiogenic and antiapoptotic responses. Ceramide inhibits Akt pathways, whereas S1P activates antiapoptotic pathways. Inflammatory cytokines, such as TNFα and IL-6, and ECMs secreted from adipose tissue also contribute to cancer progression through their receptors. These include RTKs (receptor-tyrosine kinases) and integrin-mediated pathways. Downstream mediators, such as NF-κB and focal adhesion kinase (FAK) convey the EGFR signal to the nucleus and also affect other adipokine-stimulated downstream pathways. High insulin levels frequently observed in obese subjects augment promitogenic insulin/IGF-I signaling. Proangiogenic factors, such as vascular endothelial growth factor (VEGF) secreted from adipose tissue, contribute to angiogenic capacities of endothelial cells within the tumor microenvironment. Adiponectin receptors and T-cadherin expressed in tumor endothelial cells may be associated with angiogenesis. Therefore, various signaling pathways triggered by adipokines converge upon oncogenic signaling pathways in mammary tumor cells to promote tumor progression.
Evidence supporting growth-stimulatory roles of adipocyte-derived factors (such as adipokines and lipid metabolites) in cancer progression has previously been reported in several papers (10–12) (Table 1). The levels of at least some of these factors can be altered pharmacologically. Hence, adipose tissue is emerging as a potential therapeutic target in the context of cancer studies. From this perspective, therapeutic approaches restoring normal metabolic function in adipocytes in obese patients may exert a significant beneficial effect toward the reduction of tumor progression.
Several key questions need to be addressed in this context: Is the adipocyte a direct tumor-initiating factor? Alternatively, does the adipocyte merely promote tumor growth, independent of the initial transforming event? And finally, are the potential tumor-promoting (and/or tumor-initiating) aspects of adipocytes primarily indirect effects, occurring secondarily to the general systemic metabolic changes associated with dysfunctional adipose tissue? Here, we will discuss specific changes associated with obese adipose tissue, giving rise to altered conditions within the tumor microenvironment through paracrine and endocrine signals emerging from adipocytes. We will extend the discussion to recent advances highlighting evidence for direct contributions of the adipocyte toward cancer progression. However, it is important to note that this is a field that is still in its infancy, with a solid foundation of epidemiological data, but only very limited direct mechanistic insights at this point.
II. Evidence for the Role of the Adipocyte in Cancer
Tumors are subject to both autocrine and paracrine signals, a phenomenon that Hanahan and Weinberg (13) refer to as “heterotypic signaling.” These authors further elaborated on this concept (14). The paracrine component includes interactions of tumors with stromal cells within the local microenvironment. Major stromal contributors are endothelial cells, fibroblasts, epithelial cells, macrophages, and adipocytes, all of which exert effects on the tumor cells (13) (Fig. 1A). A number of studies have underscored the physiological roles of stromal adipocytes in tumor progression, emphasizing the relevance of adipocytes on the survival rate, growth, and metastatic behavior of cancer cells. Such functions have been demonstrated both in vitro and in vivo (10, 15). In addition to these paracrine signals derived from stromal adipocytes, endocrine signals originating from adipose tissue are equally relevant for tumor behavior, either directly through endocrine effects or indirectly through their impact on whole body metabolism. Adipocytes affect glucose and lipid metabolism, can exert chronic inflammatory signals, and can coordinate the response of multiple immune cells (Fig. 1B).
Therefore, focus needs to be put on the direct effects of adipose tissue on tumor cells through paracrine signals that are released locally within the tumor microenvironment as well as the endocrine signals that originate from distal adipose tissues that may significantly disrupt local metabolic homeostasis (Fig. 1). The relative roles that paracrine vs. endocrine factors derived from adipocytes exert on tumor physiology offer a great starting point for a better understanding of the etiology of obesity-associated cancers. Beyond exerting promitogenic and antiapoptotic effects on tumor cells, adipocytes serve as key energetic buffers for the maintenance of systemic nutrient homeostasis. Cancer cells in contrast are metabolically highly active and consume vast amounts of energy to support their growth and survival. The metabolic changes associated with cancer cell metabolism involve a shift from oxidative phosphorylation to aerobic glycolysis for the generation of ATP, a phenomenon referred to as the “Warburg effect” (16). This metabolic switch ensures that sufficient levels of ATP are available to synthesize the cellular building blocks necessary for rapidly proliferating cells to survive (16). Aerobic glycolysis is associated with de novo lipogenesis to supply lipid precursors for proliferating cancer cells (16, 17). Although the primary energy source for cancer cells is glucose, under conditions of high energy demand and limited glucose availability, glutamine and/or FFA can replace glucose (17). However, we currently do not fully understand whether the increased local flux of stromal FFA derived from insulin-resistant adipocytes, and/or FFA derived from dietary fats, have a direct impact on cancer cell metabolism. At least to date, beneficial effects of low-fat diets on tumor growth and recurrence are not well established (18).
A. Dysfunction of adipose tissues in obesity
Understanding the obesity-induced changes in adipocyte metabolism is an important starting point for the understanding of the molecular mechanisms underlying obesity-related tumor development and progression. A chronic increase in energy intake leads initially to an increase of adipocyte size (hypertrophy) and eventually to an increase in adipocyte number (hyperplasia) in an attempt to accommodate excess calories (19, 20). During this expansion process, adipose tissue encounters several challenges. These include oxidative stress, stress related to the secretory pathway, and hypoxia, all of which are causally associated with reduced metabolic flexibility of adipocytes (21). This results in an increased rate of apoptosis, ultimately triggering the local accumulation of inflammatory cells into adipose tissue (4, 22–24) (Fig. 1B). Based on our current understanding of the pathogenesis of obesity, many secretory molecules derived from adipocytes (generally referred to as adipokines), such as extracellular matrix components, angiogenic factors, inflammatory cytokines, and lipid metabolites, also contribute to the development of chronic inflammation and lipotoxicity, resulting in local and ultimately systemic insulin resistance (1, 3). Notably, these adverse metabolic effects are more strongly associated with lipid accumulation in intraabdominal adipose depots as opposed to sc depots and with tissues prone to accumulate ectopic lipids under these conditions, such as muscle or liver (25) (Table 2). As such, overall body fat distribution is considered to be a better reflection of the metabolic fitness compared with overall fat mass as measured by BMI (25). In other words, the quality of adipose tissue is of much higher relevance for cancer progression than the absolute amount of adipose tissue per se. A large number of epidemiological studies have clearly highlighted that obesity promotes the growth of existing tumors. BMI measurements can easily be obtained; however, given the inherent inadequacy of the parameter, this may account for some of the conflicting epidemiological data that argues for and against positive associations between BMI and cancer progression. However, there is overwhelming epidemiological evidence for the cancer/obesity connection. One of the biggest challenges for the field is that the molecular mechanisms related to the underlying genetic or biochemical mechanisms that link obesity with tumor progression have yet to be established.
Table 2.
Comparison between sc and visceral adipose tissue from high-fat diet-exposed obese mice
| Comparison | Subcutaneous adipose tissue | Visceral adipose tissue |
|---|---|---|
| Morphology (H&E stain)a | 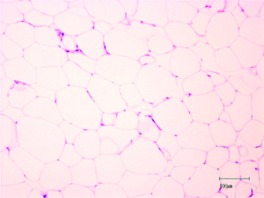 |
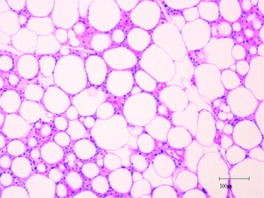 |
| Function | Regulates whole body energy metabolism | Regulates whole body energy metabolism |
| Stores and/or releases lipids during feeding and fasting | Stores and/or releases lipids during feeding and fasting | |
| Secretes various adipokines and chemokines—acts as endocrine organ | Secretes various adipokines and chemokines—acts as endocrine organ | |
| Composition | Adipocytes, endothelial cells, macrophages, immune cells, fibroblasts, etc. | Adipocytes, endothelial cells, macrophages, immune cells, fibroblasts, etc. |
| Inflammation | Less inflamed | Increased macrophage infiltration |
| Chronic inflammation | ||
| TNFα, IL-6, CCL2, and PAI-1 ↑ | ||
| Adipokines | Adiponectin ↑ | Adiponectin ↓ |
| Leptin ↑ | Leptin ↑ | |
| Insulin signals | Sensitive | Resistance |
| Hypertrophy | Healthy expansion | Increase adipocyte apoptosis/necrosis |
| Fibrosis | Fibrogenic properties of preadipocytes ↑ (194) | Less fibrotic preadipocytes |
H&E, Hematoxylin and eosin.
Scale bars, 100 μm.
B. Adipocyte and cancer biology in obesity
Although obesity-associated dysfunctional adipocytes have been implicated in promoting the growth of existing tumors at the various stages of progression, there are no emerging patterns indicating which tumor types and/or the stages of progression are more prone to be influenced by dysfunctional adipocytes. Tumor types that are observed more frequently in individuals with higher BMI are found in endometrium, kidney, thyroid, pancreas, breast, liver, and esophagus (7). The altered endocrine environment prevalent in obesity seems to affect these tumor types preferentially. In addition, ectopic lipids can accumulate in cells other than adipocytes. Such cells, which for the most part are not specialized in lipid storage, frequently experience lipid-induced stress responses, leading to an increased rate of apoptosis and consequently to the attraction of macrophages. This in turn also contributes to an altered tumor microenvironment. Hepatocytes are particularly prone to this ectopic lipid accumulation under such altered metabolic conditions. Intrahepatic lipid accumulation is frequently associated with obese conditions, which contributes significantly to the development of inflammation and, ultimately, serves as a contributing factor for tumorigenesis (26). Interestingly, a subset of cancers, such as breast, colon, and endometrial cancers, are differentially affected by BMI, depending on the menopausal status (6). Hormonal signals modulated by menopausal status, such as progesterone and estrogen, clearly influence the behavior of certain tumor types, such as those arising from breast, colon, and the endometrium (6). Because adipose tissue is the main source of aromatase (CYP19) in the postmenopausal state, it is not surprising to see a differential effect on hormone-dependent growth of transformed cells in obese vs. lean patients.
The contributions of adipocytes to tumor physiology may differ during the various stages of tumor progression. Although the tumor phenotype changes during progression, the question is whether the basic cellular physiology of the adipocyte is equally modified during tumor development, as an adaptive response to the altered requirements of the tumor. Based on histological observations, differentiated adipocytes that are present in the microenvironment of early-stage tumors are replaced by fibroblast-like preadipocytes and/or dedifferentiated adipocytes in later stages of tumor progression (27). Many studies have supported the concept of tumor-associated fibroblasts promoting tumor progression and metastasis through various signaling molecules, such as TGFβ1 and stromal cell-derived factor-1 (28, 29). To date, it is unclear whether this transition of the adipocyte to a more “fibroblastic” phenotype is induced by specific factors or whether it is simply a reflection of the high local energy demand that results in a depletion of lipid reserves within the stroma. Although we currently have limited mechanistic insights, fibroblast-like preadipocytes do indeed appear to exert a supportive role on the growth of tumor cells (27). This is particularly relevant in the context of stromal cell types that may further reactivate a latent embryonic program in tumor cells, which is referred to as “epithelial-to-mesenchymal transition” (EMT), a transition critically involved in increased motility of tumor cells (30, 31). The EMT process is induced by several cytokines and chemokines, including TGFβ, or by the expression of several developmentally important transcription factors that include Twist, Slug, and Snail. Receptor-mediated pathways, such as integrins, receptor tyrosine kinases, wnt, and TGFβ signals are crucial for the initiation of EMT (30, 31). Some of these key regulators of EMT (such as TGFβ, Twist, and Slug) are expressed by adipocytes and regulate adipogenesis under normal conditions (32–34). In this regard, it is striking that adipocyte-derived signaling molecules that regulate adipogenesis are also activated during EMT process and tumor progression. Some EMT-derived stromal cells are transdifferentiated to adipocytes, osteoblast, and chondrocyte-like mesenchymal stem cells (35). Nevertheless, direct experimental evidence to support the relevance of stromal adipocytes and/or fibroblast-like adipocyte precursor cells (preadipocytes) for the EMT process in tumor cells remains to be clarified.
From an adipose tissue physiology perspective, we understand that obesity-associated dysfunctional adipocytes behave differently relative to adipocytes in lean patients. We do not know, however, whether these lean adipocytes differentially interact with tumor cells and differentially influence immune and endothelial cell behavior over the course of tumor progression compared with obese adipocytes. We do appreciate that obese adipose tissue suffers from chronic hypoxia, which causes a major alteration in the local tumor milieu for infiltrating cancer cells (36, 37). Adipose tissues in obese individuals are hypoxic due to the excess demand for tissue expansion, which causes dysregulation of adipokine expression. This process is associated with chronic inflammation and ER stress (38). As a result of the hypoxia, fibroblast-like preadipocytes and dysfunctional inflamed stromal adipocytes become more prevalent locally. These apoptotic and/or the necrotic hypertrophic adipocytes are thus viewed as major contributors toward enhanced tumor progression and survival (Fig. 3).
Fig. 3.

Potential involvement of adipocytes in tumor progression. Metabolically healthy adipocytes are crucial to maintaining whole body energy metabolism. Obesity-induced dysregulation of adipocytes can be associated with tumor progression. Contributing factors include: 1) hypoxia due to inadequate expansion of adipocytes in obesity; 2) fibrosis, which is associated with dedifferentiation of stromal adipocytes into fibrogenic preadipocytes during the EMT process within the tumor microenvironment. Drastic remodeling of the extracellular matrix by dysfunctional adipocytes contributes to enhanced fibrosis; 3) increased adipocyte apoptosis/necrosis triggers macrophage infiltration into adipose tissues and obesity-dependent chronic inflammation; and 4) disturbance of local hormonal milieu. These changes promote a more “tumor-friendly” environment through dysregulation of adipokines and chemokines as well as hormones and lipid metabolites.
Many cancer patients suffer from cachexia at the later stages of tumor progression. The extent of this wasting syndrome depends on the tumor type and can also occur during chemotherapy. Weight loss mainly due to a loss of adipose tissue mass and to some extent muscle mass is a diagnostic marker for poor survival (39). Adipose tissue atrophy is a hallmark of cancer cachexia and causes additional comorbidities such as hyperlipidemia and insulin resistance, which may add to higher cancer mortalities and poorer outcomes. Independent of its association with cancer, we appreciate that partial (or severe) loss of body fat mass in the form of lipodystrophies can result in profound systemic metabolic abnormalities. These include chronic inflammation, hyperinsulinemia, hyperglycemia, and dyslipidemia, collectively arising as a result of the inability to properly store triglycerides (40). As such, lipodystrophies with their characteristic lack of adipose tissue share many symptoms associated with obesity. The mechanisms underlying cancer-associated lipodystrophy have not been clearly elucidated. Is this process tightly regulated during tumor progression or simply a reflection of loss of lipids due to high-energy demand?
Several attempts have been made in both mouse and human studies to explore tumor-associated adipose tissue remodeling. This process is under the control of transcriptional regulators, which involve CCAAT/enhancer binding protein α, sterol regulatory element binding protein-1c, hepatic nuclear factor-4, and proliferator-activated receptor-γ coactivator-1α. All of these factors play a role in cytoskeletal and extracellular matrix (ECM) remodeling (41, 42). In light of these observations, attempts to restore full functionality and metabolic flexibility for tumor-associated adipocytes represent an important therapeutic approach toward reducing the growth potential of tumor cells invading the stromal compartment (Fig. 3). Minimally, reducing the negative metabolic consequences of cachexia is associated with an extension of both quality and duration of life (43).
III. How Do Adipocytes Influence Cancer Biology?
A. Paracrine effects of dysregulated adipocytes
Obesity-associated alterations in paracrine signaling tend to drastically affect tumor types that have significant adipocyte content in the stromal microenvironment. Obesity-associated tumors, such as tumors originating in the colon, kidney, pancreas, esophageal, liver, thyroid, and endometrium, tend to experience the effects of local adipocytes, although they may not be in direct physical contact with the fat pads. For example, the submucosal layer of the esophagus, thyroid, and endometrium contain a respectable number of stromal adipocytes, and the kidney and pancreas are anatomically located in the retroperitoneum, close to neighboring fat pads. The colon is close to the abdominal fat; mammary epithelium is in direct contact with sc fat. Liver lipid stores increase under obese conditions, which contribute to alterations in local tumor microenvironment in the context of hepatocellular carcinoma. In contrast, cancers that have epidemiologically a less apparent association with obesity, e.g., lung cancer, are physically more distant from fat pads. Thus, interventions targeted at adipose tissue are likely to be more relevant for those tumors that have a positive association to obesity.
We consider two main adipocyte-derived paracrine pathways: ECM-mediated signaling and hypoxia-induced signaling. Both pathways are associated with chronic inflammation, fibrosis, angiogenesis, and insulin resistance at the level of adipocyte metabolism through modulation of insulin signaling, nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) pathways, TGFβ pathways, and unfolded protein response pathways (38, 44–51). Furthermore, the phenotypic transition of stromal adipocytes to fibroblast-like preadipocytes during tumor progression is an additional component of the changing tumor microenvironment (Fig. 3). The health of the stromal adipocytes (albeit not necessarily the absolute mass) is therefore a key determinant of the secretory profile of adipokines that adipocytes release upon tumor cells for either regression or growth.
1. Extracellular matrix and fibrosis
The adipose tissue ECM is an important component of the adipose tissue microenvironment that is constantly subjected to active modulation by metabolic status. Like many other organs, adipose tissue has a unique composition; it provides a scaffold for embedded blood vessels, nerve cells, fibroblasts, and immune cells. Unlike other tissues, however, the ECM of adipose tissue must remain flexible enough to accommodate rapid expansion during exposure to excess caloric intake and drastic reductions in tissue mass during caloric restriction. Adipocytes themselves secrete numerous molecules that modify the ECM, such as matrix metalloproteinases (MMPs), their tissue inhibitors, collagens, cathepsins, fibronectin, laminin, and osteonectin (52). Adipocytes actively reorganize the ECM during differentiation and periods of high demand on lipid storage, ultimately to support the hypertrophic expansion of cells. Notably, the ECM can directly regulate adipogenesis and adipose tissue metabolism (53). For instance, the global expression of ECM components and their modifiers, such as MMPs and tissue inhibitor of metalloproteinase-1in adipose tissues, are heavily modulated by obesity. As such, several of these genes are expressed differentially in an adipose tissue depot-specific manner (54, 55). This is associated with metabolic abnormalities in the obese state through a modulation of mechanical tension within tissues or signaling pathways. We have previously shown that the level of adipocyte-derived collagen type VI (COLVI) is markedly increased in obesity and diabetes. COLVI expression levels are associated with tissue fibrosis, resulting in metabolic dysfunction, such as insulin resistance (47). COLVI deficiency in mice in the background of obese animal models, such as ob/ob mice, causes improved metabolic profiles as a direct effect of a “softening” of the local ECM due to a reduction in adipose tissue fibrosis (47).
To gauge the relevance of adipose tissue-derived ECM components for tumor progression, two distinct aspects of adipose tissue ECM need to be considered. One is the direct physical impact due to increased levels of collagens, causing fibrotic stress. The other aspect relates to the crosstalk between stromal adipocytes and tumor cells, through ECM components functioning as signaling molecules. We have demonstrated that adipocyte-derived COLVI is involved in mammary tumor progression in vivo (10). Furthermore, lesion growth was significantly reduced in the absence of COLVI, presumably at least in part as a consequence of reducing the local fibrotic environment, with local fibrosis generally a condition associated with increased tumor growth (11). In addition to this, our recent work suggests that COLVI-derived cleavage products can act as potent growth stimulators for tumor cells (J. Park and P. E. Scherer, unpublished observations). The fibrotic components are particularly relevant for breast tissue where the increase in profibrotic fibroblasts is an epidemiologically well-established risk factor (56, 57). Additional epidemiological data support the involvement of tissue fibrosis in pancreatic and hepatic cancer progression, mediated by TGFβ and EMT pathways in combination with inflammation and hypoxia (58, 59). Despite the abundance of epidemiological evidence, the precise molecular mechanisms underlying the effects of fibrosis still remain to be explored. However, the metabolic changes associated with dysfunctional adipocytes in the tumor stroma contribute to local fibrosis through modulation of their ECM environment. Another example is MMP-11, whose levels are increased in obese visceral adipose tissues (54). Moreover, this up-regulation can directly promote the early tumor invasion process (60). Cancer cells induce MMP-11 secretion from adipocytes, thereby promoting tumor cell invasion due to ECM remodeling (27, 60). This highlights the importance of local, adipocyte-derived MMP for the tumor-associated remodeling process. Therefore, adipocyte-derived ECM components in the stroma seem to play a pertinent role in cancer development, primarily at the stage of invasion and stromal growth.
2. Hypoxia and angiogenesis
During adipose tissue expansion, hypoxia is prevalent; therefore, appropriate expansion of the adipose vasculature is vital for the overall integrity and health of the fat depot in both mice and humans (61). Although adipocytes can in principle induce vascularization through the secretion of angiogenic factors, such as leptin, hepatocyte growth factor, vascular endothelial growth factor, adiponectin, TGFβ, and angiopoietin (61), obese adipose tissue is frequently in a hypoxic state due to insufficient angiogenesis (38). Recent studies have suggested that obesity-associated hypoxia is the key source of several downstream events, including a decrease in insulin-signaling pathways (45) and an increase in NF-κB pathways (62), TGFβ pathways (50), and the unfolded protein response pathways (38) that are frequently observed in dysfunctional adipose tissue. This causes a direct dysregulation of adipokine secretion. Proteins such as adiponectin and plasminogen activator inhibitor type-1 (PAI-1) (38) are prominently affected in addition to alterations in glucose and lipid metabolism that eventually trigger fibrosis and chronic inflammation (38, 63). Phenomena related to hypoxia and angiogenesis are crucial for tumor growth due to the obvious impact they both have on tumor expansion and survival. Angiogenic factors and/or hypoxia-induced cytokines derived from adipocytes are actively involved in such processes. Angiogenesis is considered an attractive therapeutic area for obesity and its related metabolic disorders (64). However, this poses major therapeutic challenges: whereas proangiogenic interventions that support the expansion of adipose tissue are beneficial and lead to improvements in adipose tissue physiology, the very same approach can be counterproductive in the context of cancer treatment regimens that aim to curb the growth of a rapidly expanding tumor mass.
B. Endocrine effects of dysregulated adipocytes
Notably, endocrine signals such as adipokines, inflammatory cytokines, lipid metabolites, and other hormones are produced by adipose tissues both proximal and distal to the tumor mass. Tumor types that have less of a direct exposure to adipocytes are therefore mostly affected by the endocrine aspects of adipose tissue and less prone to paracrine effects, such as those derived from extracellular matrix constituents or hypoxia.
1. Adipokines
Adipocytes secrete different profiles of adipokines depending on the specific anatomical location (25) (Table 2). In addition, the overall health of the fat pad also affects secretory profiles (65). The quality and metabolic health of the fat pad may be a more important determinant than absolute fat mass as far as adipokines are concerned as well. Substantial epidemiological efforts have been directed toward the identification of association between adipokines and cancer incidence and progression. Critical adipokines include adiponectin, leptin, resistin, hepatocyte growth factor, TNFα, CCL2 (also known as monocyte chemotactic protein 1), and IL-6. This list is continuously expanding due to an ever-growing list of factors that are implicated in the metabolic dysregulation of adipocytes. Although none of the adipokines are exclusively produced in adipocytes, adiponectin and leptin are highly enriched in adipose tissue. Most other adipokines, such as TNFα, IL-6, and CCL2 are secreted from both adipocytes and macrophages, or from unrelated cell types, such as other immune cells. Here, we discuss the adipokines adiponectin and leptin, which are two of the most adipocyte-specific proteins. They are also the only two adipokines for which there are both epidemiological and preclinical data available in the context of tumor studies.
a. Adiponectin.
It has now been extensively documented that adiponectin expression is inversely correlated with obesity. Further studies have established that adiponectin has potent protective effects against insulin resistance, cardiovascular disease, and chronic inflammation due to its ability to improve systemic carbohydrate and lipid profiles (66). Adiponectin is secreted by adipocytes and circulates in several different complexes in plasma, which target multiple tissues expressing their receptors, including AdipoR1, AdipoR2 (67), or T-cadherin (also known as CDH13) (68). Each of the receptors displays differential affinities for the various forms of adiponectin; e.g., AdipoR1 confers a high affinity for globular adiponectin with low affinity for full-length adiponectin, whereas AdipoR2 exhibits intermediate affinity for both full-length and globular adiponectin (67). T-cadherin was identified as a receptor for hexameric (middle molecular weight oligomer) adiponectin and for the high-molecular form (high molecular weight oligomer) of adiponectin (68). Notably, the high molecular weight oligomer adiponectin complex is the major form responsible for the large number of metabolic functions observed for adiponectin (69). However, the specific contributions of the different forms of adiponectin in tumorigenesis remain to be fully elucidated.
Measurements of the plasma levels of adiponectin have been the primary focus of numerous epidemiological studies that highlight the strong correlation between insulin resistance, visceral obesity, and the potential negative associations with cancers, such as postmenopausal breast cancer, endometrial, colon, gastric, prostate, and renal cancers (70, 71). Interestingly, it has been observed that both benign and malignant tumors (for instance, colon, breast, lung, endometrial, and pancreatic cancers) all express AdipoR1 and AdipoR2 (72–76). T-Cadherin has shown to be expressed on tumor-associated endothelial cells (77), suggesting that adiponectin may exert a direct impact via the endothelium. Moreover, in a specific subset of tumors, including tumors of the colon, esophagus, and breast, several groups have emphasized the protective roles of adiponectin on tumorigenesis (78–80). Such effects have been postulated to be mediated by downstream effectors of AdipoR1 and AdipoR2, such as AMP kinase (AMPK) (78, 79) and c-Jun N-terminal kinase (80), which can activate cell proliferation, apoptosis, and inflammation in vitro. Conversely, more compelling data at the molecular level argue that in murine mammary cancer models, adiponectin and its receptors exert potent proangiogenic effects that promote tumor growth (77, 81, 82). These results highlight the differences between epidemiological associations (in which adiponectin reflects an inverse correlation to BMI) and the direct molecular effects on tumor growth, as highlighted by manipulation of adiponectin levels in vivo (77, 81, 82). An interesting new set of observations suggests that the established adiponectin receptors AdipoR1 and AdipoR2 contain (or are at least tightly associated with) a ceramidase activity (83). Upon stimulation, these receptors convert ceramides rapidly to downstream degradation products, such as sphingosines, that are rapidly converted to sphingosine-1-phosphate (S1P). Elevated S1P is associated with increased cell survival and higher local proangiogenic activity, a phenomenon that is indeed observed in mammary tumor mouse models in which adiponectin levels have been manipulated (81, 82). These exciting findings offer additional hope to overcome tumor-inherent resistance mechanisms to chemotherapeutic interventions. Widely used agents, such as doxorubicin, cause an elevation of tumor cell-associated ceramides (84). The adiponectin receptor-associated ceramidase activity critically counteracts this ceramide buildup. In the context of tumor studies, it would therefore be desirable to find small molecule inhibitors against these receptors. This is in sharp contrast to the efforts directed toward overall metabolic improvements outside the context of tumorigenesis, during which stimulation rather than inhibition of these receptors remains an attractive pharmacological goal.
b. Leptin.
Unlike adiponectin, leptin levels are directly proportional to the amount of body fat mass. Leptin plays a key role in energy homeostasis and appetite control through its action in the hypothalamus, where the leptin receptor (LEPR) is highly expressed (85). Beyond its central role on energy homeostasis, leptin is required for reproduction, plays an important role for T cells in the adaptive immune response, and positively influences angiogenesis and hematopoiesis through LEPR-mediated signaling pathways (86). Moreover, at least five different spliced forms of LEPR exist. However, it is the long form only of LEPR (LEPR-B) that can transduce downstream signaling pathways, such as activation of the phosphatidylinositol 3-kinase (PI3K), ERK1/2, and Janus kinase 2/signal transducer and activator of transcription 3 (85), all of which are well-established protumorigenic signaling pathways.
Leptin is the most extensively studied adipokine in cancer research related to obesity. The LEPR is highly expressed on many tumor cells, including those derived from mammary, pancreatic, and gastrointestinal tract, such as esophageal, gastric, and colon compared with matched normal tissue samples in which the receptor is expressed at very low levels or not at all (87, 88). Leptin has attracted ample attention over the years due to its potential function as a mitogenic, antiapoptotic, proangiogenic, and prometastatic agent, as observed in numerous in vitro studies. In contrast, in vivo studies evaluating the direct impact that leptin has on cancer development have been challenging. Genetic manipulation of leptin in mice (Lep, also known as ob) or its receptor (Lepr, also known as db) using ob/ob or db/db mutant mice as well as fa/fa Zucker rats carrying a LEPR mutant can significantly alter whole body metabolism, consequently resulting in hyperinsulinemia, hyperglycemia, and dyslipidemia (85, 89). Accordingly, the majority of studies utilizing obese rodent models such as ob/ob and db/db mice as well as fa/fa Zucker rats have not managed to differentiate between the systemic metabolic changes and the direct local effects of leptin on tumor development (90–92). Moreover, it has been demonstrated that no mammary tumors develop in the setting of ob/ob or db/db mice due to the defective, leptin-mediated mammary gland development (90, 91). A recent report showed, however, that restoration of the LEPR-B signaling pathway in the brain of db/db mice not only reverses the metabolic abnormalities of the observed original phenotype (such as obesity, diabetes, and infertility) but also restores conditions permissive for the mammary gland to develop, despite the continued absence of peripheral leptin signaling (93). In this setting, we have examined for the first time in vivo the precise role of peripheral LEPR-B signaling in relation to mammary tumor progression. Our observations indicate that LEPR-B-mediated pathways involving ERK1/2 and Janus kinase 2/signal transducer and activator of transcription 3 support tumor progression, metastasis, and cancer metabolism in the context of rat sarcoma (Ras)- and PI3K-dependent tumor progression (94). Moreover, leptin is a crucial mediator of enriched environment, an optimized condition for cerebral health through the learning and memory training-associated tumor remission (95). Enriched environment-stimulated hypothalamic brain-derived neurotropic factor selectively decreases leptin levels produced in adipocytes through β-adrenergic signaling, which results in the decrease of tumor growth (95). This suggests that the brain-adipose tissue axis through leptin signaling can play a role in tumor progression.
Although adiponectin is down-regulated in obese adipose tissues, leptin is up-regulated from the very same adipocytes. The possibilities of functional crosstalk between these two adipokines have been suggested; however, little is known about interplay between these two adipokines that could influence whole body homeostasis through systemic regulation of insulin, glucose, and lipid metabolism. Based on recent insights regarding the associations between circulating adipokine levels and cancer development, adipokines may exert their impact on cancer development in a fashion independent of obesity. For instance, adiponectin in circulation inversely correlates with obesity but promotes mammary tumor progression in vivo (81, 82). The discrepancy seen between the epidemiological association and molecular studies in adiponectin and breast cancer may be explained by proangiogenic properties of adiponectin, regardless of the level of obesity present. Effects may be augmented further by obesity due to additive effects of dysregulation of other adipokines, chronic inflammation, insulin resistance, and an up-regulation along the IGF-I axis.
2. Other hormones
a. The insulin-IGF-I axis.
An increase in insulin levels is a compensatory response to the insulin resistance frequently observed in obese subjects. Systemically, hyperinsulinemia has been proposed to have an impact on cancer cell behavior by affecting proliferation, survival, and angiogenesis through changes in the levels of sex hormones and adipokines in addition to changes along the insulin-IGF-I axis (96). The insulin-IGF-I axis has emerged as an attractive therapeutic target due to the ability of IGF-I to function as a potent growth factor contributing to tumor cell proliferation. Hyperinsulinemia augments IGF-I bioavailability through competitive binding to IGF binding proteins, which inhibit IGF-I downstream signaling (97).
b. Sex hormones.
Epidemiologically, a number of cancers (such as colon, endometrial, and breast cancer) are affected by menopausal status (6). Because sex hormone levels are altered by increasing fat mass, tumor cells derived from these cancers are particularly prone to be affected by obesity (6). After transition through menopause, the stromal preadipocyte fraction in adipose tissue becomes an important source of systemic estrogen due to the high local expression levels of aromatase (98), the key catalytic enzyme for estrogen production. This subsequently has an impact on tumor progression, in particular in the context of estrogen-dependent breast cancer (99). Tamoxifen, a competitive antagonist of estrogen, has been widely used in the treatment of hormone-responsive tumors in postmenopausal women (100). However, there are clear limits due to drug efficacy and side effects, such as thromboembolism and an increase in endometrial cancer risk (101).
The crosstalk between hormones such as estrogen, insulin/IGF-I, and adipokine-mediated signaling pathways has been considered as important target sites to treat hormone-responsive tumors in relation to obesity (Fig. 2). The crosstalk of insulin signaling with ceramide pathways and estrogen action offers many different nodes at which these signaling pathways interconnect (102). The recent description of the specific role that adiponectin and its receptors play in these pathways further suggests a strong obesity component mediated in part through adipokine action (83).
3. Chronic inflammation and the immune response
Adipocytes play a role in the innate and adaptive immune responses (65). Obesity is considered a chronic subclinical inflammatory disease (1, 103). This is due at least in part to an increase in infiltrating immune cells into dysfunctional adipose tissue, and macrophages rank prominently among those infiltrating immune cells. In particular, the visceral adipose tissue (104) is an important contributor toward higher levels of circulating proinflammatory cytokines and chemokines, such as colony-stimulating factor 1, CCL2, MIF1 (also known as HERPUD1), IL-6, TNFα, and leptin. Furthermore, adipocytes produce high levels of acute phase reactants, proteins that are released into circulation in response to local inflammatory processes. These include serum amyloid A-3 and α1 acid glycoprotein (orosomucoid). These chronic inflammatory conditions prevailing in adipose tissue are strongly associated with many aspects of the pathogenic sequelae of obesity-related metabolic disorders, including insulin resistance, type 2 diabetes, cardiovascular disease, and last but not least, cancer (105). There is no generally agreed-upon unifying model explaining the underlying initial steps leading to this inflammatory state. An increased rate of adipocyte death is a likely contributor (106–108). Once inflammation has been initiated, recent reports have revealed important links between the T-cell immune response and chronic inflammation in obesity (109–111). The CD8+ effector T cell (Teff) population is increased in visceral adipose tissues in obese subjects, whereas CD4+ regulatory T cells (Treg) are decreased, resulting in recruitment of macrophages into adipose tissue and the secretion of proinflammatory cytokines (TH1 type), rather than antiinflammatory cytokines (TH2 type) (109, 110). Thus, the net balance of the different T-cell populations plays a critical role in determining the inflammatory state of adipose tissues (110).
Similar to dysfunctional adipose tissue, tumor tissues are frequently infiltrated by multiple inflammatory cell types (9). An important question therefore relates to the possible connection between the subclinical inflammatory state of dysfunctional adipose tissue and tumor-associated inflammation. Paradoxically, inflammation is associated with both tumor suppression and tumor progression (112, 113). Inflammation is required for host immune surveillance to kill the cancer cells, whereas chronic inflammation has been shown to promote cancer progression and metastasis (114). Thus, the proper regulation of anti- vs. proinflammatory responses in the tumor microenvironment is crucial for the growth and survival of cancer cells. In this respect, the dysregulation of inflammatory cytokine production by adipose tissue has an impact on the host immune surveillance and has therefore a direct impact on tumor cell survival. These effects influence not only the local tumor microenvironment but also nonproximal tumor cells through systemic effects of endocrine signals. Moreover, the specific response of tumors to inflammation depends on the cancer cell type, location, and stage of the disease. Epidemiological studies have supported the idea that chronic systemic subclinical inflammation predisposes individuals to develop some cancer types, including colon, liver, stomach, intestinal, thyroid, prostate, and gastric cancers (115, 116). In fact, nonsteroidal antiinflammatory drugs (NSAIDs) have beneficial effects on the prevention and treatment of some cancers, such as colon and breast cancer (117, 118). Similarly, similar approaches are also effective for improvements in obesity-related metabolic diseases (119).
4. Lipid metabolites
In the obese, insulin-resistant state, an increased rate of lipolysis is observed in adipocytes. As a result, lipid metabolites such as FFA are elevated in circulation, which in turn exacerbates insulin resistance further. In addition, several lipid metabolites, such as diacylglycerol and ceramides that are released from adipocytes, serve as secondary messengers in signaling pathways that are involved in cell proliferation, growth, and apoptosis. Such key signaling pathways include the PI3K, the protein kinase C, and the NF-κB inflammatory pathways (84, 120, 121). A recent paper suggests that lipid-induced insulin resistance mediated by the Toll-like receptor 4 requires saturated fatty acid-induced ceramide biosynthesis (122), further establishing the tight link of elevated FFA and insulin resistance. However, whether an increase in systemic availability of FFA and related lipid metabolites leads to enhanced tumor progression through increased β-oxidation and reactive oxygen species generation or through the availability of key metabolites as signaling moieties remains to be established. Some recent observations do, however, link elevated FFA and tumor progression (12).
IV. Targeting Adipose Tissues in Cancer Prevention and Treatment
A. Targeting adipose tissues
1. Agents that target preadipocytes
Cloned bone marrow-derived mesenchymal stem cells can be induced to differentiate into mature cartilage, bone, muscle, or adipocytes under the appropriate conditions (123). Preadipocytes identified in all adipose tissues are thought to represent an intermediary cell type along the adipocyte differentiation axis between the bone marrow-derived mesenchymal cells and mature adipocytes. These preadipocytes are morphologically indistinguishable from fibroblasts, occur in regions of loose connective tissue, and are located in association with proliferating capillaries (124). In addition, these preadipocytes, along with other fibroblasts, may contribute to the fibrillar environment of the stroma of the mammary gland and other tissues.
a. Peroxisome proliferator-activated receptor-γ (PPARγ) agonists.
The mechanisms that drive mesenchymal stem cell commitment to a preadipocyte lineage are currently unknown; however, terminal differentiation of preadipocytes begins with growth arrest, followed by expression of a variety of genes related to glucose uptake and lipid processing (125). This later step is mediated by PPARγ signaling, a nuclear receptor that heterodimerizes with retinoid X receptor (126, 127). Pharmacological induction of preadipocyte differentiation improves systemic metabolic profiles by promoting glucose and fatty acid uptake into newly generated, insulin-sensitive adipocytes. From this perspective, similar to the phenomenon of “healthy” fat expansion leading to improvements in insulin sensitivity, an increased degree of adipogenesis may be protective in the context of tumor growth as well, provided that it is not associated with increased hypoxia and inflammation (128). PPARγ activation can be achieved pharmacologically with a variety of agents (Table 3). Furthermore, induction of adipocyte differentiation by PPARγ agonists reduces aromatase activity in cultured mammary preadipocytic stromal cells, an effect that could potentially reduce local estradiol levels (129).
Table 3.
Agents that target adipose tissues and systemic metabolism
| Agent | Target | Effects |
|---|---|---|
| Target adipose tissues to induce preadipocyte differentiation | ||
| TZDs (pioglitazone, rosiglitazone, troglitazone) | PPARγ | Induces preadipocyte differentiation, reduces inflammation, proangiogenic, antifibrotic (195–197) |
| Anandamide | PPARγ | An endogenous cannabinoid receptor ligand that can induce preadipocyte differentiation via PPARγ binding (198) |
| RWJ-348260 | PPARγ | Benzoxazinone is a non-TZD PPARγ agonist (142) |
| Angiotensin receptor blockers | PPARγ | Induced lipogenesis and adiponectin production (199) |
| Telmisartan | ||
| Irbesartan | ||
| Losartan | ||
| Cilostazol | PPARγ | A phosphodiesterase-3 inhibitor that inhibits platelet aggregation (138) |
| KR-62776 | PPARγ | Partial PPARγ agonist that interferes with subcellular localization (143) |
| Target adipose tissues to inhibit aromatase activity in preadipocytes | ||
| Aromatase inhibitor (anastrozole, letrozole, exemestane) | Aromatase | Inhibit aromatase activity to reduce estrogen production (131–135) |
| NSAID | COX2 | Inhibit COX2 results in decrease aromatase activity (130), decrease inflammation |
| Improve systemic metabolism | ||
| TZDs | PPARγ | Improve insulin sensitivity |
| Metformin | AMPK | Improve glucose (162) and lipid metabolism (166, 185); improve insulin sensitivity (161); decrease proinflammatory cytokines in diabetes (159) or polycystic ovary patients (160) |
b. Aromatase inhibitors.
Most of the circulating estradiol in postmenopausal women is derived from the peripheral conversion of androgens by the enzyme aromatase, which is highly expressed by preadipocytes and regulated by glucocorticoids, class I cytokines, and the cyclooxygenase (COX) product prostaglandin E2. As such, the use of nonsteroidal antiinflammatory agents, which inhibit COX, has been associated with reduced circulating estradiol levels in postmenopausal women (130). Type I (steroidal) and type II (nonsteroidal) aromatase inhibitors have been developed as tamoxifen replacements and undergone clinical trials. Third-generation aromatase inhibitors, such as anastrozole, letrozole, and exemestane, have shown higher efficacy and fewer side effects than tamoxifen (131–135). Aromatase activity is further increased through various adipokines, such as IL-6, TNFα, and leptin, whereas adiponectin has been shown to decrease aromatase activity through the LKB1/AMPK pathway (136, 137). An increased level of aromatase activity, in combination with a dysregulated adipokine profile, may therefore contribute to an overall environment favoring tumor progression in an obese setting. From this perspective, several additional approaches have been investigated, aimed at pharmacologically lowering aromatase activity. Experimental treatments with PPARγ agonists (129), in addition to metformin (139), have been examined in this context. In both cases, the hope was to negatively interfere with the growth stimulatory environment of hormone-sensitive tumors (100).
B. Targeting systemic metabolic effects
1. Targeting dysfunctional adipocytes
As described in the preceding sections, obesity-associated insulin resistance in adipocytes is associated with alterations in growth factors, adipokines, and inflammatory markers that can be linked to cancer development and progression. Therapeutic strategies to normalize the systemic metabolic effects due to adipocyte dysfunction in obesity have been investigated to treat the obesity-related cancers (Table 3).
2. Agents that target metabolic abnormalities
a. PPARγ agonist.
Similar to PPARγ-mediated signaling has local consequences on terminal adipocyte differentiation along with antiinflammatory properties. Among the most thoroughly studied PPARγ agonists are the thiazolidinediones (TZDs), which have been used in the treatment of type 2 diabetes since 1997, with early trials going back as far as 1994 (140). The risks and side effects of some TZDs may limit their use in cancer prevention and treatment. There are currently two Food and Drug Administration-approved TZD compounds: rosiglitazone and pioglitazone, with restrictions for the use of rosiglitazone recently put in place due to cardiovascular side effects. Newer TZDs, such as rivoglitazone (141), and non-TZD PPARγ agonists such as RWJ-348260 (142) and KR-62776 (143) are currently being evaluated.
The use and effectiveness of PPARγ agonists in the cancer setting is complex and highly dependent on tumor type and tumor stage. PPARγ can exert antiproliferative effects through interactions with cyclin D1 (144), and β-catenin (145) can induce terminal differentiation and can be potently antiinflammatory. On the other hand, through its ability to induce angiogenesis, it may enable a more proliferative response in a tumor setting. TZD monotherapy in clinical trials did not show any beneficial effects on advanced epithelial tumor cells (146–148). However, combining TZDs with platinum-based chemotherapy has been shown to increase susceptibility to cytotoxic treatments in a number of different tumor cell lines (149). Despite the complexity and nuanced response depending on the type of cancer and transformation status of the cell, PPARγ remains an attractive and widely studied pharmacological target for cancer studies. We have summarized the ongoing efforts regarding PPARγ activation in Table 3.
b. Metformin.
Metformin is also a potent insulin sensitizer, activating the insulin receptor through enhanced tyrosine phosphorylation and inhibiting insulin receptor dephosphorylation by inhibiting the key enzyme for this step, protein tyrosine phosphatase 1B (150). Metformin also activates AMPK through its effects on LKB1 (151). This may be related to effects exerted by metformin on mitochondrial function. In addition, several studies have shown that metformin affects reduction of plasma levels of proinflammatory cytokines, such as C-reactive protein levels in overweight prediabetic patients (152) and TNFα levels in polycystic ovary syndrome patients (153). Hence, a reduction of these proinflammatory markers may contribute to the antitumorigenic properties of metformin.
Recent studies have evaluated the antitumorigenic effects of metformin in the treatment of human cancers, such as pancreas, prostate, colon, and breast tumors (154, 155). A retrospective study recently suggested that diabetic breast cancer patients that were administered metformin had a significantly greater response rate to neoadjuvant chemotherapy than either diabetic women without metformin treatment or nondiabetic women (156). The inhibitory effect of metformin on tumor growth may in part be mediated by mammalian target of rapamycin, PI3K, and AMPK pathways (154, 155, 157, 158). Although metformin directly targets cancer cells through activation of the LKB1-AMPK pathway, the efficacy of metformin in human cancers in diabetic patients may be associated with the metformin-induced overall improvement of metabolism with a concomitant normalization of adipocyte function through regulation of adipokines such as leptin (159) and PAI-1 (160), the insulin signaling pathway (161), glucose uptake (162), and lipid metabolism (163, 164) mediated by ACC2 phosphorylation and proliferator-activated receptor-γ coactivator-1α up-regulation, at least in part (163–165) (Fig. 4). A more detailed analysis of these phenomena will have to be performed in preclinical models to describe the detailed molecular mechanisms of this surprising metformin-cancer connection.
Fig. 4.

Pharmacological options for modulating adipose tissue-associated hormonal and metabolic effects. Stromal preadipocytes of adipose tissue, which are derived from mesenchymal stem cells or EMT, are a major source of aromatase activity. To modulate hormonal effects of adipose tissues, two different approaches have been entertained: 1) induction of preadipocyte differentiation to healthy adipocytes; and 2) inhibition of aromatase activity. A variety of pharmacological agents are available to induce preadipocyte differentiation. Most agents that induce preadipocyte differentiation do so through PPARγ-dependent mechanisms. This results in increased adiponectin levels, reduced inflammation, and improved insulin sensitivity. Steroidal (type I) and nonsteroidal (type II) aromatase inhibitors and NSAID have been widely used to inhibit aromatase activity. From a metabolism perspective, systemic metabolic abnormalities due to dysfunctional adipocytes in obesity are key risk factors of tumor progression. Agents that normalize metabolic abnormalities, such as insulin resistance, chronic inflammation, and adipokine dysregulation, are potentially useful to treat obesity-related cancers. In this regard, PPARγ agonists and metformin, both widely used agents in the diabetes clinic, are under investigation. CRP, C-reactive protein.
V. Conclusions
In the physiological state associated with obesity, adipocytes undergo dramatic alterations due to various metabolic adaptations that occur during adipose tissue hypertrophy. The reason for the increased tumor burden in obese patients is likely related to dysfunctional metabolic pathways prevalent in the tumor directly surrounding the obese adipocytes. There is a critical need to better establish the impact that dysfunctional stromal adipocytes in the tumor microenvironment exert on tumor physiology. It is also important to distinguish between the metabolically “healthy” and the “unhealthy” obese phenotype, with less emphasis on the pure metrics strictly reflecting fat mass, such as BMI. At present, most observations suggest that obesity is likely associated with tumor progression rather than tumor initiation. Nevertheless, the chronic inflammation status prevalent in obesity may be a factor contributing to tumorigenesis. At this point, the field remains somewhat immature with many descriptive studies prevailing that generally lack detailed mechanistic insights. However, there is ample evidence establishing that adipocytes play a critical role in shaping the local tumor microenvironment as well as the systemic metabolic parameters that tumor cells are exposed to.
In summary, we propose four distinct areas in which dysfunctional, obese stromal adipose tissue exerts growth-promoting effects on tumors: 1) increased presence of hypoxic adipocytes; 2) enhanced fibrosis—ECM remodeling by dysfunctional adipocytes and/or dedifferentiation of stromal adipocytes to preadipocytes result in increased fibroblastic content with increased local expression of EMT markers, which are generally associated with enhanced carcinoma cell migration and survival, thereby facilitating malignant progression; 3) increased apoptosis/necrosis of stromal adipocytes, leading to the recruitment of macrophages and immune cells to adipose tissues, triggering chronic inflammation that offers an environment promoting tumor progression; and 4) disturbance of hormonal regulation, including insulin/IGF-I and sex hormones (Fig. 3). All four events occur much more frequently in obese fat pads and have a profound effect on local adipokine and proinflammatory cytokine profiles that contribute to tumor cell behavior through both paracrine and endocrine changes (Fig. 1). This consequently opens the door to numerous exciting experimental approaches that could indeed be used to mitigate, or potentially negate, the negative effects of dysfunctional adipose tissue on tumor growth.
Acknowledgments
We thank Christine M. Kusminski for critical comments on the manuscript.
Our work is supported by National Institutes of Health Grants R01-CA112023, R01-DK55758, P01-DK088761, and RC1 DK086629 (to P.E.S.). J.P. is supported by a fellowship from the Department of Defense (USAMRMC BC085909).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AMPK
- AMP kinase
- BMI
- body mass index
- COLVI
- collagen type VI
- COX
- cyclooxygenase
- ECM
- extracellular matrix
- EMT
- epithelial-to-mesenchymal transition
- ER
- endoplasmic reticulum
- FFA
- free fatty acid
- LEPR
- leptin receptor
- LEPR-B
- long form of LEPR
- MMP
- matrix metalloproteinase
- NF-κB
- nuclear factor κ-light-chain-enhancer of activated B cells
- NSAID
- nonsteroidal antiinflammatory drug
- PAI-1
- plasminogen activator inhibitor type-1
- PI3K
- phosphatidylinositol 3-kinase
- PPAR
- peroxisome proliferator-activated receptor
- S1P
- sphingosine-1-phosphate
- TZD
- thiazolidinedione.
References
- 1. Rajala MW, Scherer PE. 2003. Minireview: the adipocyte—at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 144:3765–3773 [DOI] [PubMed] [Google Scholar]
- 2. Lionetti L, Mollica MP, Lombardi A, Cavaliere G, Gifuni G, Barletta A. 2009. From chronic overnutrition to insulin resistance: the role of fat-storing capacity and inflammation. Nutr Metab Cardiovasc Dis 19:146–152 [DOI] [PubMed] [Google Scholar]
- 3. Guilherme A, Virbasius JV, Puri V, Czech MP. 2008. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 9:367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blüher M. 2009. Adipose tissue dysfunction in obesity. Exp Clin Endocrinol Diabetes 117:241–250 [DOI] [PubMed] [Google Scholar]
- 5. Calle EE, Kaaks R. 2004. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer 4:579–591 [DOI] [PubMed] [Google Scholar]
- 6. Reeves GK, Pirie K, Beral V, Green J, Spencer E, Bull D. 2007. Cancer incidence and mortality in relation to body mass index in the Million Women Study: cohort study. BMJ 335:1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M. 2008. Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 371:569–578 [DOI] [PubMed] [Google Scholar]
- 8. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. 2003. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 348:1625–1638 [DOI] [PubMed] [Google Scholar]
- 9. Qian BZ, Pollard JW. 2010. Macrophage diversity enhances tumor progression and metastasis. Cell 141:39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iyengar P, Combs TP, Shah SJ, Gouon-Evans V, Pollard JW, Albanese C, Flanagan L, Tenniswood MP, Guha C, Lisanti MP, Pestell RG, Scherer PE. 2003. Adipocyte- secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene 22:6408–6423 [DOI] [PubMed] [Google Scholar]
- 11. Iyengar P, Espina V, Williams TW, Lin Y, Berry D, Jelicks LA, Lee H, Temple K, Graves R, Pollard J, Chopra N, Russell RG, Sasisekharan R, Trock BJ, Lippman M, Calvert VS, Petricoin EF, 3rd, Liotta L, Dadachova E, Pestell RG, Lisanti MP, Bonaldo P, Scherer PE. 2005. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J Clin Invest 115:1163–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. 2010. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140:49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hanahan D, Weinberg RA. 2000. The hallmarks of cancer. Cell 100:57–70 [DOI] [PubMed] [Google Scholar]
- 14. Hanahan D, Weinberg RA. 2011. Hallmarks of cancer: the next generation. Cell 144:646–674 [DOI] [PubMed] [Google Scholar]
- 15. Castellot JJ, Jr, Karnovsky MJ, Spiegelman BM. 1982. Differentiation-dependent stimulation of neovascularization and endothelial cell chemotaxis by 3T3 adipocytes. Proc Natl Acad Sci USA 79:5597–5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vander Heiden MG, Cantley LC, Thompson CB. 2009. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hsu PP, Sabatini DM. 2008. Cancer cell metabolism: Warburg and beyond. Cell 134:703–707 [DOI] [PubMed] [Google Scholar]
- 18. Thomson CA, Thompson PA. 2009. Dietary patterns, risk and prognosis of breast cancer. Future Oncol 5:1257–1269 [DOI] [PubMed] [Google Scholar]
- 19. Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, Blomqvist L, Hoffstedt J, Näslund E, Britton T, Concha H, Hassan M, Rydén M, Frisén J, Arner P. 2008. Dynamics of fat cell turnover in humans. Nature 453:783–787 [DOI] [PubMed] [Google Scholar]
- 20. Rosen ED, MacDougald OA. 2006. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol 7:885–896 [DOI] [PubMed] [Google Scholar]
- 21. Kai S, Kusminski CM, Scherer PE. 2011 Adipose tissue remodeling and obesity. J Clin Invest 10.1172/JCI45887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wellen KE, Hotamisligil GS. 2005. Inflammation, stress, and diabetes. J Clin Invest 115:1111–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS. 2004. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461 [DOI] [PubMed] [Google Scholar]
- 24. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. 2004. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114:1752–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ibrahim MM. 2010 Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev 11:11–18 [DOI] [PubMed] [Google Scholar]
- 26. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. 2010. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140:197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Motrescu ER, Rio MC. 2008. Cancer cells, adipocytes and matrix metalloproteinase 11: a vicious tumor progression cycle. Biol Chem 389:1037–1041 [DOI] [PubMed] [Google Scholar]
- 28. Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. 2005 Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121:335–348 [DOI] [PubMed] [Google Scholar]
- 29. Oft M, Heider KH, Beug H. 1998. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol 8:1243–1252 [DOI] [PubMed] [Google Scholar]
- 30. Thiery JP, Sleeman JP. 2006. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 7:131–142 [DOI] [PubMed] [Google Scholar]
- 31. Peinado H, Olmeda D, Cano A. 2007. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 7:415–428 [DOI] [PubMed] [Google Scholar]
- 32. Pérez-Mancera PA, Bermejo-Rodríguez C, González-Herrero I, Herranz M, Flores T, Jiménez R, Sánchez-García I. 2007. Adipose tissue mass is modulated by SLUG (SNAI2). Hum Mol Genet 16:2972–2986 [DOI] [PubMed] [Google Scholar]
- 33. Zamani N, Brown CW. 20 December 2010 Emerging roles for the transforming growth factor-β superfamily in regulating adiposity and energy expenditure. Endocr Rev 10.1210/er.2010-0018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pettersson AT, Mejhert N, Jernås M, Carlsson LM, Dahlman I, Laurencikiene J, Arner P, Rydén M. 2011. Twist1 in human white adipose tissue and obesity. J Clin Endocrinol Metab 96:133–141 [DOI] [PubMed] [Google Scholar]
- 35. Battula VL, Evans KW, Hollier BG, Shi Y, Marini FC, Ayyanan A, Wang RY, Brisken C, Guerra R, Andreeff M, Mani SA. 2010. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells 28:1435–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yin J, Gao Z, He Q, Zhou D, Guo Z, Ye J. 2009. Role of hypoxia in obesity-induced disorders of glucose and lipid metabolism in adipose tissue. Am J Physiol Endocrinol Metab 296:E333–E342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tajima R, Kawaguchi N, Horino Y, Takahashi Y, Toriyama K, Inou K, Torii S, Kitagawa Y. 2001. Hypoxic enhancement of type IV collagen secretion accelerates adipose conversion of 3T3–L1 fibroblasts. Biochim Biophys Acta 1540:179–187 [DOI] [PubMed] [Google Scholar]
- 38. Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, Shimomura I. 2007. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56:901–911 [DOI] [PubMed] [Google Scholar]
- 39. Tisdale MJ. 2002. Cachexia in cancer patients. Nat Rev Cancer 2:862–871 [DOI] [PubMed] [Google Scholar]
- 40. Simha V, Garg A. 2006. Lipodystrophy: lessons in lipid and energy metabolism. Curr Opin Lipidol 17:162–169 [DOI] [PubMed] [Google Scholar]
- 41. Bing C, Russell S, Becket E, Pope M, Tisdale MJ, Trayhurn P, Jenkins JR. 2006. Adipose atrophy in cancer cachexia: morphologic and molecular analysis of adipose tissue in tumour-bearing mice. Br J Cancer 95:1028–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dahlman I, Mejhert N, Linder K, Agustsson T, Mutch DM, Kulyte A, Isaksson B, Permert J, Petrovic N, Nedergaard J, Sjölin E, Brodin D, Clement K, Dahlman-Wright K, Rydén M, Arner P. 2010. Adipose tissue pathways involved in weight loss of cancer cachexia. Br J Cancer 102:1541–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Coss CC, Bohl CE, Dalton JT. 2011. Cancer cachexia therapy: a key weapon in the fight against cancer. Curr Opin Clin Nutr Metab Care 14:268–273 [DOI] [PubMed] [Google Scholar]
- 44. Kammoun HL, Hainault I, Ferré P, Foufelle F. 2009. Nutritional related liver disease: targeting the endoplasmic reticulum stress. Curr Opin Clin Nutr Metab Care 12:575–582 [DOI] [PubMed] [Google Scholar]
- 45. Regazzetti C, Peraldi P, Grémeaux T, Najem-Lendom R, Ben-Sahra I, Cormont M, Bost F, Le Marchand-Brustel Y, Tanti JF, Giorgetti-Peraldi S. 2009. Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes 58:95–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Elsharkawy AM, Mann DA. 2007. Nuclear factor-κB and the hepatic inflammation-fibrosis-cancer axis. Hepatology 46:590–597 [DOI] [PubMed] [Google Scholar]
- 47. Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, Scherer PE. 2009. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol 29:1575–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Q, Hata A, Kosugi C, Kataoka N, Funaki M. 2010. The density of extracellular matrix proteins regulates inflammation and insulin signaling in adipocytes. FEBS Lett 584:4145–4150 [DOI] [PubMed] [Google Scholar]
- 49. Huang G, Ge G, Wang D, Gopalakrishnan B, Butz DH, Colman RJ, Nagy A, Greenspan DS. 2011. α3(V) Collagen is critical for glucose homeostasis in mice due to effects in pancreatic islets and peripheral tissues. J Clin Invest 121:769–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou S, Lechpammer S, Greenberger JS, Glowacki J. 2005. Hypoxia inhibition of adipocytogenesis in human bone marrow stromal cells requires transforming growth factor-β/Smad3 signaling. J Biol Chem 280:22688–22696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Spencer M, Yao-Borengasser A, Unal R, Rasouli N, Gurley CM, Zhu B, Peterson CA, Kern PA. 2010. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. Am J Physiol Endocrinol Metab 299:E1016–E1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Halberg N, Wernstedt-Asterholm I, Scherer PE. 2008. The adipocyte as an endocrine cell. Endocrinol Metab Clin North Am 37:753–768, x-xi [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Taleb S, Cancello R, Clément K, Lacasa D. 2006. Cathepsin s promotes human preadipocyte differentiation: possible involvement of fibronectin degradation. Endocrinology 147:4950–4959 [DOI] [PubMed] [Google Scholar]
- 54. Maquoi E, Munaut C, Colige A, Collen D, Lijnen HR. 2002. Modulation of adipose tissue expression of murine matrix metalloproteinases and their tissue inhibitors with obesity. Diabetes 51:1093–1101 [DOI] [PubMed] [Google Scholar]
- 55. Chavey C, Mari B, Monthouel MN, Bonnafous S, Anglard P, Van Obberghen E, Tartare-Deckert S. 2003. Matrix metalloproteinases are differentially expressed in adipose tissue during obesity and modulate adipocyte differentiation. J Biol Chem 278:11888–11896 [DOI] [PubMed] [Google Scholar]
- 56. Boyd NF, Rommens JM, Vogt K, Lee V, Hopper JL, Yaffe MJ, Paterson AD. 2005. Mammographic breast density as an intermediate phenotype for breast cancer. Lancet Oncol 6:798–808 [DOI] [PubMed] [Google Scholar]
- 57. Van den Eynden GG, Colpaert CG, Couvelard A, Pezzella F, Dirix LY, Vermeulen PB, Van Marck EA, Hasebe T. 2007. A fibrotic focus is a prognostic factor and a surrogate marker for hypoxia and (lymph) angiogenesis in breast cancer: review of the literature and proposal on the criteria of evaluation. Histopathology 51:440–451 [DOI] [PubMed] [Google Scholar]
- 58. Kaminska B, Wesolowska A, Danilkiewicz M. 2005. TGFβ signalling and its role in tumour pathogenesis. Acta Biochim Pol 52:329–337 [PubMed] [Google Scholar]
- 59. Radisky DC, Kenny PA, Bissell MJ. 2007. Fibrosis and cancer: do myofibroblasts come also from epithelial cells via EMT? J Cell Biochem 101:830–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Andarawewa KL, Motrescu ER, Chenard MP, Gansmuller A, Stoll I, Tomasetto C, Rio MC. 2005. Stromelysin-3 is a potent negative regulator of adipogenesis participating to cancer cell-adipocyte interaction/crosstalk at the tumor invasive front. Cancer Res 65:10862–10871 [DOI] [PubMed] [Google Scholar]
- 61. Cao Y. 2007. Angiogenesis modulates adipogenesis and obesity. J Clin Invest 117:2362–2368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ye J, Gao Z, Yin J, He Q. 2007. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab 293:E1118–E1128 [DOI] [PubMed] [Google Scholar]
- 63. Wood IS, de Heredia FP, Wang B, Trayhurn P. 2009. Cellular hypoxia and adipose tissue dysfunction in obesity. Proc Nutr Soc 68:370–377 [DOI] [PubMed] [Google Scholar]
- 64. Bråkenhielm E, Cao R, Gao B, Angelin B, Cannon B, Parini P, Cao Y. 2004. Angiogenesis inhibitor, TNP-470, prevents diet-induced and genetic obesity in mice. Circ Res 94:1579–1588 [DOI] [PubMed] [Google Scholar]
- 65. Tilg H, Moschen AR. 2006. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 6:772–783 [DOI] [PubMed] [Google Scholar]
- 66. Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. 2001. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 7:947–953 [DOI] [PubMed] [Google Scholar]
- 67. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. 2003. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423:762–769 [DOI] [PubMed] [Google Scholar]
- 68. Hug C, Wang J, Ahmad NS, Bogan JS, Tsao TS, Lodish HF. 2004. T-cadherin is a receptor for hexameric and high- molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci USA 101:10308–10313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pajvani UB, Hawkins M, Combs TP, Rajala MW, Doebber T, Berger JP, Wagner JA, Wu M, Knopps A, Xiang AH, Utzschneider KM, Kahn SE, Olefsky JM, Buchanan TA, Scherer PE. 2004. Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. J Biol Chem 279:12152–12162 [DOI] [PubMed] [Google Scholar]
- 70. Kelesidis I, Kelesidis T, Mantzoros CS. 2006. Adiponectin and cancer: a systematic review. Br J Cancer 94:1221–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Spyridopoulos TN, Petridou ET, Skalkidou A, Dessypris N, Chrousos GP, Mantzoros CS. 2007. Low adiponectin levels are associated with renal cell carcinoma: a case-control study. Int J Cancer 120:1573–1578 [DOI] [PubMed] [Google Scholar]
- 72. Yoneda K, Tomimoto A, Endo H, Iida H, Sugiyama M, Takahashi H, Mawatari H, Nozaki Y, Fujita K, Yoneda M, Inamori M, Nakajima N, Wada K, Nagashima Y, Nakagama H, Uozaki H, Fukayama M, Nakajima A. 2008. Expression of adiponectin receptors, AdipoR1 and AdipoR2, in normal colon epithelium and colon cancer tissue. Oncol Rep 20:479–483 [PubMed] [Google Scholar]
- 73. Petridou ET, Mitsiades N, Gialamas S, Angelopoulos M, Skalkidou A, Dessypris N, Hsi A, Lazaris N, Polyzos A, Syrigos C, Brennan AM, Tseleni-Balafouta S, Mantzoros CS. 2007. Circulating adiponectin levels and expression of adiponectin receptors in relation to lung cancer: two case-control studies. Oncology 73:261–269 [DOI] [PubMed] [Google Scholar]
- 74. Takahata C, Miyoshi Y, Irahara N, Taguchi T, Tamaki Y, Noguchi S. 2007. Demonstration of adiponectin receptors 1 and 2 mRNA expression in human breast cancer cells. Cancer Lett 250:229–236 [DOI] [PubMed] [Google Scholar]
- 75. Takemura Y, Osuga Y, Yamauchi T, Kobayashi M, Harada M, Hirata T, Morimoto C, Hirota Y, Yoshino O, Koga K, Yano T, Kadowaki T, Taketani Y. 2006. Expression of adiponectin receptors and its possible implication in the human endometrium. Endocrinology 147:3203–3210 [DOI] [PubMed] [Google Scholar]
- 76. Dalamaga M, Migdalis I, Fargnoli JL, Papadavid E, Bloom E, Mitsiades N, Karmaniolas K, Pelecanos N, Tseleni-Balafouta S, Dionyssiou-Asteriou A, Mantzoros CS. 2009. Pancreatic cancer expresses adiponectin receptors and is associated with hypoleptinemia and hyperadiponectinemia: a case-control study. Cancer Causes Control 20:625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hebbard LW, Garlatti M, Young LJ, Cardiff RD, Oshima RG, Ranscht B. 2008. T-cadherin supports angiogenesis and adiponectin association with the vasculature in a mouse mammary tumor model. Cancer Res 68:1407–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kim KY, Baek A, Hwang JE, Choi YA, Jeong J, Lee MS, Cho DH, Lim JS, Kim KI, Yang Y. 2009. Adiponectin-activated AMPK stimulates dephosphorylation of AKT through protein phosphatase 2A activation. Cancer Res 69:4018–4026 [DOI] [PubMed] [Google Scholar]
- 79. Fujisawa T, Endo H, Tomimoto A, Sugiyama M, Takahashi H, Saito S, Inamori M, Nakajima N, Watanabe M, Kubota N, Yamauchi T, Kadowaki T, Wada K, Nakagama H, Nakajima A. 2008. Adiponectin suppresses colorectal carcinogenesis under the high-fat diet condition. Gut 57:1531–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rubenstein JH, Kao JY, Madanick RD, Zhang M, Wang M, Spacek MB, Donovan JL, Bright SD, Shaheen NJ. 2009. Association of adiponectin multimers with Barrett's oesophagus. Gut 58:1583–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Landskroner-Eiger S, Qian B, Muise ES, Nawrocki AR, Berger JP, Fine EJ, Koba W, Deng Y, Pollard JW, Scherer PE. 2009. Proangiogenic contribution of adiponectin toward mammary tumor growth in vivo. Clin Cancer Res 15:3265–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Denzel MS, Hebbard LW, Shostak G, Shapiro L, Cardiff RD, Ranscht B. 2009. Adiponectin deficiency limits tumor vascularization in the MMTV-PyV-mT mouse model of mammary cancer. Clin Cancer Res 15:3256–3264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, Wade MR, Tenorio VM, Kuo MS, Brozinick JT, Zhang BB, Birnbaum MJ, Summers SA, Scherer PE. 2011. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 17:55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Reynolds CP, Maurer BJ, Kolesnick RN. 2004. Ceramide synthesis and metabolism as a target for cancer therapy. Cancer Lett 206:169–180 [DOI] [PubMed] [Google Scholar]
- 85. Friedman JM, Halaas JL. 1998. Leptin and the regulation of body weight in mammals. Nature 395:763–770 [DOI] [PubMed] [Google Scholar]
- 86. Muoio DM, Lynis Dohm G. 2002. Peripheral metabolic actions of leptin. Best Pract Res Clin Endocrinol Metab 16:653–666 [DOI] [PubMed] [Google Scholar]
- 87. Howard JM, Pidgeon GP, Reynolds JV. 2010. Leptin and gastro-intestinal malignancies. Obes Rev 11:863–874 [DOI] [PubMed] [Google Scholar]
- 88. Ishikawa M, Kitayama J, Nagawa H. 2004. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin Cancer Res 10:4325–4331 [DOI] [PubMed] [Google Scholar]
- 89. Truett GE, Bahary N, Friedman JM, Leibel RL. 1991. Rat obesity gene fatty (fa) maps to chromosome 5: evidence for homology with the mouse gene diabetes (db). Proc Natl Acad Sci USA 88:7806–7809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cleary MP, Juneja SC, Phillips FC, Hu X, Grande JP, Maihle NJ. 2004. Leptin receptor-deficient MMTV-TGF-α/Lepr(db)Lepr(db) female mice do not develop oncogene-induced mammary tumors. Exp Biol Med (Maywood) 229:182–193 [DOI] [PubMed] [Google Scholar]
- 91. Cleary MP, Phillips FC, Getzin SC, Jacobson TL, Jacobson MK, Christensen TA, Juneja SC, Grande JP, Maihle NJ. 2003. Genetically obese MMTV-TGF-α/Lep(ob)Lep(ob) female mice do not develop mammary tumors. Breast Cancer Res Treat 77:205–215 [DOI] [PubMed] [Google Scholar]
- 92. Hakkak R, Holley AW, Macleod SL, Simpson PM, Fuchs GJ, Jo CH, Kieber-Emmons T, Korourian S. 2005. Obesity promotes 7,12-dimethylbenz(a)anthracene-induced mammary tumor development in female zucker rats. Breast Cancer Res 7:R627–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. de Luca C, Kowalski TJ, Zhang Y, Elmquist JK, Lee C, Kilimann MW, Ludwig T, Liu SM, Chua SC., Jr 2005. Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J Clin Invest 115:3484–3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Park J, Kusminski CM, Chua SC, Scherer PE. 2010. Leptin receptor signaling supports cancer cell metabolism through suppression of mitochondrial respiration in vivo. Am J Pathol 177:3133–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cao L, Liu X, Lin EJ, Wang C, Choi EY, Riban V, Lin B, During MJ. 2010. Environmental and genetic activation of a brain-adipocyte BDNF/leptin axis causes cancer remission and inhibition. Cell 142:52–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Pollak M. 2008. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer 8:915–928 [DOI] [PubMed] [Google Scholar]
- 97. Renehan AG, Frystyk J, Flyvbjerg A. 2006. Obesity and cancer risk: the role of the insulin-IGF axis. Trends Endocrinol Metab 17:328–336 [DOI] [PubMed] [Google Scholar]
- 98. Simpson ER, Mahendroo MS, Means GD, Kilgore MW, Hinshelwood MM, Graham-Lorence S, Amarneh B, Ito Y, Fisher CR, Michael MD, Mendelson CR, Bulun SE. 1994. Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocr Rev 15:342–355 [DOI] [PubMed] [Google Scholar]
- 99. Simpson ER, Misso M, Hewitt KN, Hill RA, Boon WC, Jones ME, Kovacic A, Zhou J, Clyne CD. 2005. Estrogen—the good, the bad, and the unexpected. Endocr Rev 26:322–330 [DOI] [PubMed] [Google Scholar]
- 100. Johnston SR, Dowsett M. 2003. Aromatase inhibitors for breast cancer: lessons from the laboratory. Nat Rev Cancer 3:821–831 [DOI] [PubMed] [Google Scholar]
- 101. Cuzick J, Powles T, Veronesi U, Forbes J, Edwards R, Ashley S, Boyle P. 2003. Overview of the main outcomes in breast-cancer prevention trials. Lancet 361:296–300 [DOI] [PubMed] [Google Scholar]
- 102. Holland WL, Summers SA. 2008. Sphingolipids, insulin resistance, and metabolic disease: new insights from in vivo manipulation of sphingolipid metabolism. Endocr Rev 29:381–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Scherer PE. 2006. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes 55:1537–1545 [DOI] [PubMed] [Google Scholar]
- 104. Wellen KE, Hotamisligil GS. 2003. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest 112:1785–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Rocha VZ, Libby P. 2009. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol 6:399–409 [DOI] [PubMed] [Google Scholar]
- 106. Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, Wang S, Fortier M, Greenberg AS, Obin MS. 2005. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res 46:2347–2355 [DOI] [PubMed] [Google Scholar]
- 107. Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, Cinti S. 2008. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res 49:1562–1568 [DOI] [PubMed] [Google Scholar]
- 108. Alkhouri N, Gornicka A, Berk MP, Thapaliya S, Dixon LJ, Kashyap S, Schauer PR, Feldstein AE. 2010. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J Biol Chem 285:3428–3438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T, Nagai R. 2009. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15:914–920 [DOI] [PubMed] [Google Scholar]
- 110. Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, Dorfman R, Wang Y, Zielenski J, Mastronardi F, Maezawa Y, Drucker DJ, Engleman E, Winer D, Dosch HM. 2009. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 15:921–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D. 2009. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15:930–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Markiewski MM, Lambris JD. 2009. Is complement good or bad for cancer patients? A new perspective on an old dilemma. Trends Immunol 30:286–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bui JD, Schreiber RD. 2007. Cancer immunosurveillance, immunoediting and inflammation: independent or interdependent processes? Curr Opin Immunol 19:203–208 [DOI] [PubMed] [Google Scholar]
- 114. Pollard JW. 2004. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4:71–78 [DOI] [PubMed] [Google Scholar]
- 115. Coussens LM, Werb Z. 2002. Inflammation and cancer. Nature 420:860–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Mantovani A, Allavena P, Sica A, Balkwill F. 2008. Cancer-related inflammation. Nature 454:436–444 [DOI] [PubMed] [Google Scholar]
- 117. Gupta RA, Dubois RN. 2001. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer 1:11–21 [DOI] [PubMed] [Google Scholar]
- 118. Ulrich CM, Bigler J, Potter JD. 2006. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer 6:130–140 [DOI] [PubMed] [Google Scholar]
- 119. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE. 2001. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkβ. Science 293:1673–1677 [DOI] [PubMed] [Google Scholar]
- 120. Blobe GC, Obeid LM, Hannun YA. 1994. Regulation of protein kinase C and role in cancer biology. Cancer Metastasis Rev 13:411–431 [DOI] [PubMed] [Google Scholar]
- 121. Schütze S, Machleidt T, Krönke M. 1994. The role of diacylglycerol and ceramide in tumor necrosis factor and interleukin-1 signal transduction. J Leukoc Biol 56:533–541 [DOI] [PubMed] [Google Scholar]
- 122. Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, Pagliassotti MJ, Scherer PE, Summers SA. 2011. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest 121:1858–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. 1999. Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147 [DOI] [PubMed] [Google Scholar]
- 124. Wassermann F. 1926. The fat organs of man: development, structure, and systematic place of the so-called adipose tissue. Z Zellforsch Mikroskop Anat Abt Histochem 3:325 [Google Scholar]
- 125. Rosen ED, Spiegelman BM. 2000 Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol 16:145–171 [DOI] [PubMed] [Google Scholar]
- 126. Vernochet C, Milstone DS, Iehlé C, Belmonte N, Phillips B, Wdziekonski B, Villageois P, Amri EZ, O'Donnell PE, Mortensen RM, Ailhaud G, Dani C. 2002. PPARγ-dependent and PPARγ-independent effects on the development of adipose cells from embryonic stem cells. FEBS Lett 510:94–98 [DOI] [PubMed] [Google Scholar]
- 127. Sato M, Yajima Y, Kawashima S, Tanaka K, Kagechika H. 2001. Synergistic potentiation of thiazolidinedione-induced ST 13 preadipocyte differentiation by RAR synergists. Biochem Biophys Res Commun 280:646–651 [DOI] [PubMed] [Google Scholar]
- 128. Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE. 2007. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 117:2621–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Rubin GL, Zhao Y, Kalus AM, Simpson ER. 2000. Peroxisome proliferator-activated receptor γ ligands inhibit estrogen biosynthesis in human breast adipose tissue: possible implications for breast cancer therapy. Cancer Res 60:1604–1608 [PubMed] [Google Scholar]
- 130. Hudson AG, Gierach GL, Modugno F, Simpson J, Wilson JW, Evans RW, Vogel VG, Weissfeld JL. 2008. Nonsteroidal anti-inflammatory drug use and serum total estradiol in postmenopausal women. Cancer Epidemiol Biomarkers Prev 17:680–687 [DOI] [PubMed] [Google Scholar]
- 131. Baum M, Budzar AU, Cuzick J, Forbes J, Houghton JH, Klijn JG, Sahmoud T. 2002. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet 359:2131–2139 [DOI] [PubMed] [Google Scholar]
- 132. Geisler J, Haynes B, Anker G, Dowsett M, Lønning PE. 2002. Influence of letrozole and anastrozole on total body aromatization and plasma estrogen levels in postmenopausal breast cancer patients evaluated in a randomized, cross-over study. J Clin Oncol 20:751–757 [DOI] [PubMed] [Google Scholar]
- 133. Smith IE. 2003. Letrozole versus tamoxifen in the treatment of advanced breast cancer and as neoadjuvant therapy. J Steroid Biochem Mol Biol 86:289–293 [DOI] [PubMed] [Google Scholar]
- 134. Mouridsen H, Gershanovich M, Sun Y, Pérez-Carrión R, Boni C, Monnier A, Apffelstaedt J, Smith R, Sleeboom HP, Jänicke F, Pluzanska A, Dank M, Becquart D, Bapsy PP, Salminen E, Snyder R, Lassus M, Verbeek JA, Staffler B, Chaudri-Ross HA, Dugan M. 2001. Superior efficacy of letrozole versus tamoxifen as first-line therapy for postmenopausal women with advanced breast cancer: results of a phase III study of the International Letrozole Breast Cancer Group. J Clin Oncol 19:2596–2606 [DOI] [PubMed] [Google Scholar]
- 135. Bonneterre J, Buzdar A, Nabholtz JM, Robertson JF, Thürlimann B, von Euler M, Sahmoud T, Webster A, Steinberg M. 2001. Anastrozole is superior to tamoxifen as first-line therapy in hormone receptor positive advanced breast carcinoma. Cancer 92:2247–2258 [DOI] [PubMed] [Google Scholar]
- 136. Purohit A, Newman SP, Reed MJ. 2002. The role of cytokines in regulating estrogen synthesis: implications for the etiology of breast cancer. Breast Cancer Res 4:65–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Brown KA, Simpson ER. 2010. Obesity and breast cancer: progress to understanding the relationship. Cancer Res 70:4–7 [DOI] [PubMed] [Google Scholar]
- 138. Park SY, Lee JH, Kim KY, Kim EK, Yun SJ, Kim CD, Lee WS, Hong KW. 2008. Cilostazol increases 3T3–L1 preadipocyte differentiation with improved glucose uptake associated with activation of peroxisome proliferator-activated receptor-γ transcription. Atherosclerosis 201:258–265 [DOI] [PubMed] [Google Scholar]
- 139. Brown KA, Hunger NI, Docanto M, Simpson ER. 2010. Metformin inhibits aromatase expression in human breast adipose stromal cells via stimulation of AMP-activated protein kinase. Breast Cancer Res Treat 123:591–596 [DOI] [PubMed] [Google Scholar]
- 140. Nolan JJ, Ludvik B, Beerdsen P, Joyce M, Olefsky J. 1994. Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N Engl J Med 331:1188–1193 [DOI] [PubMed] [Google Scholar]
- 141. Rohatagi S, Carrothers TJ, Jin J, Jusko WJ, Khariton T, Walker J, Truitt K, Salazar DE. 2008. Model-based development of a PPARγ agonist, rivoglitazone, to aid dose selection and optimize clinical trial designs. J Clin Pharmacol 48:1420–1429 [DOI] [PubMed] [Google Scholar]
- 142. Chen X, Osborne MC, Rybczynski PJ, Zeck R, Yang M, Xu J, Zhou L, Cryan E, Tang Y, Demarest KT. 2005. Pharmacological profile of a novel, non-TZD PPARγ agonist. Diabetes Obes Metab 7:536–546 [DOI] [PubMed] [Google Scholar]
- 143. Kim J, Han DC, Kim JM, Lee SY, Kim SJ, Woo JR, Lee JW, Jung SK, Yoon KS, Cheon HG, Kim SS, Hong SH, Kwon BM. 2009. PPAR γ partial agonist, KR-62776, inhibits adipocyte differentiation via activation of ERK. Cell Mol Life Sci 66:1766–1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wang C, Pattabiraman N, Zhou JN, Fu M, Sakamaki T, Albanese C, Li Z, Wu K, Hulit J, Neumeister P, Novikoff PM, Brownlee M, Scherer PE, Jones JG, Whitney KD, Donehower LA, Harris EL, Rohan T, Johns DC, Pestell RG. 2003. Cyclin D1 repression of peroxisome proliferator-activated receptor γ expression and transactivation. Mol Cell Biol 23:6159–6173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Sharma C, Pradeep A, Wong L, Rana A, Rana B. 2004. Peroxisome proliferator-activated receptor γ activation can regulate β-catenin levels via a proteasome-mediated and adenomatous polyposis coli-independent pathway. J Biol Chem 279:35583–35594 [DOI] [PubMed] [Google Scholar]
- 146. Burstein HJ, Demetri GD, Mueller E, Sarraf P, Spiegelman BM, Winer EP. 2003. Use of the peroxisome proliferator-activated receptor (PPAR) γ ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res Treat 79:391–397 [DOI] [PubMed] [Google Scholar]
- 147. Kulke MH, Demetri GD, Sharpless NE, Ryan DP, Shivdasani R, Clark JS, Spiegelman BM, Kim H, Mayer RJ, Fuchs CS. 2002. A phase II study of troglitazone, an activator of the PPARγ receptor, in patients with chemotherapy-resistant metastatic colorectal cancer. Cancer J 8:395–399 [DOI] [PubMed] [Google Scholar]
- 148. Smith MR, Manola J, Kaufman DS, George D, Oh WK, Mueller E, Slovin S, Spiegelman B, Small E, Kantoff PW. 2004. Rosiglitazone versus placebo for men with prostate carcinoma and a rising serum prostate-specific antigen level after radical prostatectomy and/or radiation therapy. Cancer 101:1569–1574 [DOI] [PubMed] [Google Scholar]
- 149. Girnun GD, Naseri E, Vafai SB, Qu L, Szwaya JD, Bronson R, Alberta JA, Spiegelman BM. 2007. Synergy between PPARγ ligands and platinum-based drugs in cancer. Cancer Cell 11:395–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Holland W, Morrison T, Chang Y, Wiernsperger N, Stith BJ. 2004. Metformin (glucophage) inhibits tyrosine phosphatase activity to stimulate the insulin receptor tyrosine kinase. Biochem Pharmacol 67:2081–2091 [DOI] [PubMed] [Google Scholar]
- 151. Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. 2005. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310:1642–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Bulcão C, Giuffrida FM, Ribeiro-Filho FF, Ferreira SR. 2007. Are the beneficial cardiovascular effects of simvastatin and metformin also associated with a hormone-dependent mechanism improving insulin sensitivity? Braz J Med Biol Res 40:229–235 [PubMed] [Google Scholar]
- 153. Jakubowska J, Bohdanowicz-Pawlak A, Milewicz A, Szymczak J, Bednarek-Tupikowska G, Demissie M. 2008. Plasma cytokines in obese women with polycystic ovary syndrome, before and after metformin treatment. Gynecol Endocrinol 24:378–384 [DOI] [PubMed] [Google Scholar]
- 154. Li D, Yeung SC, Hassan MM, Konopleva M, Abbruzzese JL. 2009. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology 137:482–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. 2008. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 27:3576–3586 [DOI] [PubMed] [Google Scholar]
- 156. Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM. 2009. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer [see comment]. J Clin Oncol 27:3297–3302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. 2007. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res 67:10804–10812 [DOI] [PubMed] [Google Scholar]
- 158. Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. 2006. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res 66:10269–10273 [DOI] [PubMed] [Google Scholar]
- 159. Mueller WM, Stanhope KL, Gregoire F, Evans JL, Havel PJ. 2000. Effects of metformin and vanadium on leptin secretion from cultured rat adipocytes. Obes Res 8:530–539 [DOI] [PubMed] [Google Scholar]
- 160. He G, Pedersen SB, Bruun JM, Lihn AS, Richelsen B. 2003. Metformin, but not thiazolidinediones, inhibits plasminogen activator inhibitor-1 production in human adipose tissue in vitro. Horm Metab Res 35:18–23 [DOI] [PubMed] [Google Scholar]
- 161. Jacobs DB, Hayes GR, Truglia JA, Lockwood DH. 1986. Effects of metformin on insulin receptor tyrosine kinase activity in rat adipocytes. Diabetologia 29:798–801 [DOI] [PubMed] [Google Scholar]
- 162. Matthaei S, Greten H. 1991. Evidence that metformin ameliorates cellular insulin-resistance by potentiating insulin-induced translocation of glucose transporters to the plasma membrane. Diabete Metab 17:150–158 [PubMed] [Google Scholar]
- 163. Daval M, Foufelle F, Ferré P. 2006. Functions of AMP-activated protein kinase in adipose tissue. J Physiol 574:55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. 2002. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8:1288–1295 [DOI] [PubMed] [Google Scholar]
- 165. Towler MC, Hardie DG. 2007. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 100:328–341 [DOI] [PubMed] [Google Scholar]
- 166. Ren T, He J, Jiang H, Zu L, Pu S, Guo X, Xu G. 2006. Metformin reduces lipolysis in primary rat adipocytes stimulated by tumor necrosis factor-α or isoproterenol. J Mol Endocrinol 37:175–183 [DOI] [PubMed] [Google Scholar]
- 167. Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, Anania FA. 2007. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res 67:2497–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Sharma D, Wang J, Fu PP, Sharma S, Nagalingam A, Mells J, Handy J, Page AJ, Cohen C, Anania FA, Saxena NK. 2010. Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology 52:1713–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Somasundar P, Yu AK, Vona-Davis L, McFadden DW. 2003. Differential effects of leptin on cancer in vitro. J Surg Res 113:50–55 [DOI] [PubMed] [Google Scholar]
- 170. Ogunwobi O, Mutungi G, Beales IL. 2006. Leptin stimulates proliferation and inhibits apoptosis in Barrett's esophageal adenocarcinoma cells by cyclooxygenase-2-dependent, prostaglandin-E2-mediated transactivation of the epidermal growth factor receptor and c-Jun NH2-terminal kinase activation. Endocrinology 147:4505–4516 [DOI] [PubMed] [Google Scholar]
- 171. Konturek PC, Burnat G, Rau T, Hahn EG, Konturek S. 2008. Effect of adiponectin and ghrelin on apoptosis of Barrett adenocarcinoma cell line. Dig Dis Sci 53:597–605 [DOI] [PubMed] [Google Scholar]
- 172. Rubenstein JH, Dahlkemper A, Kao JY, Zhang M, Morgenstern H, McMahon L, Inadomi JM. 2008. A pilot study of the association of low plasma adiponectin and Barrett's esophagus. Am J Gastroenterol 103:1358–1364 [DOI] [PubMed] [Google Scholar]
- 173. Ratke J, Entschladen F, Niggemann B, Zänker KS, Lang K. 2010. Leptin stimulates the migration of colon carcinoma cells by multiple signaling pathways. Endocr Relat Cancer 17:179–189 [DOI] [PubMed] [Google Scholar]
- 174. Jaffe T, Schwartz B. 2008. Leptin promotes motility and invasiveness in human colon cancer cells by activating multiple signal-transduction pathways. Int J Cancer 123:2543–2556 [DOI] [PubMed] [Google Scholar]
- 175. Hardwick JC, Van Den Brink GR, Offerhaus GJ, Van Deventer SJ, Peppelenbosch MP. 2001. Leptin is a growth factor for colonic epithelial cells. Gastroenterology 121:79–90 [DOI] [PubMed] [Google Scholar]
- 176. Jain SS, Bird RP. 2010. Elevated expression of tumor necrosis factor-α signaling molecules in colonic tumors of Zucker obese (fa/fa) rats. Int J Cancer 127:2042–2050 [DOI] [PubMed] [Google Scholar]
- 177. Fenton JI, Birmingham JM, Hursting SD, Hord NG. 2008. Adiponectin blocks multiple signaling cascades associated with leptin-induced cell proliferation in Apc Min/+ colon epithelial cells. Int J Cancer 122:2437–2445 [DOI] [PubMed] [Google Scholar]
- 178. Kim AY, Lee YS, Kim KH, Lee JH, Lee HK, Jang SH, Kim SE, Lee GY, Lee JW, Jung SA, Chung HY, Jeong S, Kim JB. 2010. Adiponectin represses colon cancer cell proliferation via AdipoR1- and -R2-mediated AMPK activation. Mol Endocrinol 24:1441–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Horiguchi A, Sumitomo M, Asakuma J, Asano T, Zheng R, Asano T, Nanus DM, Hayakawa M. 2006. Leptin promotes invasiveness of murine renal cancer cells via extracellular signal-regulated kinases and rho dependent pathway. J Urol 176:1636–1641 [DOI] [PubMed] [Google Scholar]
- 180. Attoub S, Noe V, Pirola L, Bruyneel E, Chastre E, Mareel M, Wymann MP, Gespach C. 2000. Leptin promotes invasiveness of kidney and colonic epithelial cells via phosphoinositide 3-kinase-, rho-, and rac-dependent signaling pathways. FASEB J 14:2329–2338 [DOI] [PubMed] [Google Scholar]
- 181. Pinthus JH, Kleinmann N, Tisdale B, Chatterjee S, Lu JP, Gillis A, Hamlet T, Singh G, Farrokhyar F, Kapoor A. 2008. Lower plasma adiponectin levels are associated with larger tumor size and metastasis in clear-cell carcinoma of the kidney. Eur Urol 54:866–873 [DOI] [PubMed] [Google Scholar]
- 182. Onuma M, Bub JD, Rummel TL, Iwamoto Y. 2003. Prostate cancer cell-adipocyte interaction: leptin mediates androgen-independent prostate cancer cell proliferation through c-Jun NH2-terminal kinase. J Biol Chem 278:42660–42667 [DOI] [PubMed] [Google Scholar]
- 183. Park HY, Kwon HM, Lim HJ, Hong BK, Lee JY, Park BE, Jang Y, Cho SY, Kim HS. 2001. Potential role of leptin in angiogenesis: leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp Mol Med 33:95–102 [DOI] [PubMed] [Google Scholar]
- 184. Bub JD, Miyazaki T, Iwamoto Y. 2006. Adiponectin as a growth inhibitor in prostate cancer cells. Biochem Biophys Res Commun 340:1158–1166 [DOI] [PubMed] [Google Scholar]
- 185. Zhang T, He J, Xu C, Zu L, Jiang H, Pu S, Guo X, Xu G. 2009. Mechanisms of metformin inhibiting lipolytic response to isoproterenol in primary rat adipocytes. J Mol Endocrinol 42:57–66 [DOI] [PubMed] [Google Scholar]
- 186. Sharma D, Saxena NK, Vertino PM, Anania FA. 2006. Leptin promotes the proliferative response and invasiveness in human endometrial cancer cells by activating multiple signal-transduction pathways. Endocr Relat Cancer 13:629–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Yin N, Wang D, Zhang H, Yi X, Sun X, Shi B, Wu H, Wu G, Wang X, Shang Y. 2004. Molecular mechanisms involved in the growth stimulation of breast cancer cells by leptin. Cancer Res 64:5870–5875 [DOI] [PubMed] [Google Scholar]
- 188. Lam JB, Chow KH, Xu A, Lam KS, Liu J, Wong NS, Moon RT, Shepherd PR, Cooper GJ, Wang Y. 2009. Adiponectin haploinsufficiency promotes mammary tumor development in MMTV-PyVT mice by modulation of phosphatase and tensin homolog activities. PLoS One 4:e4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Wang Y, Lam JB, Lam KS, Liu J, Lam MC, Hoo RL, Wu D, Cooper GJ, Xu A. 2006. Adiponectin modulates the glycogen synthase kinase-3β/β-catenin signaling pathway and attenuates mammary tumorigenesis of MDA-MB-231 cells in nude mice. Cancer Res 66:11462–11470 [DOI] [PubMed] [Google Scholar]
- 190. Liu J, Lam JB, Chow KH, Xu A, Lam KS, Moon RT, Wang Y. 2008. Adiponectin stimulates Wnt inhibitory factor-1 expression through epigenetic regulations involving the transcription factor specificity protein 1. Carcinogenesis 29:2195–2202 [DOI] [PubMed] [Google Scholar]
- 191. Gao J, Tian J, Lv Y, Shi F, Kong F, Shi H, Zhao L. 2009. Leptin induces functional activation of cyclooxygenase-2 through JAK2/STAT3, MAPK/ERK, and PI3K/AKT pathways in human endometrial cancer cells. Cancer Sci 100:389–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Catalano S, Giordano C, Rizza P, Gu G, Barone I, Bonofiglio D, Giordano F, Malivindi R, Gaccione D, Lanzino M, De Amicis F, Andò S. 2009. Evidence that leptin through STAT and CREB signaling enhances cyclin D1 expression and promotes human endometrial cancer proliferation. J Cell Physiol 218:490–500 [DOI] [PubMed] [Google Scholar]
- 193. Cong L, Gasser J, Zhao J, Yang B, Li F, Zhao AZ. 2007. Human adiponectin inhibits cell growth and induces apoptosis in human endometrial carcinoma cells, HEC-1-A and RL95 2. Endocr Relat Cancer 14:713–720 [DOI] [PubMed] [Google Scholar]
- 194. Keophiphath M, Achard V, Henegar C, Rouault C, Clément K, Lacasa D. 2009. Macrophage-secreted factors promote a profibrotic phenotype in human preadipocytes. Mol Endocrinol 23:11–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Hutley LJ, Newell FM, Joyner JM, Suchting SJ, Herington AC, Cameron DP, Prins JB. 2003. Effects of rosiglitazone and linoleic acid on human preadipocyte differentiation. Eur J Clin Invest 33:574–581 [DOI] [PubMed] [Google Scholar]
- 196. Sandouk T, Reda D, Hofmann C. 1993. The antidiabetic agent pioglitazone increases expression of glucose transporters in 3T3–F442A cells by increasing messenger ribonucleic acid transcript stability. Endocrinology 133:352–359 [DOI] [PubMed] [Google Scholar]
- 197. Kim YC, Gomez FE, Fox BG, Ntambi JM. 2000. Differential regulation of the stearoyl-CoA desaturase genes by thiazolidinediones in 3T3–L1 adipocytes. J Lipid Res 41:1310–1316 [PubMed] [Google Scholar]
- 198. Bouaboula M, Hilairet S, Marchand J, Fajas L, Le Fur G, Casellas P. 2005. Anandamide induced PPARγ transcriptional activation and 3T3–L1 preadipocyte differentiation. Eur J Pharmacol 517:174–181 [DOI] [PubMed] [Google Scholar]
- 199. Janke J, Schupp M, Engeli S, Gorzelniak K, Boschmann M, Sauma L, Nystrom FH, Jordan J, Luft FC, Sharma AM. 2006. Angiotensin type 1 receptor antagonists induce human in-vitro adipogenesis through peroxisome proliferator-activated receptor-γ activation. J Hypertens 24:1809–1816 [DOI] [PubMed] [Google Scholar]