

Abstract
Heme-regulated eIF2α kinase (Hri) is necessary for balanced synthesis of heme and globin. In addition, Hri deficiency exacerbates the phenotypic severity of β-thalassemia intermedia in mice. Activation of Hri during heme deficiency and in β-thalassemia increases eIF2α phosphorylation and inhibits globin translation. Under endoplasmic reticulum stress and nutrient starvation, eIF2α phosphorylation also induces the Atf4 signaling pathway to mitigate stress. Although the function of Hri in regulating globin translation is well established, its role in Atf4 signaling in erythroid precursors is not known. Here, we report the role of the Hri-activated Atf4 signaling pathway in reducing oxidative stress and in promoting erythroid differentiation during erythropoiesis. On acute oxidative stress, Hri−/− erythroblasts suffered from increased levels of reactive oxygen species (ROS) and apoptosis. During chronic iron deficiency in vivo, Hri is necessary both to reduce oxidative stress and to promote erythroid differentiation. Hri−/− mice developed ineffective erythropoiesis during iron deficiency with inhibition of differentiation at the basophilic erythroblast stage. This inhibition is recapitulated during ex vivo differentiation of Hri−/− fetal liver erythroid progenitors. Importantly, the Hri-eIF2αP-Atf4 pathway was activated and required for erythroid differentiation. We further demonstrate the potential of modulating Hri-eIF2αP-Atf4 signaling with chemical compounds as pharmaceutical therapies for β-thalassemia.
Introduction
Heme-regulated eIF2α kinase (Hri) was the first discovered member of the family of eIF2α kinases, which control translational initiation under diverse stress conditions by phosphorylation of the α-subunit of eIF2 (eIF2αP).1,2 Hri is necessary to coordinate translation of globin mRNAs with the availability of heme for the production of large amounts of hemoglobin during erythroid maturation.3 In Hri deficiency, excessive globins synthesized during heme deficiency precipitate and form inclusions causing proteotoxicity.3,4 Beyond heme deficiency, Hri is also activated by oxidative stress and denatured proteins,5 both of which occur in thalassemia.6 Indeed, Hri is required to reduce the phenotypic severity of the Hbb−/− mouse model of β-thalassemia intermedia lacking both copies of β-globin major.4
Besides Hri, there are 3 additional mammalian eIF2α kinases: the double-stranded RNA-dependent eIF2α kinase (Pkr), the Gcn2 protein kinase, and the Pkr-like ER resident kinase (Perk).1 Each kinase elicits a major physiologic response in vivo: Pkr responds to viral infection; Gcn2 senses nutrient starvations; Perk is activated by endoplasmic reticulum (ER) stress; and Hri is regulated by heme. In addition to inhibition of global protein synthesis, the second function of eIF2α phosphorylation is to reprogram translation and transcription necessary for adaptation to stress7 as illustrated in Figure 7A. In mammalian cells, translation of activating transcriptional factor 4 (Atf4) is selectively increased by eIF2αP amid general inhibition of protein synthesis during ER stress and amino acid starvation via upstream open reading frames in the 5′-UTR (untranslated region).8,9 A major target gene of Atf4 is the transcription factor, C/EBP Homologous Protein-10 (Chop), which is up-regulated on stress.10 In ER stress, induction of Chop leads to expression of Gadd34, which recruits eIF2αP for dephosphorylation by PPase1.11,12 This feedback mechanism of Gadd34 in regenerating active eIF2 is necessary for the recovery of protein synthesis during the late stage of the stress response.13,14
Figure 7.
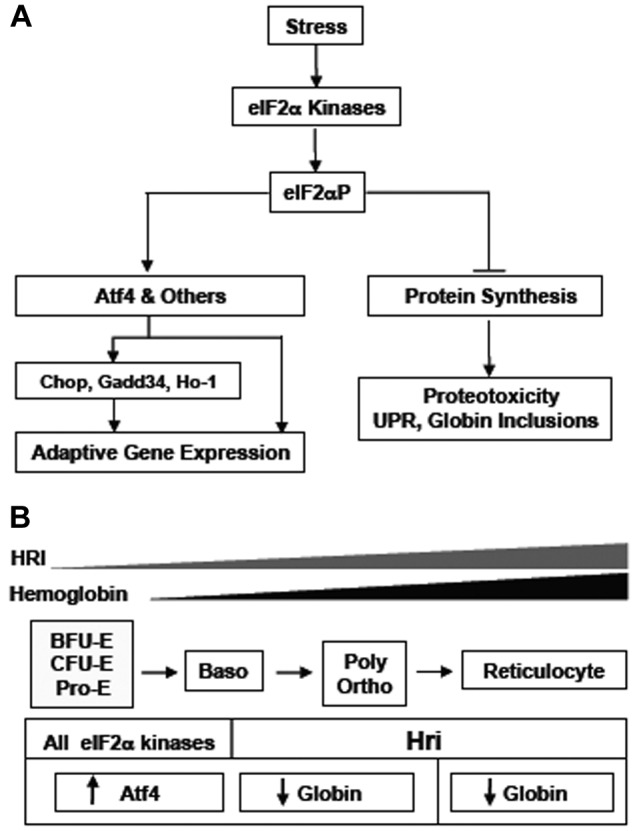
A schematic illustration of Hri-eIF2αP-Atf4 signaling pathway in mitigating stress and during erythropoiesis. (A) Inhibition of general protein synthesis and enhancement of Atf4 translation by eIF2α. On various stress conditions, eIF2α kinases are activated and phosphorylate eIF2α. The first order of action by eIF2α is to inhibit protein synthesis to prevent proteotoxicity resulted from accumulation of excessive unfolded proteins. In the erythroid precursors, Hri is necessary to inhibit globin synthesis in heme deficiency to prevent accumulation of denatured heme-free globins. The second action by eIF2α is to selectively enhance the translation of some mRNAs with open reading frames in their 5′-UTR. At present, Atf4 mRNA is the only known target of this regulation. However, Atf4 is not the only target; there are yet to be identified mRNAs. Induction of Atf4 leads to a signaling pathway for adaptive gene expression as described in the “Introduction.” (B) Dual roles of Hri in erythropoiesis. Expression of Hri is increased during erythropoiesis from basophilic erythroblasts to reticulocytes. Both roles of Hri in inhibition of globin translation and in activation of Atf4 during erythroid differentiation contribute to reduced anemic phenotypes.
Although we demonstrated the necessity of Hri in inhibiting globin translation during iron/heme deficiency and in β-thalassemia, the role of Hri under these stress conditions is more than just inhibition of translation. Hri is also necessary to reduce ineffective erythropoiesis.3,4 To further understand the molecular mechanism by which Hri mitigates stress and reduces ineffective erythropoiesis, we investigated the eIF2αP-Atf4 signaling pathway in the stress response of the erythroid lineage.
In this article we demonstrate that Hri activates the Atf4 stress response pathway in nucleated erythroid precursors for adaptation to oxidative stress. Significantly, this Hri-dependent eIF2αP-Atf4 pathway is also operative and necessary for erythroid differentiation, especially under stress conditions. Our study reveals the function of Hri in translational activation of Atf4 mRNA and the subsequent regulation of gene expression necessary for stress adaptation. We also demonstrate the feasibility of targeting the Hri-eIF2αP-Atf4 pathway for the treatment of β-thalassemia with the small chemical salubrinal, which inhibits dephosphorylation of eIF2αP and promotes the Atf4 signaling pathway.15
Methods
Animals
Mice were maintained at the Massachusetts Institute of Technology (MIT) animal facility, and all experiments were carried out using the protocols approved by the Division of Comparative Medicine at MIT. Hri−/− mice were generated in our laboratory.3 The generations of Atf4+/− and Hri+/−Hbb−/− mice were as previously described.4,16 All mice used for this study were of C57BL/6J genetic background. Mice were maintained in an iron-deficient diet (Harlan Teklad) on weaning for 2 months before analysis.
Fetal liver cell isolation and ex vivo differentiation
Ter119− cells were isolated from E14.5 fetal liver (FL) and cultured for ex vivo differentiation as described.17 Morphologic examination of cells was carried out on cytospin slides stained with Giemsa and benzidine.17
Sodium arsenite and H2O2 treatments
E14.5 FL cells from Hri +/+ and −/− mice were recovered for 1 hour in Iscove medium supplemented with cytokines17 before sodium arsenite treatment as indicated. Peripheral blood (2 μL) was resuspended in 1 mL of Hanks balanced salt solution (HBSS). Red blood cells (RBCs) were washed once with HBBS, resuspended in 200 μL of HBSS, and recovered for 20 minutes at 37°C incubator. The RBCs were then preincubated with 5μM of 5 and 6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA; Invitrogen) for 30 minutes followed by treatment with H2O2 for 20 minutes.
Analyses of ROS, erythroid differentiation, and apoptosis by flow cytometry
All the flow cytometric analyses were performed at the MIT Core Facility, which provides excellent technical support for gating, analyzing and sorting cell populations. For cellular reactive oxygen species (ROS) measurements, cells were resuspended in prewarmed phosphate-buffered saline (PBS) supplemented with 5% fetal bovine serum (FBS) and incubated with 5μM 2′, 7′-dichlorodihydrofluorescein diacetate (DCF; Invitrogen) in the dark for 30 minutes at room temperature, and then were analyzed immediately by flow cytometry.18 Erythroid differentiation was analyzed as previously described.17 Reticulocyte formation during ex vivo culture was measured by enucleation.19 Apoptosis was measured by annexin V (AnV) binding.3 Propidium iodide (BD Pharmingen) positive cells that contain late stage AnV+ apoptotic and necrotic cells were excluded from these analyses.
Western blot analysis
Cell extracts were prepared as previously described.18 Proteins (5-20 μg) were separated by 7% (for Hri) and 10% (for other proteins) SDS-PAGE. Blots were incubated overnight at 4°C with primary antibodies against Hri (1:1000),18 eIF2α (1:1000; BioSource), phosphorylated eIF2α (1:1000; BioSource), Chop (1:500; Santa Cruz Biotechnology), Ho-1 (1:1000; Stressgen), or Atf4 (1:500; Abnova).
Quantitative real time PCR
RNAs were isolated using the Promega SV Total RNA Isolation kit. The reverse transcription of mRNA and polymerase chain reaction (PCR) were performed using Quantitect kits (QIAGEN RT:205311, PCR:204143). Primer sequences for quantitative PCR are listed in supplemental Table 1 (available on the Blood Web site; see the Supplemental Materials link at the top of the online article). eIF2α was used as internal control for normalization. Raw data were processed through LinRegPCR for baseline correction to determine C(t) values. Relative expression of mRNA was determined by the Δ[ΔC(t)] method.
Knockdown of Atf4 expression
Knockdown of Atf4 expression in differentiating mouse erythroid leukemic (MEL) cells was performed with Sigma-Aldrich Mission small interfering RNAs (siRNAs) using the protocol described by Paradkar et al.20 Atf4 knockdown siRNA sequences were shown in supplemental Table 2. Mission siRNA Universal Negative Control #1 (Sigma-Aldrich) was used as a negative control.
Salubrinal treatment and measurement of protein synthesis
Blood samples from adult Hri+/−Hbb−/− mice, which contained approximately 30% reticulocytes, were recovered in complete DMEM medium as described.3 Cells were then treated with salubrinal as indicated. E14.5 FL cell were pretreated with 5μM of arsenite for 2 hours to enhance eIF2αP levels. Cells were washed with PBS to remove arsenite and then treated with various concentrations of salubrinal as indicated. Splenic Ter119+ cells from Hbb−/− mice were isolated using magnetic-activated cell sorting MACS column purification (Miltenyi Biotec). Cells were then recovered for 30 minutes before salubrinal treatment at concentrations indicated for 3.5 hours. The rate of globin synthesis was determined by S35-methionine/cysteine incorporation into globin proteins.3
Data analysis
The statistical analyses among the various groups were performed by the 2-tailed Student t test with a P value of < .05 for statistical significance.
Results
Activation of Hri-eIF2αP-Atf4 pathway in erythroid precursors on arsenite-induced oxidative stress
We have shown earlier that Hri is the eIF2α kinase activated by treatment with sodium arsenite in erythroid precursors and that the activation of Hri by arsenite is prevented by pretreatment with the ROS scavenger N-acetylcysteine.5 To gain further insight into mechanistic details of ROS production and Hri activation, ROS levels in E14.5 FL cells, which are highly enriched in nucleated erythroid precursors, were measured after arsenite treatment. As shown in Figure 1A, ROS levels increased rapidly within 15 minutes and peaked at 30 minutes on arsenite treatment of FL cells. Concomitantly, activation of Hri also peaked at 30 minutes after arsenite treatment. Hri remained activated for at least 60 minutes as shown by the hyperphosphorylation of Hri and the increased levels of eIF2αP (Figure 1B).
Figure 1.

Induction of the Hri-eIF2αP-Atf4 signaling pathway and protection against oxidative stress in FL erythroblasts. (A) ROS levels. Hri+/+ FL cells were treated with 25μM sodium arsenite. At times indicated, cellular ROS levels were measured by DCF fluorescence. (B) Hyperphosphorylation and activation of Hri. Hyperphosphorylation of Hri was examined by the slower migration in SDS-PAGE. eIF2α was used as a loading control. (C) Induction of the Hri-dependent Atf4 signaling pathway. Cells were treated with 5μM As. At times indicated, samples were analyzed for expression of eIF2αP, Atf4, Chop, and eIF2α proteins. (D) Relative expression of mRNAs of the Hri-Atf4 signaling pathway components. Atf4, Chop, Gadd34, and Crep mRNA levels from FL cells treated with 5μM As for 3 hours were analyzed by qPCR. Expression of each of the mRNA in untreated Hri+/+ cells was defined as 1. Data are presented as mean ± SD (n = 3). P values denote the comparison between arsenite-treated Hri+/+ and Hri−/− samples; P < .001 for Atf4 and Gadd34; P < .005 for Chop. (E) ROS levels. Hri+/+ and Hri−/− FL cells treated with 5 or 25μM arsenite for 9 hours before ROS measurement.
Arsenite treatments also resulted in the Hri-dependent induction of the eIF2αP-Atf4 signaling pathway as shown by increased levels of eIF2αP, Atf4 and Chop proteins in Hri+/+ FL cells at 1 to 6 hours of exposure to arsenite (Figure 1C). There was no increase in eIF2αP, Atf4, or Chop protein levels in Hri−/− cells on arsenite stress. Importantly, the Atf4 mRNA level did not change significantly on arsenite stress in either Hri+/+ or Hri−/− cells (Figure 1D), consistent with the immediate translational regulation of Atf4 by increased eIF2αP.9 Both Chop and Gadd34 mRNA levels were increased in Hri+/+ cells on arsenite stress, consistent with their transcriptional induction by Atf4. In contrast to Gadd34, the constitutively expressed regulator of eIF2αP dephosphorylation Crep21 was not affected by arsenite stress as would be expected (Figure 1D). These results establish that activation of Hri by arsenite stress elicits the eIF2αP signaling pathway of up-regulating Atf4, Chop, and Gadd34 in primary erythroid precursors.
Increased oxidative stress in Hri−/− erythroid precursors on arsenite stress
To investigate whether this up-regulation of the Hri-Atf4 signaling pathway might contribute to adaptation to oxidative stress, levels of ROS in Hri+/+ and Hri−/− FL cells were measured at different time points after arsenite treatment. ROS levels in Hri−/− cells were significantly higher than those in Hri+/+ cells at 3, 6, and 9 hours after treatment with 25μM arsenite (supplemental Figure 1A). Furthermore, ROS levels declined to baseline in Hri+/+ cells at 9 hours of sodium arsenite treatments whereas ROS levels in Hri−/− cells continued to rise (Figure 1E, supplemental Figure 1A). These results demonstrate that Hri is required to mitigate acute oxidative stress induced by arsenite. In addition, Hri is necessary for recovery from oxidative stress after exposure to arsenite for 3 hours (supplemental Figure 1B-C). Hri−/− cells not only failed to recover from oxidative stress induced by arsenite treatment, but also suffered further increase in oxidative stress during the recovery phase. This increased oxidative stress of Hri−/− FL cells on arsenite exposure resulted in increased apoptosis accompanied with elevated activation of caspases 3 and 9 (supplemental Figure 2).
Hri and Atf4 dependent expression of Ho-1 and antioxidant genes
To further investigate the mechanism by which Hri protects against oxidative stress, we examined the expression of heme oxygenase 1 (Ho-1), which catabolizes heme to biliverdin, CO, and Fe, and plays a pivotal role in reducing oxidative stress.22 We found that Ho-1 protein in Hri+/+ erythroblasts was increased by treatment with arsenite from 0.1 to 3μM (Figure 2A). Interestingly, the increase of Ho-1 expression by arsenite in Hri−/− cells was less prominent and required higher concentrations compared with Hri+/+ cells (Figure 2A). Furthermore, the level of Ho-1 mRNA was significantly increased on arsenite treatment in Hri+/+ cells, but not in Hri−/− cells (Figure 2C).
Figure 2.

Hri and Atf4 dependent induction of antioxidant genes. (A) Hri-dependent Ho-1 protein expression. Hri+/+ and Hri−/− FL cells were treated for 5 hours with increasing concentrations of arsenite as indicated. (B) Atf4-dependent Ho-1 protein expression. Atf4 +/+, +/−, and −/− FL cells were treated with 5μM arsenite for 3 hours. (C-D) Relative expression of mRNA of antioxidant genes. Hri+/+, Hri−/−, Atf4+/+, and Atf4−/− FL cells were treated with 5μM arsenite for 3 hours. mRNA levels of treated and untreated cells were analyzed by qPCR. Data are presented as mean ± SD (n = 3). P values denote the comparison between arsenite-treated Hri+/+ and Hri−/− samples; P < .01 for Ho-1 and Nqo1, P < .05 for Gstμ, and P = .06 for Sod2; or between the comparison of arsenite-treated Atf4+/+ and Atf4−/− samples; P < .005 for Chop, P < .05 for Ho-1, P < .001 for Nqo1. (E) Elevated ROS levels in Hri−/− and Atf4−/− RBCs upon H2O2 treatment. Blood samples were treated with 50μM H2O2. (F) ROS levels in RBCs of Hri+/+ (wt) and Hri−/− (Ko) mice in iron deficiency.
We then examined whether the induction of Ho-1 by arsenite required Atf4, a downstream target of Hri signaling pathway (Figure 1C). As shown in Figure 2B, there is less Ho-1 protein in Atf4−/− FL cells compared with Atf4+/−or Atf4+/+ cells at the baseline without arsenite treatment. On arsenite treatment, Ho-1 protein expression was increased in FL cells from all the 3 genotypes. However, there was still less Ho-1 protein in Atf4−/− cells (Figure 2B). These results demonstrate that Ho-1 expression in primary erythroid precursors is dependent on both Hri and Atf4.
Besides Ho-1, arsenite treatment also induced expression of other antioxidant genes (Figure 2C), such as glutathione S-tranferase μ (Gstμ), NAD(P)H quinone oxidoreductase 1 (Nqo1) and superoxide dismutase 2 (Sod2) in Hri+/+, but not in Hri−/− FL cells. Similarly, induction of Ho-1 and Nqo1 mRNA was observed in Atf4+/+, but not in Atf4−/− cells (Figure 2D). These results demonstrate that both Hri and Atf4 are necessary to induce some antioxidant genes to mitigate oxidative stress incurred by arsenite treatment. Consistent with decreased expression of Ho-1 and Nqo1, RBCs from Hri−/− and Atf4−/− mice were more sensitive to H2O2-induced oxidative stress and exhibited increased ROS levels compared with RBCs from wild-type (WT) and Atf4+/− mice (Figure 2E; quantitative data of ROS levels are shown in supplemental Figure 3A). These results indicate that Hri-Atf4 signaling may also be required during erythroid development. Impairment of this pathway may generate RBCs that are more sensitive to oxidative insults.
Increased oxidative stress in Hri−/− erythroid cells during chronic iron deficiency in vivo
As shown above, Hri is required for reducing acute oxidative stress in vitro (Figure 1E). We investigated whether Hri also participated in reducing oxidative stress of erythroid cells in vivo during chronic iron deficiency. Levels of ROS were measured in RBCs from WT and Hri−/− (KO) mice maintained on either a normal or iron-deficient diet. Representative histograms of ROS levels by FACS analysis are shown in Figure 2F. The quantitative data of ROS levels are shown in supplemental Figure 3B. It is important to note that iron deficiency alone (WT-Fe) did not significantly increase ROS levels in RBCs. However, during iron deficiency, the absence of Hri (KO-Fe) resulted in dramatically elevated ROS levels in RBCs compared with those of WT-Fe cells (Figure 2F). These results demonstrate that Hri is necessary to mitigate oxidative stress in erythroid cells under chronic stress of iron deficiency in vivo. These findings are also consistent with our gene profiling data indicating that expression of many redox genes, such as catalase, Sod2, Gsts, thioredoxin reductase 2, peroxiredoxins, ferritins, and Ho-1 is significantly lower in KO-Fe FL cells compared with WT-Fe cells.23 Together, these results indicate that both Hri-mediated inhibition of globin translation and up-regulation of the Atf4 signaling pathway contribute in mitigating oxidative stress during iron deficiency.
Inhibition of erythroid differentiation in Hri−/− mice during chronic iron deficiency in vivo
We have shown earlier that fetal definitive erythropoiesis in iron deficiency is inhibited at the basophilic erythroblast stage and that this inhibition is more severe in Hri deficiency.23 Here, we examined erythroid differentiation in adult mice during iron deficiency. We found that there was no statistically significant increase of CD71highTer119high erythroblasts in adult WT mice during iron deficiency (Figure 3A). However, there was a mild inhibition of erythroid differentiation at the basophilic stage along with mild splenomegaly in approximately 20% of WT-Fe mice. The more severe inhibition of erythroid differentiation during fetal definitive erythropoiesis in response to iron deficiency is probably attributable to the much greater need for iron to accommodate a higher rate of erythropoiesis during embryonic development.
Figure 3.

Inhibition of erythroid differentiation of Hri−/− mice during iron deficiency and apoptosis of splenic Ter119+ erythroblasts in iron deficiency. (A) Erythroid differentiation of the spleen of Hri+/+ and Hri−/− adult mice maintained on a normal diet or an iron-restricted diet. (B) Benzidine-Giemsa–stained cells from sorted P3 and P4+P5 populations. Images were taken with 40× objective using Leitz microscope equipped with Pixelink digital imaging system. (C) Density plots of apoptosis of Ter119+ erythroblasts. Splenic Ter119+ erythroid cells were gated first. Apoptosis of these cells was then analyzed by their CD71 expression and AnV binding. (D) Quantitation of percentages of AnV+ cells in CD71+Ter119+ cells only. Data are presented as mean ± SD (n = 3 for each group of mice; **P < .01).
Most importantly, there was a profound inhibition of erythroid differentiation with a substantial accumulation of CD71highTer119high erythroblasts concomitant with diminished CD71lowTer119high erythroid cells in the spleens of Hri−/− mice during iron deficiency (Figure 3A, P < .005). Furthermore, these CD71highTer119high erythroblasts exhibited basophilic morphology (Figure 3B). The inhibition of erythroid differentiation was also observed in the bone marrow of KO-Fe mice (supplemental Figure 4). Further analysis of erythroid differentiation using CD44 described by Chen et al24 showed a significant accumulation of CD44highFSChigh cells in the bone marrow and spleens of Hri−/− mice under iron deficient conditions (supplemental Figure 5). Thus, these results indicate that erythroid expansion in the spleen and bone marrow of KO-Fe mice is associated with severe inhibition of erythroid differentiation.
Apoptosis of CD71+ erythroblasts during iron deficiency in vivo
We have previously shown that the percentage of apoptotic cells in Ter119+ erythroid precursors from FL, spleen, and bone marrow of Hri−/− mice increased during iron deficiency.3,23 We investigated apoptosis more closely by examining the degree to which erythroid precursors at different stages of differentiation undergo apoptosis during iron deficiency. Representative density plots of the apoptosis of Ter119+ erythroid precursors are shown in Figure 3C, whereas the quantitative data analysis is shown in Figure 3D. We found that the percentage of AnV+ apoptotic cells in total splenic Ter119+ erythroid precursors (Figure 3C right top and bottom quadrants combined) significantly increased during iron deficiency (P < .05), and further increased on combined deficiencies of Hri and iron (P < .05). In addition, this increase in apoptosis occurred mostly in CD71+Ter119+ erythroblasts (top right quadrants of Figure 3C); there were few AnV+ cells among CD71−Ter119+ erythroblasts (bottom right quadrants of Figure 3C). These results also confirm an expansion of splenic CD71+ erythroblasts among Ter119+ erythroid precursors from KO-Fe mice (top left and right quadrants of Figure 3C).
It is possible that the increase in percentage of AnV+ apoptotic cells in KO-Fe splenic Ter119+ erythroid precursors might simply be because of the expansion of CD71+Ter119+ erythroblasts in the spleens of KO-Fe mice. We therefore analyzed the percentage of AnV+ apoptotic cells in CD71+Ter119+ erythroblasts (Figure 3D). The percentage of apoptotic cells in CD71+Ter119+ erythroblasts was increased in WT-Fe spleens, but was not further increased in KO-Fe spleens (Figure 3D). Similar observations were made with bone marrow samples (data not shown).
Collectively, these results demonstrate that ineffective erythropoiesis of KO-Fe mice is primarily because of the profound inhibition of erythroid differentiation at the CD71+Ter119+ erythroblast stage. The increase of apoptotic cells in total Ter119+ erythroid precursors observed in KO-Fe mice is the consequence of the expansion and inhibition of erythroid differentiation of CD71+Ter119+ erythroblasts, which undergo apoptosis normally.25
Hri is necessary for differentiation of erythroid progenitors ex vivo
Ter119− cells in E14.5 FLs are primarily erythroid progenitors, which undergo proliferation and differentiation in ex vivo culture.17 We used this system to further investigate the role of Hri in erythroid differentiation. Differentiation of Hri−/− Ter119− erythroid progenitors progressed more slowly than that of Hri+/+ cells (Figure 4). Compared with Hri+/+ cells, Hri−/− cells had a greatly reduced population in the P3 basophilic erythroblast stage at 20 hours of culture (top panels, P < .001, supplemental Figure 6A). Whereas the percentage of cells in the P3 stage appeared similar at 40 hours, Hri−/− cells had higher CD71 and lower Ter119 expression, indicative of retarded erythroid differentiation (middle panels). At 60 hours of differentiation (bottom panels), there was a significantly lower percentage of Hri−/− cells in the P4 polychromatophilic and orthochromatophilic erythroblast stage compared with Hri+/+ cells (P < .005, supplemental Figure 6A).
Figure 4.
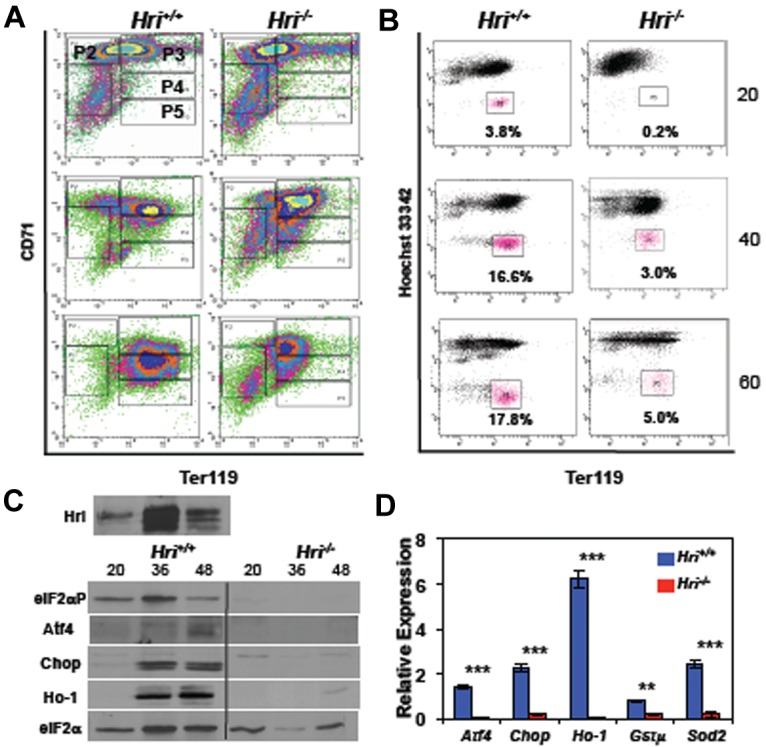
Differentiation of Ter119− FL erythroid progenitors ex vivo and the activation of the Hri signaling pathway. (A) Erythroid differentiation, and (B) reticulocyte production of Hri +/+ and −/− FL Ter119− erythroid progenitors at 20, 40, and 60 hours of ex vivo culture. (C) Activation of Hri and protein expression of its downstream targets. Vertical lines have been inserted to indicate a repositioned gel lane. The middle 3 lanes, which contains Hbb−/− samples, were removed of this Western blot because these results is not necessary for this figure. (D) qPCR analysis of the mRNA expression at 36 hours of ex vivo culture. Data are presented as relative expression normalized to eIF2α control with mean ± SD (n = 3; ***P < .005; **P < .01). Triplicate of ex vivo differentiation were carried out using Hri +/+ or −/− FL erythroid progenitors isolated from embryos of the same mother. This set of experiment was repeated 3 times with similar results.
To further substantiate the inhibition of erythroid differentiation of Hri−/− erythroblasts, the formation of reticulocytes during ex vivo differentiation was measured. Reticulocytes, which are devoid of nuclei and are characterized as the Hoechst 33 342lowTer119high population, are shown in magenta in Figure 4B. Reticulocyte formation was greatly reduced in the differentiation of Hri−/− Ter119− erythroid progenitors compared with Hri+/+ cells at 20, 40 and 60 hours (Figure 4B). This inhibition of differentiation was further corroborated by observations of cell morphologies and hemoglobinization (supplemental Figure 6B). Taken together, these results demonstrate that Hri is required for differentiation of erythroid progenitors ex vivo, similar to the requirement of Hri in erythroid differentiation of adult spleens in vivo during iron deficiency (Figure 3A). Thus, this ex vivo erythroid differentiation system recapitulates the stress erythropoiesis observed in vivo, and provides an excellent in vitro system to investigate the molecular mechanism by which Hri regulates erythroid differentiation.
Activation of the Hri signaling pathway during erythroid differentiation
We then examined the activation of the Hri-eIF2αP-Atf4 signaling pathway during ex vivo differentiation of Hri+/+ and Hri−/− cells. Hri protein expression was increased at 36 and 48 hours. Furthermore, Hri was activated by hyperphosphorylation (Hri-P; Figure 4C). Most notably, there was Hri-dependent eIF2α phosphorylation and induction of Atf4, Chop, and Ho-1 protein expression during erythroid differentiation of Hri+/+, but not Hri−/− Ter119− erythroid progenitors (Figure 4C). Furthermore, we found that at 36 hours of ex vivo differentiation, Hri−/− cells also had significantly lower levels of Atf4, Chop, Ho-1, Gstμ, and Sod2 mRNAs (Figure 4D). Together, these results demonstrate the activation of the Hri signaling pathway during erythroid differentiation ex vivo.
MEL cell is an established cell line, which undergoes erythroid differentiation on treatment with dimethylsulfoxide (DMSO). We have shown that Hri expression is up-regulated on induction of erythroid differentiation of MEL cells,26 and that overexpression of the dominant-negative Hri mutant inhibits erythroid differentiation.27 We found that the Hri-eIF2αP-Atf4 pathway was also activated during DMSO-induced erythroid differentiation of MEL cells (supplemental Figure 7).
Requirement of Atf4 for erythroid differentiation
To determine whether Atf4 is necessary for erythroid differentiation, siRNA knockdown of Atf4 expression was performed in MEL cells. Three Atf4 siRNAs, S1, S2, and S3 were used individually and in combination. Both S1 and S2 siRNA significantly reduced Atf4 mRNA, wheras S3 was less effective (Figure 5A). The combination of all 3 siRNAs (S123) worked most effectively and reduced the Atf4 mRNA level to 34.5% of the control siRNA (Figure 5A). Importantly, S123 knockdown samples also had a lower mRNA level of β-globin major compared with the control siRNA (Figure 5B), indicating an inhibition of differentiation by knocking down Atf4 expression. Erythroid differentiation of MEL cells after treatment with control and S123 siRNAs was further examined by benzidine-Giemsa staining. Cells at different stages of differentiation were shown in Figure 5C and were scored morphologically (Figure 5D). These results demonstrated that knockdown of Atf4 expression in MEL cells resulted in inhibition of erythroid differentiation at the basophilic stage and in reduction of benzidine positive cells. Furthermore, S123-treated cells were less hemoglobinized as assessed by benzidine staining (Figure 5C-D), consistent with decreased β-globin major mRNA (Figure 5B). Together, these results support the requirement of Atf4 for erythroid differentiation.
Figure 5.

Inhibition of erythroid differentiation of MEL cells by knockdown of the Atf4 expression. (A) Knockdown of Atf4 expression in MEL cells by siRNAs. (B) Decreased expression of β-globin major mRNA in Atf4 knockdown cells. Data are presented as mean ± SD (n = 3, 3 separate transfections were performed). (C-D) Inhibition of erythroid differentiation in Atf4 knockdown cells. At 5 days after siRNA transfection, differentiating cells were stained with Giemsa and benzidine. Representative cells at different stages of differentiation stained are shown in panel C. Cell images were obtained with 40× objectives as describe for Figure 3B. (D) Percentages of cells at the basophilic erythroblast (Baso), polychromatic erythroblast (Poly), and orthochromatic erythroblast (Ortho) stages as well as benzidine-positive cells were determined by scoring the Giemsa-benzidine stained cells.
It was reported that ablation of the Atf4 gene in mice results in growth retardation and transient fetal anemia, which is attributed to a proliferation deficit in erythroid burst-forming unit (BFU-E) and erythrocyte colony-forming unit (CFU-E) erythroid progenitors.16 The effect of Atf4 knockout on erythroid differentiation has not been investigated. We found that the in vivo erythroid differentiation of E14.5 Atf4−/− FLs was modestly inhibited at the P1 stage with an increased percentage of cells at P1 and a decreased percentage of cells at the P3 stage (supplemental Figure 8B). This difference in erythroid differentiation between the Atf4−/− and Hri−/− FLs23 suggests the involvement of other eIF2α kinases at erythroid progenitor and proerythroblast stages because Atf4 expression is regulated by all 4 eIF2 α kinases.2 In addition, we found that Hri activation and eIF2αP were not altered in Atf4 deficiency (supplemental Figure 8C), consistent with Atf4 being downstream of the Hri signaling pathway (Figure 1B-C).
Modulation of Hri signaling pathway in β-thalassemic erythroid precursors by salubrinal
Increased oxidative stress and decreased erythroid differentiation exacerbate severity of β-thalassemia.28 We have shown earlier that Hri−/−Hbb−/− embryos died of severe anemia at E18.5 and Hri+/−Hbb−/− mice had more severe adult β-thalassemic phenotype.4 We investigated here the proof of concept for Hri and its signaling pathway as possible potential novel pharmaceutical targets for treatment of β-thalassemia by reducing ineffective erythropoiesis.
We used salubrinal, a small chemical that selectively inhibits the dephosphorylation of eIF2αP, to test its capability to enhance the Hri signaling pathway in Hri+/−Hbb−/− β-thalassemic erythroid precursors. As shown in Figure 6A, salubrinal treatment of Hri+/−Hbb−/− reticulocytes resulted in increased eIF2αP as well as in a significant decrease in the rate of globin protein synthesis (Figure 6B). It is important to note that phosphorylation of only 20%–30% of eIF2α is sufficient to completely inhibit protein synthesis.29 Furthermore, there was less 35S-globin in the insoluble pellet fractions of salubrinal-treated reticulocytes compared with the DMSO-treated control (Figure 6B). Salubrinal treatment also increased the levels of eIF2αP and Chop in Hri+/−Hbb−/− FL cells (Figure 6C).
Figure 6.

Modulation of Hri signaling by salubrinal in β-thalassemic erythroid precursors. (A) eIF2αP levels in Hri+/−Hbb−/− reticulocytes. Cells were treated for 6 hours with concentrations of salubrinal indicated. (B) Globin protein synthesis in Hri+/−Hbb−/− reticulocytes. After treatment with 100μM salubrinal for 2 hours, globin protein synthesis at times indicated was measured in the supernatant and pellet fractions. Vertical lines have been inserted to indicate a repositioned gel lane. The middle lane between Control and Sal in the bottom rows (pellet), which was the salubrinal treated sample at time zero before 35S-Met/Cys labeling, was removed. (C) eIF2αP and Chop in Hri+/−Hbb−/− FL erythroid precursors. Cells were treated for 12 hours with salubrinal at concentrations indicated. Numbers in panels A and C denote the ratio of eIF2αP/ eIF2α or Chop/eIF2α. Numbers in panel B denote the ratio of 35S-globin/total globin. (D) eIF2αP levels and protein synthesis in Ter119+ cells from Hri+/+Hbb−/− spleen. Cells were treated for 3.5 hours with salubrinal as indicated, and labeled with 35S-Met/Cys for the last 3 hours. Total protein syntheses in the cell lysates are shown in the middle panel. Newly synthesized 35S-Atf4 immunoprecipitated with antibody (Abnova) is shown in bottom panel. Numbers indicate ratio of eIF2αP/eIF2α or globin and Atf4 syntheses relative to 0 μM controls.
Similarly, salubrinal treatment of Hri+/+Hbb−/− Ter119+ erythroid precursors increased eIF2αP and inhibited protein synthesis in a concentration-dependent manner (Figure 6D). It is to be noted that Atf4 protein was synthesized in Hri+/+Hbb−/− Ter119+ erythroid precursors, consistent with activation of Hri in β-thalassemia.4 Importantly, at 10μM salubrinal, Atf4 mRNA translation (as determined by immunoprecipitation of newly synthesized 35S-Atf4 protein) was increased by 21%, whereas globin mRNA translation was decreased by 17%. At 25μM salubrinal, eIF2αP was further increased resulting in the shut-off of protein synthesis including Atf4. This is expected, as no eIF2 can be recycled when eIF2αP sequesters all eIF2B.
Together, these results demonstrate that salubrinal is effective in increasing eIF2αP and reducing denatured globins in β-thalassemic reticulocytes. Furthermore, salubrinal also enhances Atf4 translation and its subsequent signaling pathway in β-thalassemic erythroid precursors. These observations provide the foundation for exploiting the Hri-eIF2αP signaling pathway for treatment of thalassemia.
Discussion
Regulation of ROS levels and oxidative stress is extremely important in erythropoiesis. Starting at the basophilic erythroblast stage, erythroid precursors synthesize large amounts of hemoglobin, which requires heme as a prosthetic group. Thus, iron uptake for heme biosynthesis also increases, potentially generating ROS through the iron-catalyzed Fenton reaction.30 To date, 2 nonredundant Foxo3 and Nrf2-mediated pathways to combat oxidative stress have been identified in erythroid cells.31–33 We show here for the first time that the Hri-eIF2αP-Atf4 pathway is also required in the erythroid lineage for the adaptation to oxidative stress.
As illustrated in Figure 7A, phosphorylation of eIF2α by activated Hri not only leads to inhibition of globin translation but also leads to the selective enhanced translation of Atf4 and subsequent expression of redox genes (ie, Ho-1, Gstμ, and Nqo1) for adaptation to acute and chronic oxidative stress. During chronic iron deficiency of Hri−/− mice, both reduced antioxidant gene expression (Figure 2 and Liu et al23) and heme-free globin precipitates3 probably contribute to the increased ROS levels in Hri−/− erythroid cells. Although the exact mechanisms by which misfolded proteins in the cytosol generate oxidative stress have yet to be defined, there are 2 possibilities. Denatured heme-free globin aggregates may overwhelm and compromise capacities of molecular chaperons and proteosomal degradation. Both of these processes have been shown to mitigate ROS during ER stress. Reduction of improper disulfide bonds in misfolded proteins by g lutathione (GSH) can deplete GSH level. In addition, degradation of misfolded proteins is necessary to prevent ROS accumulation during ER stress.34
Furthermore, this Hri-eIF2αP-Atf4 pathway is also necessary for erythroid differentiation (Figure 7B). In nucleated erythroblasts, Hri not only inhibits globin translation, but also increases Atf4 translation to mitigate oxidative stress and to promote erythroid differentiation. At the enucleated reticulocyte stage, the role of Hri is only to regulate globin translation to prevent excessive globin synthesis. Both of these processes are necessary for optimal erythroid maturation to prevent anemia.
Hri signaling in Ho-1 expression and heme homeostasis
Induction of Ho-1 is critical for the cell survival response on oxidative stress.35 We found that induction of Ho-1 on arsenite stress in erythroblasts is dependent on Hri and Atf4 (Figure 2). Furthermore, Ho-1 induction during ex vivo differentiation of FL erythroid progenitors is also dependent on Hri (Figure 4). These results underscore a novel role of Hri in maintaining heme homeostasis and mitigating heme cytotoxicity through regulation of Ho-1 expression. Thus, Hri can control heme homeostasis not only by translational regulation of globin synthesis in accordance with heme availability,3 but also by Ho-1 induction during oxidative stress.
The transcription of Ho-1 and a large number of other antioxidant genes is regulated by Nrf2 through its binding to an antioxidant response element in the regulatory regions of these genes.36 Atf4 has been shown to interact with Nrf2 to regulate Ho-1 gene expression at the basal level as well as on induction by CdCl2.37 Thus, increased expression of Ho-1 and other Nrf2 target genes by Hri may be mediated by increased the translation of Atf4 and its subsequent interaction with Nrf2.
Hri signaling in erythropoiesis
The ineffective erythropoiesis in β-thalassemia is thought to be because of increased proliferation and accelerated apoptosis of erythroid precursors.38 However, a recent study by Libani et al28 demonstrates that decreased differentiation exacerbates the ineffective erythropoiesis in β-thalassemia. We show that the ineffective erythropoiesis occurring in Hri−/− mice during iron deficiency is primarily because of the profound inhibition of erythroid differentiation at the basophilic erythroblast stage (Figure 3). Inhibition of erythroid differentiation is also observed in several other mouse models of stress erythropoiesis, such as in Rb deficiency,39,40 and in Stat5a/5b deficiency.41
Although Hri−/− FL cells displayed a mild defect in erythroid differentiation in vivo,23 Ter119− Hri−/− FL erythroid progenitors showed a significant inhibition of erythroid differentiation at the basophilic erythroblast stage when cultured and differentiated ex vivo (Figure 4), recapitulating the inhibition of erythroid differentiation in vivo during iron deficiency.23 We demonstrate that the Hri-dependent eIF2αP-Atf4 pathway is activated during ex vivo differentiation of Ter119− erythroid precursors (Figure 4), and during erythroid differentiation of MEL cells. Furthermore, knockdown of Atf4 in MEL cells resulted in inhibition of erythroid differentiation (Figure 5). Thus, the Hri-Atf4 signaling pathway may be necessary for inducing transcription of genes required for erythropoiesis starting at the early erythroblast stage (Figure 7B).
Targeting the Hri-eIF2αP-Atf4 pathway for treatment of thalassemia
Accumulation of unpaired α-globin, oxidative stress, and ineffective erythropoiesis are well documented in β-thalassemia.6 We have shown earlier that Hri deficiency exacerbates the pathologic severity of Hbb−/− β-thalssemic mice.4 We demonstrate here the feasibility of augmenting the eIF2αP signaling pathway with salubrinal in erythroid precursors from β-thalassemic mice (Figure 6). Elevated eIF2αP achieved by salubrinal treatment resulted in inhibition of globin translation and reduction of insoluble globin protein. Furthermore, salubrinal treatment also enhances Atf4 signaling in erythroid precursors, which may be important in reducing the oxidative stress encountered in this disease.
In summary, the Hri-eIF2αP-Atf4 signaling pathway is necessary not only for mitigating oxidative stress, but also necessary for erythroid differentiation. This study reveals the novel function of Hri in erythroid differentiation beyond its established function in coordinating globin translation with heme availability.
Supplementary Material
Acknowledgments
The authors thank Drs Albert Liau, Julian Down, and Jane M. Yang, Harvard-MIT Division of Health Sciences and Technology, for their critical reading of the paper.
This research was supported in part by grants from the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases to J.-J.C. (DK 87984 and DK78442). R.N.V.S.S. and S.L. were supported in part by postdoctoral fellowships from the Cooley's Anemia Foundation.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: R.N.V.S.S., R.S.Z., S.L., and J.-J.C. designed experiments; R.N.V.S.S., R.S.Z., and J.G.V. performed experiments; C.-W.S. and T.M.T. provided the Atf4+/− mice; R.N.V.S.S., R.S.Z., and J.-J.C. wrote the paper; and all authors participated in the editing of the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for R.N.V.S.S. is Acceleron Pharma Inc, Cambridge, MA.
Correspondence: Jane-Jane Chen, Harvard-MIT Division of Health Sciences and Technology, Massachusetts Institute of Technology, E25-421A, 77 Massachusetts Ave, Cambridge, MA 02139; e-mail: j-jchen@mit.edu.
References
- 1.Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood. 2007;109(7):2693–2699. doi: 10.1182/blood-2006-08-041830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136(4):731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han AP, Yu C, Lu L, et al. Heme-regulated eIF2alpha kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001;20(23):6909–6918. doi: 10.1093/emboj/20.23.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han AP, Fleming MD, Chen JJ. Heme-regulated eIF2alpha kinase modifies the phenotypic severity of murine models of erythropoietic protoporphyria and beta-thalassemia. J Clin Invest. 2005;115(6):1562–1570. doi: 10.1172/JCI24141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu L, Han AP, Chen JJ. Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol. 2001;21(23):7971–7980. doi: 10.1128/MCB.21.23.7971-7980.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rivella S. Ineffective erythropoiesis and thalassemias. Curr Opin Hematol. 2009;16(3):187–194. doi: 10.1097/MOH.0b013e32832990a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harding HP, Zhang Y, Zeng H, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11(3):619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 8.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 9.Harding HP, Novoa II, Zhang Y, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 10.Wang XZ, Lawson B, Brewer JW, et al. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol Cell Biol. 1996;16(8):4273–4280. doi: 10.1128/mcb.16.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153(5):1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Connor JH, Weiser DC, Li S, Hallenbeck JM, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol Cell Biol. 2001;21(20):6841–6850. doi: 10.1128/MCB.21.20.6841-6850.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP, Ron D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003;22(5):1180–1187. doi: 10.1093/emboj/cdg112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kojima E, Takeuchi A, Haneda M, et al. The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. Faseb J. 2003;17(11):1573–1575. doi: 10.1096/fj.02-1184fje. [DOI] [PubMed] [Google Scholar]
- 15.Boyce M, Bryant KF, Jousse C, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307(5711):935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- 16.Masuoka HC, Townes TM. Targeted disruption of the activating transcription factor 4 gene results in severe fetal anemia in mice. Blood. 2002;99(3):736–745. doi: 10.1182/blood.v99.3.736. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Socolovsky M, Gross AW, Lodish HF. Role of Ras signaling in erythroid differentiation of mouse fetal liver cells: functional analysis by a flow cytometry-based novel culture system. Blood. 2003;102(12):3938–3946. doi: 10.1182/blood-2003-05-1479. [DOI] [PubMed] [Google Scholar]
- 18.Liu S, Suragani RN, Wang F, et al. The function of heme-regulated eIF2alpha kinase in murine iron homeostasis and macrophage maturation. J Clin Invest. 2007;117(11):3296–3305. doi: 10.1172/JCI32084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji P, Jayapal SR, Lodish HF. Enucleation of cultured mouse fetal erythroblasts requires Rac GTPases and mDia2. Nat Cell Biol. 2008;10(3):314–321. doi: 10.1038/ncb1693. [DOI] [PubMed] [Google Scholar]
- 20.Paradkar PN, Zumbrennen KB, Paw BH, Ward DM, Kaplan J. Regulation of mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol Cell Biol. 2009;29(4):1007–1016. doi: 10.1128/MCB.01685-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jousse C, Oyadomari S, Novoa I, et al. Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. J Cell Biol. 2003;163(4):767–775. doi: 10.1083/jcb.200308075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006;70(3):432–443. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 23.Liu S, Bhattacharya S, Han A, et al. Haem-regulated eIF2alpha kinase is necessary for adaptive gene expression in erythroid precursors under the stress of iron deficiency. Br J Haematol. 2008;143(1):129–137. doi: 10.1111/j.1365-2141.2008.07293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen K, Liu J, Heck S, Chasis JA, An X, Mohandas N. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc Natl Acad Sci U S A. 2009;106(41):17413–17418. doi: 10.1073/pnas.0909296106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Pop R, Sadegh C, Brugnara C, Haase VH, Socolovsky M. Suppression of Fas-FasL coexpression by erythropoietin mediates erythroblast expansion during the erythropoietic stress response in vivo. Blood. 2006;108(1):123–133. doi: 10.1182/blood-2005-11-4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crosby JS, Lee K, London IM, Chen J-J. Erythroid expression of the heme-regulated eIF-2alpha kinase. Mol Cell Biol. 1994;14:3906–3914. doi: 10.1128/mcb.14.6.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crosby JS, Chefalo PJ, Yeh I, et al. Regulation of hemoglobin synthesis and proliferation of differentiating erythroid cells by heme-regulated eIF-2alpha kinase. Blood. 2000;96(9):3241–3247. [PubMed] [Google Scholar]
- 28.Libani IV, Guy EC, Melchiori L, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood. 2008;112(3):875–885. doi: 10.1182/blood-2007-12-126938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinnebusch AG. Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes. In: Sonenberg N, Hershey JWB, Mathews MB, editors. Translational Control of Gene Expression. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2000. pp. 185–243. [Google Scholar]
- 30.Ghaffari S. Oxidative stress in the regulation of normal and neoplastic hematopoiesis. Antioxid Redox Signal. 2008;10(11):1923–1940. doi: 10.1089/ars.2008.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawatani Y, Suzuki T, Shimizu R, Kelly VP, Yamamoto M. Nrf2 and selenoproteins are essential for maintaining oxidative homeostasis in erythrocytes and protecting against hemolytic anemia. Blood. 2011;117(3):986–996. doi: 10.1182/blood-2010-05-285817. [DOI] [PubMed] [Google Scholar]
- 32.Marinkovic D, Zhang X, Yalcin S, et al. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J Clin Invest. 2007;117(8):2133–2144. doi: 10.1172/JCI31807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu D, dos Santos CO, Zhao G, et al. miR-451 protects against erythroid oxidant stress by repressing 14-3-3zeta. Genes Dev. 2010;24(15):1620–1633. doi: 10.1101/gad.1942110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell. 2004;15(5):767–776. doi: 10.1016/j.molcel.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 35.Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol. 2007;36(2):166–174. doi: 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- 36.Itoh K, Mimura J, Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal. 2010;13(11):1665–1678. doi: 10.1089/ars.2010.3222. [DOI] [PubMed] [Google Scholar]
- 37.He CH, Gong P, Hu B, et al. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem. 2001;276(24):20858–20865. doi: 10.1074/jbc.M101198200. [DOI] [PubMed] [Google Scholar]
- 38.Schrier SL. Pathophysiology of thalassemia. Curr Opin Hematol. 2002;9(2):123–126. doi: 10.1097/00062752-200203000-00007. [DOI] [PubMed] [Google Scholar]
- 39.Sankaran VG, Orkin SH, Walkley CR. Rb intrinsically promotes erythropoiesis by coupling cell cycle exit with mitochondrial biogenesis. Genes Dev. 2008;22(4):463–475. doi: 10.1101/gad.1627208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spike BT, Dirlam A, Dibling BC, et al. The Rb tumor suppressor is required for stress erythropoiesis. EMBO J. 2004;23(21):4319–4329. doi: 10.1038/sj.emboj.7600432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood. 2001;98(12):3261–3273. doi: 10.1182/blood.v98.12.3261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.