

Abstract
Single particle electron microscopy (EM), of both negative stained or frozen hydrated biological samples, has become a versatile tool in structural biology 1. In recent years, this method has achieved great success in studying structures of proteins and macromolecular complexes 2, 3. Compared with electron cryomicroscopy (cryoEM), in which frozen hydrated protein samples are embedded in a thin layer of vitreous ice 4, negative staining is a simpler sample preparation method in which protein samples are embedded in a thin layer of dried heavy metal salt to increase specimen contrast 5. The enhanced contrast of negative stain EM allows examination of relatively small biological samples. In addition to determining three-dimensional (3D) structure of purified proteins or protein complexes 6, this method can be used for much broader purposes. For example, negative stain EM can be easily used to visualize purified protein samples, obtaining information such as homogeneity/heterogeneity of the sample, formation of protein complexes or large assemblies, or simply to evaluate the quality of a protein preparation.
In this video article, we present a complete protocol for using an EM to observe negatively stained protein sample, from preparing carbon coated grids for negative stain EM to acquiring images of negatively stained sample in an electron microscope operated at 120kV accelerating voltage. These protocols have been used in our laboratory routinely and can be easily followed by novice users.
Keywords: Bioengineering, Issue 58, Electron Microscopy, EM, cryoEM, protein, negative stain, 3D structures
Protocol
1. Preparation of EM grids for carbon coating
Fill a large glass beaker with distilled water.
Use a glass pipette to drop a single drop of collodion (~20μl, 2% in amyl acetate, Electron Microscopy Sciences, PA, USA) on the surface of the water. Collodion will spread to form a thin plastic layer at the water/air surface; Note: A first drop of collodion may be used to clear the water surface from any dust particles. After removing of the film from the first drop, the water surface is clear of dust.
Place EM grids, 400 or 200 mesh Gilder Cu grids purchased from Ted Pella Inc. (USA), one by one onto the plastic layer floating on the water surface; We usually place 50 ~ 100 grids in a close packing pattern, with the shiny side of the grid facing down into the plastic layer (Fig. 1A). Note: 400 mesh grids are good for routine negative stain EM grids; whereas 200 mesh grids are good for collecting tilt pair images.
Cut a piece of paper with dimensions slightly larger than the area where grids are placed on the plastic layer. Place the paper carefully on top of the grid (Fig. 1B), and wait until the paper becomes completely wet (Fig. 1C). Note: Different types of paper have different water absorption rates. It is important that the paper does not absorb water too fast. For example, regular filter paper is not good for this purpose because it absorbs water too fast and makes a lot of wrinkles. The carbon film made from wrinkled paper is not flat, and thus not good for collecting tilt pair images for random conical tilt 3D reconstruction. One has to test different papers to find a suitable one. In our laboratory, we have been using papers from notepads bought from local grocery store. Sometimes the newspaper paper was also found to be suitable. Another option is to use the relative thick rice paper used for Chinese/Japanese calligraphy.
Rapidly make incisions on the plastic film around and close to the soaked paper. Immediately afterwards, use a pair of forceps to pick up the paper with the grids sandwiched between the plastic film and paper. Place the paper in a petri dish with the grids facing up for air drying (Fig. 1D); Note: It is important to let the paper air-dry. Drying too fast generates many wrinkles in the paper.
2. Carbon coating grids
The carbon-coating machine used in our laboratory is a Cressington 208C High Vacuum Turbo Carbon Coater (Ted Pella). Our system is equipped with a Cressington film thickness monitor, which measures the thickness of coated carbon film based on the principle that the oscillating frequency of a quartz crystal is changed by the mass of a deposited film on its surface. However, the protocol described here does not require a thickness monitor, since it is often not available in every laboratory.
Sharpen a carbon rod using a sharpener (Ted Pella) and load carbon rods in carbon coating machine (Fig. 2A). Two rods, one with a sharp tip and the other with a flat end are placed against each other.
Applying vacuum grease on half of the frosted area of a glass slide and place it next to the grids (Fig. 2B). Only half of the frosted area of the glass slide should be covered with vacuum grease. Note: The area with vacuum grease will not change color after carbon coating. The contrast between the adjacent areas with and without vacuum grease provides an estimation of thickness of coated carbon.
Place the paper with grids on the stage of carbon coating machine. Place the glass slide next to the grids (Fig. 2C). Note: One can also first transfer grids coated with the plastic film to a glass slide before coating, in case many grids fall off from the paper.
Pump the vacuum chamber, and wait until the vacuum reaches a level of 10-5 Torr. Note: coating with lower vacuum often generates many sparks, resulting to many large carbon particles on the grid.
Increase the current very slowly. The sharp tip of the carbon rod will first become red and then white as the current increases. Do not watch the high brightness tip directly with the naked eye. Stop increasing current when the coating starts. The glass slide will change color from white to light grey and then dark grey. The darkness of the slides correlates the thickness of the carbon film being coated. Turn the current off to stop coating when desired thickness has been achieved (Fig. 2D). Note: 1) After repeated carbon coating processes, coated carbon from the inside of the glass bell jar could darken its color. This makes the coated carbon film appear thicker than it actually is. 2) One sharpened carbon rod can be used for several coating. The sharpness of the tip is gradually reduced after each coating. The maximum current is influenced by the sharpness of the carbon rod. A freshly sharpened rod with smaller diameter requires a relative lower current to evaporate carbon. Later coating may require slightly higher current.
Vent the vacuum chamber and remove coated grids for later use. Note 1. The more accurate method of controlling the carbon thickness is by using a film thickness monitor. Or one can qualitatively calibrate color against the accurate thickness measured by the monitor. A useful carbon thickness for this technique is between 3 and 7 nm.
This is a simple and easy method to prepare a large quantity of carbon coated grids, usually ~100 grids per preparation. The grids contain a thin plastic layer, which usually does not affect the quality of the negatively stained image. However, grids prepared in this method are not suitable for cryoEM. This layer of plastic can be removed by soaking the grids into chloroform for several hours.
3. Preparing uranyl formate solution (0.75%) for negative stain EM. The detailed protocol was described previously 5
Weigh out 37.5mg of uranyl formate into a small beaker;
Add 5 ml of boiling water and stir for 5 minutes in the dark;
Add 10μl 5M NaOH, continue stir for 5 minutes;
Filter the solution using a syringe filter (Fisherbrand, 13mm syringe filter, 0.2μm, Nylon non-sterile, catalog number 09-720-5), and store the filtered solution in a 8ml Falcon tube covered with aluminum foil; Note. Uranyl Formate powder can be purchased from Electron Microscopy Sciences (Hatfield, PA), catalog number 22451. Uranyl formate solution is light sensitive and should be kept away from direct light, for example by covering the Falcon tube with aluminum foil. A freshly prepared solution has a light yellow color (Fig. 3A). Stain solution can be kept on the bench for one to two weeks before it starts precipitating. Uranyl formate is acidic with a pH ~4. Adding NaOH increases the pH, but adding it too quickly or in excess can cause the stain to precipitate. In addition to generating contrast, uranyl formate also fixes the protein sample. Because the fixation rate is so rapid, the low pH usually does not affect the overall protein conformations or protein complex formation 7.
4. Protocol for preparing negative stain EM grid. This protocol was described in detail previously 5
Glow-discharge carbon coated grid, using PELCO easiGlow GlowDischarge system (Ted Pella Inc., USA). The current used is 15 mA and grids are glow discharged for 30 sec. Note. Glow-discharged grids should be used shortly after glow discharging. We often use the grids in less than 2 hours after the glow discharge.
Place two 20μl drops of distilled water and two 25μl drops of uranyl formate solution on a piece of parafilm (Fig. 3B).
Hold a glow-discharged grid using reverse-force anti-capillary forceps with the carbon-coated side facing up. Note: It is necessary to use anti-capillary forceps. Otherwise, liquid sample will be sucked away from grids by the capillary force between the forceps.
Apply 2μl protein sample to the glow-discharged grid, and wait for 30 sec. Note: The waiting time and the length of glow discharge influence the concentration of proteins adsorbed in the grid. Very diluted samples require longer adsorption times. However, prolonged adsorption may change the buffer concentration since water vapors. To achieve a proper particle distribution in grid requires somewhat trial and error approach.
Blot off the sample using filter paper.
Touch the grid surface to a water drop and then blot off the water with filter paper. Repeat with the 2nd drop of water.
Touch the grid surface to the first drop of stain briefly. Blot off the stain solution with filter paper.
Touch the grid surface to the second drop of stain for 20 seconds. Blot off stain solution with filter paper.
Dry the grid using a vacuum. Note: one can also leave grids on the bench or in a chemical hood to air dry. The benefit of vacuum dry is that one can observe the grids in the EM immediately. The possible artifact generated by quick drying is uneven stain.
5. Transmission electron microscope
The microscope used in this demonstration is a Tecnai T12 (FEI Company) equipped with a Gatan 4K x 4K CCD camera.
Briefly check the alignment of the microscope before inserting sample; Note: This is only needed once per EM session. In a model microscope, near perfect alignment can be saved earlier. Loading such an alignment file should restore the microscope alignment to a near perfect condition.
Load the EM grid in the single tilt standard specimen holder.
Insert holder into CompuStage of the T12. Wait for the pumping cycle to complete.
Insert the holder into column.
Focus specimen and set a desired defocus, usually -1.5um. Note: Focusing can be achieved either by manually focusing using the minimum contrast method or using CCD camera to capture live images and compute live Fourier power spectrum.
Record an image using CCD camera;
In images of negatively stained proteins samples, proteins are often seen with white contrast, i.e. white density against a darker background. Note that while the protein concentrations in different areas of the same grid are usually quite similar, one does observe different levels of stain in different areas of the same grid. Sometimes areas could have uneven staining or even positive staining (protein appears dark on a light background), whereas some other areas have perfect stain. It is often necessary to search the grid to locate a good area for imaging.
6. Representative Results
Examples of typical good images of negatively stained samples, horse spleen ferritin (Figure 4A), Fab (Fig. 4B), archaeal 20S proteasome (Fig. 4C) and nucleosome (Fig. 4D), are shown.
 Figure 1. Preparation of carbon coated grids for negative stain EM. A) Placing EM grids one-by-one in a closed packing pattern on the surface of collodion film. B) Placing a piece of paper slightly larger than the area of the grids. C) Wait until the paper gets completely wet. D) Pick up the paper together with the grids for air-drying.
Figure 1. Preparation of carbon coated grids for negative stain EM. A) Placing EM grids one-by-one in a closed packing pattern on the surface of collodion film. B) Placing a piece of paper slightly larger than the area of the grids. C) Wait until the paper gets completely wet. D) Pick up the paper together with the grids for air-drying.
 Figure 2. Coating EM grids with carbon. A) Setup the carbon evaporator: place the two carbon rods in the coating apparatus, with one rod (on the left) sharpened to a point tip. The other rod (on the right) has a flat surface and come in contact with the point tip of the one in the opposite side. B) Place a small amount of vacuum grease in one side of a fostered glass slide. The area within the black frame has vacuum grease. C) Setup the grids for coating. The glass slides are used to hold the paper in position. The arrowhead points the area with vacuum grease. D) After coating, the color of the slide with vacuum grease covered does not change the color. The contrast indicates the thickness of coated carbon.
Figure 2. Coating EM grids with carbon. A) Setup the carbon evaporator: place the two carbon rods in the coating apparatus, with one rod (on the left) sharpened to a point tip. The other rod (on the right) has a flat surface and come in contact with the point tip of the one in the opposite side. B) Place a small amount of vacuum grease in one side of a fostered glass slide. The area within the black frame has vacuum grease. C) Setup the grids for coating. The glass slides are used to hold the paper in position. The arrowhead points the area with vacuum grease. D) After coating, the color of the slide with vacuum grease covered does not change the color. The contrast indicates the thickness of coated carbon.
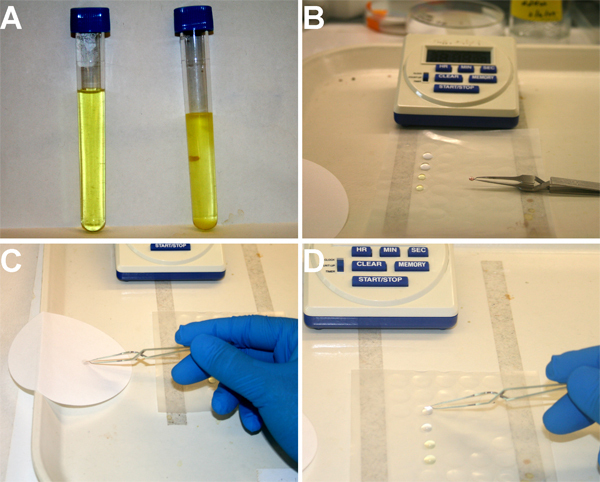 Figure 3. Illustrating the negative staining procedure. A) Freshly prepared (left) uranyl formate solution is transparent without sign of precipitates. After 1~2 weeks, precipitates start accumulating in the bottom in the bottom of the tube. B) Place two drops (~25μl) of distilled water and two drops of uranyl formate solution in a piece of parafilm. Use a pair of anti-capillary forceps to hold a glow discharged carbon-coated EM grid with carbon side up. A drop of sample (~2μl) is placed in the grid. C) After waiting for 30 second, blot the solution with a filter paper. D) Flip the grid to touch the sample side of the grid with the first drop of water. Repeat the step C and D to complete the process of staining.
Figure 3. Illustrating the negative staining procedure. A) Freshly prepared (left) uranyl formate solution is transparent without sign of precipitates. After 1~2 weeks, precipitates start accumulating in the bottom in the bottom of the tube. B) Place two drops (~25μl) of distilled water and two drops of uranyl formate solution in a piece of parafilm. Use a pair of anti-capillary forceps to hold a glow discharged carbon-coated EM grid with carbon side up. A drop of sample (~2μl) is placed in the grid. C) After waiting for 30 second, blot the solution with a filter paper. D) Flip the grid to touch the sample side of the grid with the first drop of water. Repeat the step C and D to complete the process of staining.
 Figure 4. Examples of EM images of negatively stained protein samples. A) Horse spleen ferritin, B) fragment of antigen binding (Fab), C) archaeal 20S proteasome and D) nucleosome. All images were recorded with the same magnification. The scale bar is 20nm.
Figure 4. Examples of EM images of negatively stained protein samples. A) Horse spleen ferritin, B) fragment of antigen binding (Fab), C) archaeal 20S proteasome and D) nucleosome. All images were recorded with the same magnification. The scale bar is 20nm.
Discussion
Negative stain EM is a powerful tool that can be used to study 3D structures of purified protein samples, such as using random conical tilt 8 method, which requires collecting a pair of images with one from specimen tilted to 60° and one from the same area but untilted, to calculate 3D reconstructions of macromolecular complexes. In addition, negative staining EM can have many other applications, such as screening for two-dimensional (2D) crystals of membrane proteins 9, visualizing protein complexes in different conformations 10, or simply for visualizing homogeneity and/or heterogeneity of purified proteins sample thus providing feedbacks to improve protein purification procedures, etc. It can be a powerful assay for biochemists. In this article, we presented the protocols ranging from preparing the carbon-coated grids to collecting images of negatively stained protein samples in an electron microscope. As illustrated here, the procedure is rather simple and easy to follow. If attempting to calculate 3D reconstructions of the sample being observed by random conical tilt method, it is necessary to acquire tilt pair images. However, if performing single particle analysis is the purpose, multiple untilted images will suffice. In both cases, further image processing is required and this is beyond the scope of this video article.
As with any method, there are variations to the protocols. Uranyl formate works very well as a negative stain reagent in most cases. Uranyl acetate is another commonly used negative stain reagent. It works very similar as uranyl formate, but with a larger grain size and, in many cases, lower contrast than uranyl formate. The advantage of using uranyl acetate is that the solution lasts longer than uranyl formate. One does not need to prepare a fresh solution every one or two weeks. Both uranyl formate and acetate are acidic. An obvious concern is that the low pH could induce changes in the protein sample being studied. Both stain solutions also function as protein fixatives. An earlier study has shown that both fix protein samples quickly 7. This rapid fixation makes the low pH less important. During the process of staining, a protein sample becomes fixed before the low pH can induce any changes. Nevertheless, other negative stain reagents, such as ammonium molybdate and phosphotungstic acid, may yield better results for staining samples when low pH is a concern 7. Both ammonium molybdate and phosphotungstic acid have an approximately neutral pH, but the contrast of protein samples generated by these stains is usually inferior to uranyl formate.
Disclosures
Production and Free Access of this video-article is sponsored by FEI.
Acknowledgments
Electron microscopy work at Cheng laboratory is partly support by grants from NIH (1R01GM082893, 1R01GM098672, 1S10RR026814-01 and P50GM082250 (to A. Frankel)), and a grant from UCSF Program of Breakthrough Biomedical Research, Opportunity Award and New Technology Award.
References
- Cheng Y, Walz T. The advent of near-atomic resolution in single-particle electron microscopy. Annu. Rev. Biochem. 2009;78:723–742. doi: 10.1146/annurev.biochem.78.070507.140543. [DOI] [PubMed] [Google Scholar]
- Grigorieff N, Harrison SC. Near-atomic resolution reconstructions of icosahedral viruses from electron cryo-microscopy. Curr. Opin. Struct. Biol. 2011;21(2):265–273. doi: 10.1016/j.sbi.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZH. Atomic resolution cryo electron microscopy of macromolecular complexes. Adv. Protein Chem. Struct. Biol. 2011;82:1–35. doi: 10.1016/B978-0-12-386507-6.00001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrian M, Dubochet J, Lepault J, McDowall AW. Cryo-electron microscopy of viruses. Nature. 1984;308(5954):32–36. doi: 10.1038/308032a0. [DOI] [PubMed] [Google Scholar]
- Ohi M, Li Y, Cheng Y, Walz T. Negative Staining and Image Classification - Powerful Tools in Modern Electron Microscopy. Biol. Proced. Online. 2004;6:23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl J. Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol. Cell. 2008;30(3):360–368. doi: 10.1016/j.molcel.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao FQ, Craig R. Capturing time-resolved changes in molecular structure by negative staining. J. Struct. Biol. 2003;141(1):43–52. doi: 10.1016/s1047-8477(02)00546-4. [DOI] [PubMed] [Google Scholar]
- Radermacher M, Wagenknecht T, Verschoor A, Frank J. Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli. J. Microsc. 1987;146(Pt. 2):113–136. doi: 10.1111/j.1365-2818.1987.tb01333.x. [DOI] [PubMed] [Google Scholar]
- Zhao G. Two-dimensional crystallization conditions of human leukotriene C4 synthase requiring adjustment of a particularly large combination of specific parameters. J. Struct. Biol. 2010;169(3):450–454. doi: 10.1016/j.jsb.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida N. Activation of leukocyte beta2 integrins by conversion from bent to extended conformations. Immunity. 2006;25(4):583–594. doi: 10.1016/j.immuni.2006.07.016. [DOI] [PubMed] [Google Scholar]