

Abstract
Sprouty proteins are established modifiers of receptor tyrosine kinase (RTK) signaling and play important roles in vasculogenesis, bone morphogenesis, and renal uteric branching. Little is understood, however, concerning possible roles for these molecular adaptors during hematopoiesis. Within erythroid lineage, Spry1 was observed to be selectively and highly expressed at CFU-e to erythroblast stages. In analyses of possible functional roles, an Mx1-Cre approach was applied to conditionally delete Spry1. At steady state, Spry1 deletion selectively perturbed erythroid development and led to reticulocytosis plus heightened splenic erythropoiesis. When challenged by hemolysis, Spry1-null mice exhibited worsened anemia and delayed recovery. During short-term marrow transplantation, Spry1-null donor marrow also failed to efficiently rescue the erythron. In each anemia model, however, hyperexpansion of erythroid progenitors was observed. Spry function depends on phosphorylation of a conserved N-terminal PY motif. Through an LC-MS/MS approach, Spry1 was discovered to be regulated via the erythropoietin receptor (EPOR), with marked EPO-induced Spry1-PY53 phosphorylation observed. When EPOR signaling pathways were analyzed within Spry1-deficient erythroid progenitors, hyperactivation of not only Erk1,2 but also Jak2 was observed. Studies implicate Spry1 as a novel regulator of erythropoiesis during anemia, transducer of EPOR signals, and candidate suppressor of Jak2 activity.
Introduction
Sprouty proteins (or Spry proteins) are best known as receptor tyrosine kinase (RTK)–activated modulators of fibroblast growth factor (FGF) and epidermal growth factor (EGF) signaling.1–3 Drosophila Sprouty (dSprouty) first was described as an FGF receptor (FGFR) antagonist, and modulator of apical branching within embryonic airways.4 For dSprouty, negative-feedback roles within EGFR and torso RTK systems also have been described (with Gap as one implicated target).5 Within vertebrates, 4 Spry proteins have evolved.1–3 This provides capacities for extended effects via varied spatial-temporal expression among tissues and via interactions with alternate molecular partners. One recent illustration is provided by dysregulation resulting from Spry2 disruption of diastema formation during tooth development.6 In this context, epithelial Spry2 plus mesenchymal Spry4 act in concert to enforce FGF refractivity and tooth bud regression.
With regards to molecular mechanisms that support the effects of Spry proteins, several studies emphasize negative actions on an Ras/Raf/Mek/Erk axis. Here, Raf is one prime candidate target for Spry binding.7 As recently studied in a T-cell receptor context, this also may involve Spry effects on PLC-γ.8 In Xenopus, such actions of Spry proteins have been extended to include regulation of the duration of ERK signaling, and consequentially dorsoventral patterning.9 Another candidate target for Spry proteins is PTEN. In particular, levels of this phosphoinositide phosphatase are increased on ectopic Spry2 expression, with accompanying observed decreases in Akt activation.10 Notably, Spry proteins also can exert apparent positive roles. This is perhaps best characterized in the EGFR system, and one mechanism involves Spry2 binding and sequestration of the E3 ubiquitin ligase, Cbl.11 When Cbl is sequestered, EGFR ubiquitinylation and turnover are attenuated, and signaling is sustained.12 Underlying these diverse effects of Spry proteins are several major structural features.1–3 These include an essential N-terminal PY motif for RTK/PTK-mediated phosphorylation (as well as Cbl and PTK binding), a regulatory serine-rich motif, and a C-terminal cysteine-rich domain, which mediates plasma membrane translocation and also encodes proposed docking sites for several cofactors, including Grb2 and Shp protein tyrosine phosphatases.3,13,14
Within hematopoietic contexts, comparably little attention has been paid to possible roles for Spry proteins. This is despite an intriguing prior set of experiments by Eckfeldt et al in which Spry proteins were identified as candidate markers of hematopoietic stem cells.15 Through RNAi experiments, zebrafish Spry4 further was indicated to regulate blood cell development15; and in murine gain-of-function experiments, Spry1 was suggested to modulate reconstitution of the hematopoietic system during bone marrow transplantation (possibly by affecting engraftment).16 In addition, Spred1 and Spred2 (as more distantly related orthologs of Sprys)17 recently have been implicated as mediators of eosinophilia and neutrophilia.18,19 For Spry2, epigenetic silencing interestingly has also been associated with B-cell lymphomagenesis.20
Through global transcriptome profiling of defined cohorts of developing murine bone marrow (pro)erythroblasts, we observed selective high-level expression of Spry1 during CFU-e to erythroblast development. This prompted us to study the role of Spry1 in hematopoiesis and erythropoiesis via a conditional Mx1-Cre–mediated disruption of Spry1. As one finding observed at steady state, Spry1 proved to be involved in normal erythroid homeostasis. Specifically, the loss of Spry1 in hematopoietic precursors led to reticulocytosis and the engagement of splenic stress erythropoiesis. After experimentally induced hemolysis, Spry1-deficient mice furthermore exhibited sustained, worsened anemia. This initially suggested positive erythropoietic roles for Spry1. In short-term bone marrow transplant experiments, Spry1-deficient donor cells also failed to efficiently reconstitute the erythron. In each anemia model, however, an overexpansion of proerythroblast progenitor pools was observed (together with an apparent faltering in progression to late-stage erythroblast formation). In related analyses, roles for Spry1 in modulating erythropoietin (EPO)–dependent phases of erythropoiesis were implicated. Specifically, via global PY-phospho-proteomic analyses, Spry1 was discovered to be a clear target for EPO/EPO receptor (EPOR)–mediated PY phosphorylation. Finally, within primary bone marrow proerythroblasts, Spry1 deficiency led to heightened EPO/EPOR activation of not only Erk1/2, but also Jak2. Collectively, these data reveal novel nonredundant roles for Spry1 as an erythropoietic regulator, a new target of the EPOR, and a potential suppressor of Jak2 activity. Findings therefore may point to candidate actions of Spry1 within the contexts of polycythemia and additional myeloproliferative neoplasms.21,22
Methods
Mouse and anemia models
Mice harboring a floxed Spry1 allele were as detailed previously23 and were backcrossed to C57BL/6 mice for more than 10 generations. Mx1-Cre mice are a fully C57BL/6 background (#005673; The Jackson Laboratory). Cre expression was activated via poly dIdC dosing (22.5 mg/kg, 5 doses at 2-day intervals followed by a 2-week no-treatment interval). Mice were studied at age 2 to 5 months. Control mice typically were Spry1+/flox:Mx1-Cre (or if limiting, age- and sex-matched wild-type C57BL/6 mice). Steady-state peripheral blood cell populations were determined using an Advia 2120 hematology analyzer (The Jackson Laboratory). To generate a hemolytic anemia, mice were dosed with phenylhydrazine (75 mg/kg, single intraperitoneal injection). In short-term bone marrow transplantations, 5 × 105 donor bone marrow was transplanted (retro-orbital injection) to irradiated B6 Ptprca recipients (450 cGy at 4 and 1 hour before transplantation). Hematocrits and reticulocyte levels also were determined by capillary microcentrifugation and thiazole-orange staining (via flow cytometry). Burst-forming units-erythroid (BFU-e) and colony-forming units-erythroid (CFU-e) assays were as per manufacturer-defined protocols (StemCell Technologies). All procedures used were approved by the Institutional Animal Care and Use Committee of the Maine Medical Center Research Institute.
Primary hematopoietic cell preparations
Marrow from thoroughly cleaned femurs and tibiae was expelled into IMDM (Invitrogen) plus 2% bovine serum. Cells then were passed through 21-gauge needles and a 40-μm strainer, and resuspended in PBS (#14190-144; Invitrogen). Before culture, red cells were lysed by exposure to buffered 0.8% ammonium chloride.24 Washed cells were used to initiate cultures at 1.8 × 106 cells/mL in StemPro34 medium (Invitrogen) supplemented with 2.5 U/mL EPO (Epoietin-alpha; Amgen), 100 ng/mL murine stem cell factor (PeproTech), 0.5μM dexamethasone, 1.5μM β-estradiol, 75 μg/mL holo-transferrin (T-0665; Sigma-Aldrich), 0.5% BSA (#9300; StemCell Technologies), 0.1mM 2-mercaptoethanol, 1.5mM l-glutamine (Invitrogen), 100 U/mL penicillin G, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B (ie, “SP34-ex” medium; Invitrogen). At 24 and 48 hours, medium was refreshed.24–26
At day 3 of expansion, stage E1, E2, and E3 cell populations were isolated via combined magnetic-activated selective cell depletion, plus retrieval procedures (Miltenyi Biotec). For E1 plus E2 populations, 2 rounds of lineage depletion (biotinylated CD5, CD45R/B220, CD11b, Ter119, and Ly6G antibodies; StemCell Technologies) first were applied (followed by an additional round of Ter119+ cell depletion). Stage E1 cells then were isolated via CD117+ cell retrieval with stringent washing. Remaining stage E2 cells were isolated via CD71 MACS retrieval. Stage E3 erythroblasts were isolated via MACS-based isolation of Ter119+ cells at an early point of formation. In EPO challenge experiments, erythroid progenitor cells (as specified) were isolated from SP34ex expansion cultures and were cultured for 5.5 hours in IMDM, 0.5% BSA, holo-transferrin (50 μg/mL), and insulin (15 ng/mL). Cells were then exposed to EPO as indicated before the direct preparation of cell lysates,24 or RNA25,26 (for use in transcriptome and/or RT-PCR analyses).
Flow cytometry
In flow cytometry, 1 × 106 cells were collected, washed, and incubated at 4°C in 0.2 mL PBS, 0.1% BSA plus 1 μg rat IgG for 15 minutes. Cells were then stained for 40 minutes at 4°C with allophycocyanin-CD117 (2 μg/mL), FITC-CD71 (4 μg/mL) plus PE-Ter119 (4 μg/mL; BD Biosciences). Washed cells then were analyzed (FACScalibur cytometer; CellQuest Version 3.2.1 software). For direct flow cytometry analyses, bone marrow or spleen cells were mechanically dispersed in IMDM, 2% FBS, sieved (40-μm strainer), washed in PBS (Invitrogen), and analyzed without ammonium chloride exposure (but with gating off red blood cells and reticulocytes).25 Allophycocyanin–annexin-V binding assays were performed in 140mM NaCl, 2.5mM CaCl2, 10mM HEPES, pH 7.4, for 30 minutes at 23°C. Washed cells were analyzed via flow cytometry as equivalent numbers of gated events in all experiments.
Transcriptome analyses and RT-PCR
RNA was isolated directly and was used to synthesize biotin-cRNA. Hybridizations were to Affymetrix 430 Version 2.0 arrays (GeneChip scanner 3000, and GCOS Version 1.0 software). GeneSpring software was used in data analyses (GC-RMA). Quantitative RT-PCR was performed as previously described25,26 using the following primer pairs (from SA Bioscience): Epo, Spry1, Spry2, Spry3, Spry4, and β-actin.
PY phosphoproteome analyses
In tandem LC-MS/MS analyses of EPOR-regulated protein tyrosine phosphorylation events, EPO-dependent erythromegakaryocytic UT7epo cells were used. Specifically, exponentially growing cells were washed and cultured for 8 hours in IMDM containing holo-transferrin (20 μg/mL), 0.5% BSA, 0.1mM 2-mercaptoethanol, and 20mM HEPES (pH 7.5). Cells then were exposed to EPO (± 4 U/mL). At 15 minutes, 9M urea lysates were prepared (in the presence of 1mM orthovanadate, 1mM β-glycerol phosphate, 2.5mM sodium pyrophosphate, and 20mM HEPES, pH 8.0). Cells were sonicated 3 times (15-W output) for 20-second intervals, with 1 minute cooling on ice between sonications. Homogenized lysates (2 × 108 cells per sample) were cleared by centrifugation for 15 minutes at 20 000g. Extracts (40 mg total protein each) were then reduced with DTT (4.5mM), carboxamidomethylated using iodoacetamide (10mM), and subsequently digested with trypsin (1:100 weight, trypsin to total protein). Peptides were separated from nonpeptide material by solid-phase extraction with Sep-Pak C18 cartridges. Lyophilized peptides were redissolved, and phosphorylated peptides were isolated using a slurry of immobilized phosphotyrosine antibody (#9411; Cell Signaling Technologies) conjugated to protein-G agarose beads (Roche Diagnostics). Peptides were eluted from antibody-resin into a total volume of 100 μL in 0.15% TFA. Eluted peptides were concentrated with PerfectPure C18 tips immediately before LC-MS analysis. Peptides were loaded directly onto a 10-cm by 75-μm PicoFrit capillary column packed with Magic C18 AQ reversed-phase resin. Reversed-phase columns were developed with a 45-minute linear gradient of acetonitrile in 0.125% formic acid (280 nL/min). Tandem mass spectra were collected with an LTQ-Orbitrap hybrid mass spectrometer using a top-10 method, a dynamic exclusion repeat count of 1, and a repeat duration of 30 seconds. MS spectra were collected in the Orbitrap component of the mass spectrometer, and MS/MS spectra were collected in the LTQ. MS/MS spectra were evaluated using TurboSequest in the Proteomics Browser package (Version 27, rev13). The false-positive assignment rate was approximated by taking the ratio of the reversed database assignments to the forward database assignments after filtering the initial SEQUEST search results based on XCorr (≥ 1.5), mass accuracy (± 10 ppm), and on the presence of the expected sequence motif (Y*). The mass accuracy range was narrowed further based on the XCorr-versus-mass error plot for each experiment. The average false-positive assignment rate obtained from the SEQUEST search for the 2 LC-MS/MS experiments (4 total LC-MS/MS runs) was determined to be approximately 0.8%. The ratios of the integrated peak height intensities for phosphopeptide quantification were obtained using the XCalibur Version 2.0.7 software (ThermoFinnigan). A reduction in peak intensity in EPO-treated cells compared with control was expressed as a negative fold change value. All integrated peak intensity calculations also were manually reviewed to ensure proper integration of consistently shaped, coeluting peaks. Changes in phosphorylated peptide levels were measured by taking the ratio of raw intensities between control and treated cells, with the untreated sample as the reference (denominator) in each case. Raw intensity ratios were normalized using a median adjustment technique whereby the log2 ratios comprising each binary comparison (EPO-treated vs control) was independently and globally adjusted such that the normalized median log2 ratio is zero. The normalized log2 ratios were then converted to their corresponding normalized fold-changes.
Retroviruses
To prepare retroviruses, mSpry1 cDNA was first cloned to pMSCVneo, and a C-terminal in-frame FLAG-tag cassette was added. The codon encoding Y53 (TAC) was then mutated to encode F53 (TTT). Retroviruses encoding each were then prepared by transfection of Clontech GP2-293 cells with these templates, plus VSV-G encoding constructs.27 Viruses were concentrated using Retro-Concentin reagent (SBI #RV100A-1) and titered in National Institutes of Health3T3 cells. Viruses then were used to stably express Spry1 (Flag) and Spry1Y53F(Flag) in UT7epo cells.28
Western blotting and immunopreciptations
Cell extracts were prepared as recently detailed.24–26 Cleared extracts were assayed for protein content, denatured, electrophoresed, transferred to polyvinylidene difluoride membranes, and blotted. Primary antibodies were from Cell Signaling: anti-Erk1,2 and anti-PT202, PY204, Erb1,2; anti-Jak2 and anti-PY1007, PY1008- Jak2; anti-Akt and anti-PS473Akt; anti-Stat5 and anti-Y694Stat5; anti-Stat1 and anti–Y701-Stat1; and anti–β-tubulin. HRP-conjugated antibodies and ECL reagents were as described previously.24–26 In immunoprecipitations, Spry1(Flag) and Spry1-Y53(Flag) were immunoprecipitated using Sepharose-immobilized anti-Flag epitope antibodies (Sigma-Aldrich) and eluted using a FLAG epitope peptide. In quantitative analyses of Western blot signals, ImageJ Version 1.41 software was used (http://rsb.info.nih.gov/ij/).
Results
Spry1 is highly expressed in developing (pro)erythroblasts, and conditional Spry1 deletion activates reticulocytosis plus splenic erythropoiesis
In the course of investigating the in vivo actions of EPO,24–26,29 our laboratory recently characterized 3 substages of bone marrow erythroid progenitor development as Kit+CD71highTer119− CFUe-like cells, Kit−CD71highTer119− proerythroblasts, and Kit− CD71highTer119+ erythroblasts (termed stages E1, E2, and E3; supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article).25 When these subpopulations were isolated and subjected to expression array profiling, a strong and developmentally sustained expression of select signal transduction factors was apparent, including Spry1. By comparison, no other Spry genes were expressed at elevated levels within these bone marrow (pro)erythroblasts (Figure 1A). Mining of extant transcriptome databases further showed that Spry1 levels were depressed in (erythro)megakaryocytic cells from Gata1-knockdown mice,30 suggesting a role for Gata1 in Spry1 expression (Figure 1B). Within a series of human CD34-derived developing erythroid progenitors,31 Spry1 levels also were observed to be maximal at an erythroblast stage (Figure 1C).
Figure 1.

Spry1 is selectively up-modulated in developing (pro)erythroblasts and is subject to Gata1 modulation. (A) Selective representation of Spry1 in developing murine bone marrow–derived erythroid progenitors. After short-term expansion in SP34ex medium, CFUe-like Kit+CD71highTer119− cells (stage E1), Kit−CD71highTer119− proerythroblasts (stage E2), and Kit−CD71highTer119+ erythroblasts (stage E3) were purified. Via gene profiling and RT-PCR analyses, Spry1 to Spry4 expression levels at each stage were then determined (mean ± SE; n = 3). Bottom panels: Giemsa-May-Grunwald–stained cytospins for isolated stage E1, E2, and E3 cells. (B) In erythromegakaryocytic splenocytes from Gata1-knockdown mice, Spry1 levels are decreased to approximately 25% of wild-type controls (GEO accession no. GSE2527).30 (C) In human CD34+-derived developing erythroid progenitors, Spry1 is also selectively elevated at a maturing erythroblast stage. Here, values are relative expression intensities (mean ± SE) derived from Keller et al31 (and GEO accession no. GDS2431).
Based on predominant (pro)erythroblast expression of Spry1 and on previously suggested roles for Spry proteins as regulators of hematopoiesis,15,16 a loss-of-function approach was used to determine the functional significance of Spry1 expression during erythropoiesis. To conditionally delete Spry1 in hematopoietic cells, C57BL/6 Mx1-Cre mice were crossed with C57BL/6 mice harboring a Spry1flox/flox allele. Mx1Cre:Spry1+/flox progeny were then crossed with Spry1flox/flox mice to yield Mx1Cre1:Spry1flox/flox mice. In adult mice, poly dIdC then was used to activate Cre expression and delete Spry1. Control mice (with the genotype Spry1flox/flox or Mx1Cre:Spry1+/flox) received poly dIdC in parallel. Efficient deletion of Spry1 was confirmed via RT-PCR of peripheral blood cells (Figure 2A).
Figure 2.
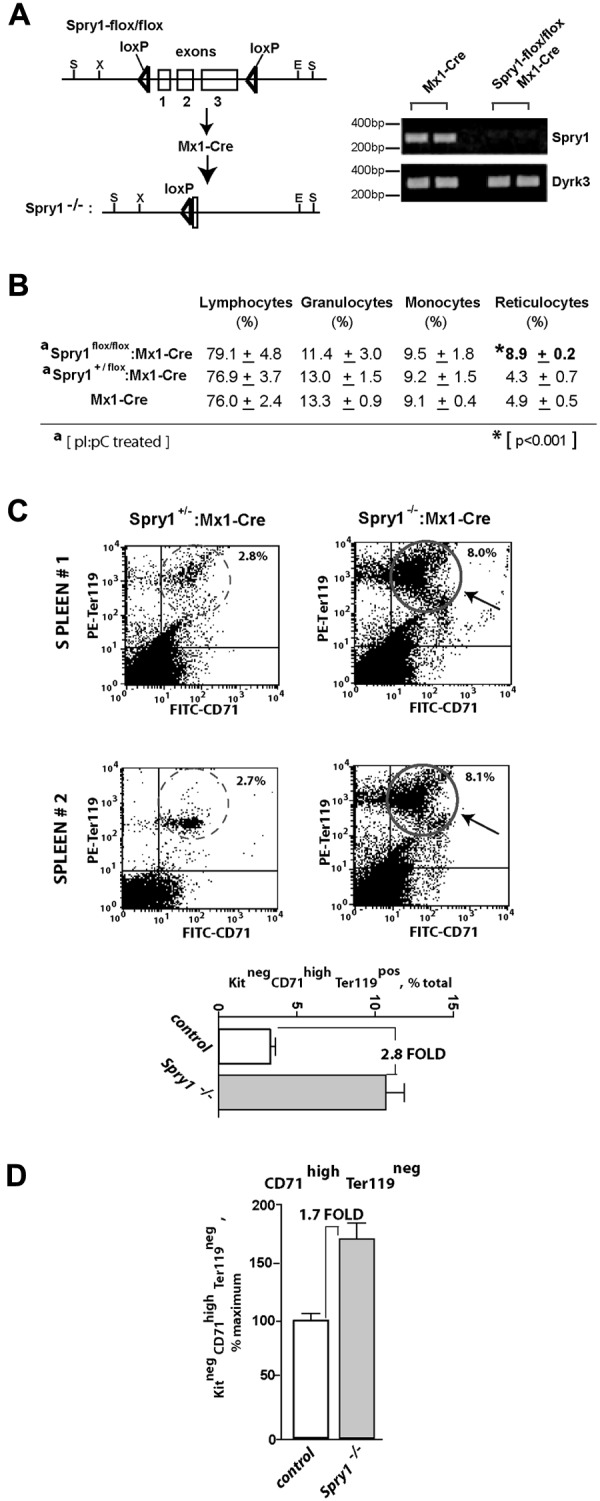
Conditional deletion of Spry1 leads to reticulocytosis and engagement of splenic erythropoiesis. (A) Mx1-Cre mediated deletion of Spry1. The floxed Spry1 allele used in these studies is illustrated, together with its efficient deletion after polyinosinic acid-polycytidylic acid activation of Mx1-Cre. In assessments of deletion, peripheral blood cells were analyzed by RT-PCR. (B) At steady state, Spry1 deletion selectively alters (and elevates) reticulocyte production. (C) Spry1 deletion induces splenic erythropoiesis. Flow cytometric analyses of erythroid progenitors within spleens of conditional Spry1−/− and control mice revealed an approximate 280% increase in erythroblast frequencies resulting from Spry1 deletion. Top panels: representative primary flow data. Bottom panel: mean (± SE) erythroblast frequencies (n = 4). (D) For Spry1−/− mice, flow analyses also identified an approximate 170% elevation of splenic stage E2 proerythroblasts.
At steady state, Spry1 deletion did not lead to major skewing of myeloid of lymphoid peripheral blood cell populations but did selectively elevate reticulocyte counts (Figure 2B, supplemental Table 1, supplemental Figure 2). In addition, flow cytometric analyses of splenic erythroid cells revealed increased erythropoiesis within the spleen. Specifically, Spry1 deletion elevated spleen-resident erythroblasts approximately 280% (Figure 2C). Levels of splenic stage E2 proerythroblasts also were elevated by approximately 170% (Figure 2D). Possible effects on Epo expression levels also were assessed via quantitative RT-PCR of kidney Epo levels.32 Levels in conditional Spry1-null mice were elevated 2.1- ± 0.3-fold (n = 4). This suggested selective modest roles for Spry1 during steady-state erythropoiesis. BFU-e and CFU-e levels within bone marrow of control versus Spry1-deleted mice, however, did not differ significantly at steady state (supplemental Table 2). In Mx1-Cre conditional Spry1-null mice, an apparent trend of increased platelet levels also was observed (supplemental Table 1). In the present study, platelets were not investigated further (but by speculation this trend might involve dysregulation of megakaryocyte-erythroid progenitors and/or possible roles for Spry1 during megakaryopoiesis).
During hemolytic anemia, and after short-term bone marrow transplantation, Spry1-deficient progenitors falter in repopulating the erythron but hyperexpand at proerythroblast stages
Based on the results of Figure 2, we next determined how mice harboring a conditional deletion of Spry1 responded to anemia incurred by phenylhydrazine-mediated hemolysis. In this context, Spry1 deficiency unexpectedly proved to significantly worsen anemia, with an observed nadir in hematocrit of 28.0% ± 0.57% compared with 33.84% ± 0.44% among controls (Figure 3A). Recovery from this severe anemia also was delayed, with a δ in hematocrit at day 9 of approximately 11 points. These results therefore are consistent with possible proerythropoietic roles for Spry1 during red cell formation. Interestingly, however, flow cytometric analyses of erythroid progenitors in spleen (after phenylhydrazine) also revealed increases in a subpopulation of Kit+ stage E1 CFUe-like progenitors resulting from Spry1 deletion (Figure 3B). At this time point of analysis, no substantial differences were observed in E2 or E3 pools (supplemental Table 3 central panel).
Figure 3.

Mx-Cre deletion of Spry1 worsens, and prolongs, hemolysis-induced anemia. (A) Conditional Spry1−/− and wild-type control mice were challenged by hemolysis (phenylhydrazine [PHZ]; 70 mg/kg). At the indicated intervals, red cell mass was assayed (hematocrits). Values are mean ± SE (n = 4). (B) Analyses of erythroid cells in spleen at day 3.5 after phenylhydrazine revealed an approximate 270% elevation in frequencies of Spry1-deficient stage E1 progenitors.
To better assess intrinsic properties of Spry1-deficient erythroid progenitors in the absence of hemolysis and oxidative stress, bone marrow transplant experiments were performed using marrow from Spry1-deleted versus control mice. At a time point when hematocrits were largely restored in irradiated recipients by transplanted control donor cells (day 16), hematocrits in mice transplanted with Spry1-deficient bone marrow failed to efficiently rebound (Figure 4A). In particular, recoveries in red cell mass were limited to approximately 54% of wild-type controls. In these experiments, relative frequencies of Kit−CD71high Ter119− stage E2 proerythroblasts also were analyzed within spleens of transplanted mice. Notably, E2 proerythroblasts proved to be over-represented (Figure 4C-D). This heightened frequency of splenic E2 cells (and to some extent E1 progenitors) also was reflected by an observed increase in absolute numbers within spleens of recipient mice (supplemental Table 3 bottom panel). This further suggested that Spry1 deficiency also might dysregulate proerythroblast expansion (and that sustained hyperexpansion might limit conversion of these cells to late stage E3 erythroblasts).
Figure 4.

Spry1-null hematopoietic progenitors fail to efficiently repopulate the erythron during short-term bone marrow transplantation. (A) Bone marrow cells from conditional Spry1-null mice, or wild-type controls, were transplanted to irradiated congenic recipients. At day 16, rebound hematocrits were determined. Values are mean ± SE (n = 4). Results of 2 independent experiments are illustrated. (B) Time course of erythron repopulation after short-term bone marrow transplantation of wild-type recipients as transplanted with control versus Spry1-null donor bone marrow cells. Graphed values are mean hematocrits (± SE) for n = 4 mice per time point. (C) Elevated stage E2 and E1 proerythroblast levels in mice transplanted with Spry1-null donor cells. In transplanted mice, analyses of erythroid progenitors in spleen revealed multifold increases in frequencies of Spry1- null donor-derived stage E2 proerythroblasts. Bottom panels: representative primary flow cytometry data. Bottom panel: composite data are graphed: mean ± SE (n = 4).
In an aim to assess possible effects of Spry1 deletion in erythroid progenitor cells (EPCs), at a cellular level, we also used an optimized ex vivo expansion (and development) system for murine bone marrow EPCs.25 Specifically, control and conditional Spry1-null EPCs were expanded (from bone marrow), and whether Spry1 deficiency might affect cell survival and/or cell cycle properties then was assessed. For survival potential, no advantages (or disadvantages) were detected among E1 or E2 EPCs (using YoPro3 as a sensitive agent for detecting compromised cells). Spry1-deficient stage E3 erythroblasts, however, exhibited moderately compromised survival profiles (supplemental Figure 3). These results suggest that, within this late EPC compartment, Spry1 may exert at least limited prosurvival effects.
EPOR mediates rapid phosphorylation of Spry1 at Y53, and Spry1 deficiency potentiates Erk1/2 plus Jak2 activation
Taken together, the outcomes from hemolysis-induced anemia and short term BMT suggested that one action of Spry1 might be to limit erythroid progenitor expansion during anemia (in both bone marrow and spleen). Within RTK receptor systems, Spry action depends on the induced phosphorylation of a conserved N-terminal tyrosine 53 residue.33 This especially is established for the FGFR.1–3 FGFs previously have been reported to modulate hematopoiesis34 and perhaps erythroid development.35 In transcriptome-based analyses of possible FGFR expression among stage E1, E2, and E3 (pro)erythroblasts, however, expression was very limited (to undetectable; Figure 5A).
Figure 5.
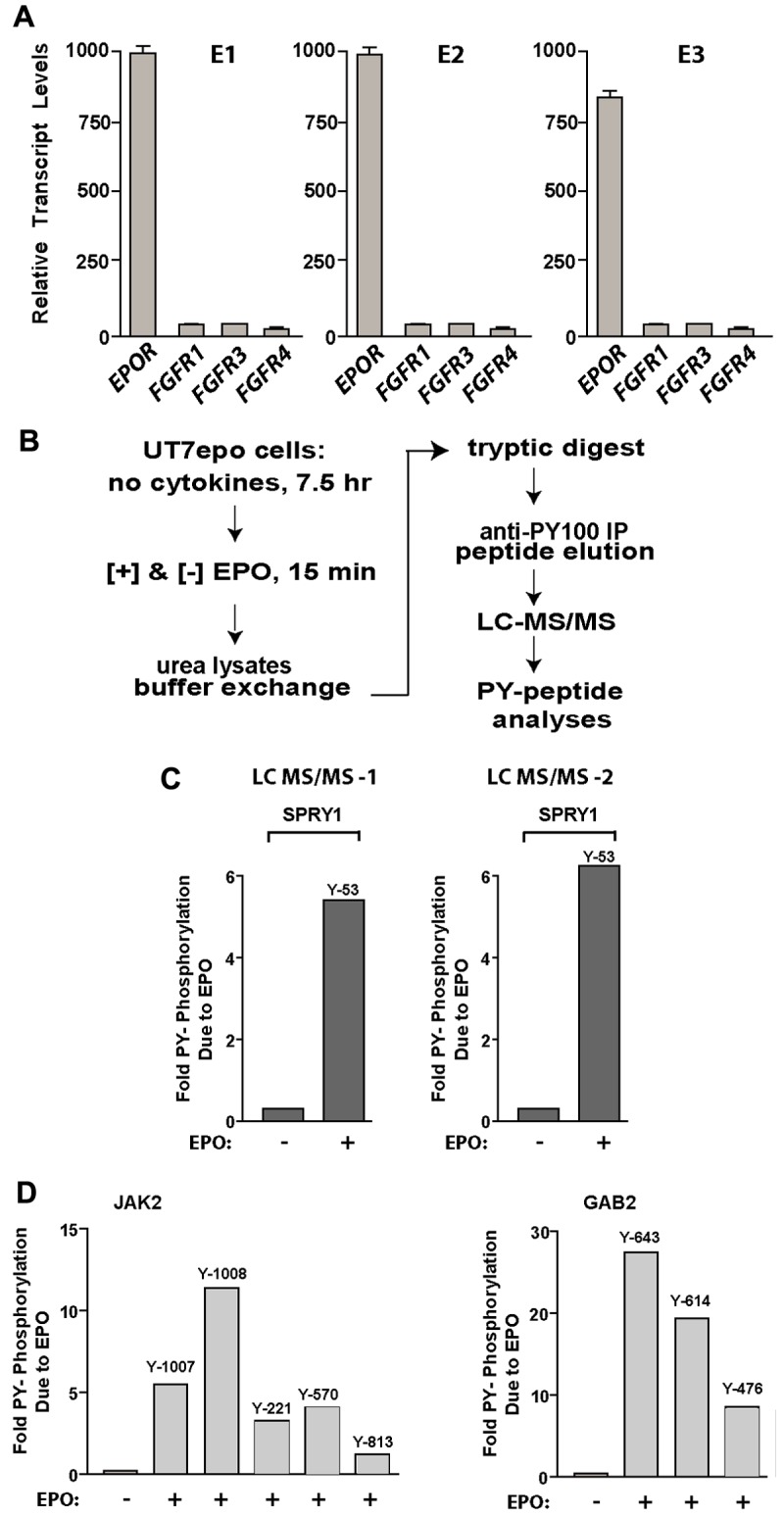
Spry1 is strongly and rapidly PY-phosphorylated on EPOR activation. (A) Primary bone marrow–derived erythroid progenitors do not express substantial levels of FGF receptors. In purified erythroid stage E1, E2, and E3 cells, transcriptome and RT-PCR analyses were applied to assess FGFR-1, -3, and -4 levels. (B) The LC-MS/MS approach used to discover novel EPOR PY-modulated signal transduction factors (including Spry1) in erythromegakaryocytic UT7epo cells is outlined. (C) Multifold EPO/EPOR stimulation of Spry1 phosphorylation at PY-Y53. Data shown illustrate results for duplicate LC-MS/MS analyses. (D) Verification of LC-MS/MS sensitivity and specificity based on EPO/EPOR regulation of JAK2 PY phosphorylation (PY sites Y-1007, Y-1008, Y-221, Y570, and Y-813) and GAB2 PY phosphorylation (PY sites Y643, Y614, and Y476).
Based on the dependency of late CFU-e and proerythroblasts for EPO, we next investigated whether Spry proteins might represent targets for EPOR/Jak2-mediated tyrosine phosphorylation. To provide residue-specific insight, an LC-MS/MS approach was applied (as outlined in Figure 5B). To provide sufficient cells for such analyses, EPO-dependent erythromegakaryocytic UT7epo cells were used. In these analyses (and for independent duplicate samples), Spry1 proved to be modulated in its Y53 phosphorylation more than 5.5-fold within 15 minutes of EPO exposure (Figure 5C, supplemental Figures 4, 5B-D). By direct comparison (and as validating examples), tyrosine phosphorylation of Jak2 was observed (in duplicate) at each of 5 known sites. As a known EPO/EPOR-regulated adaptor, Gab2 tyrosine phosphorylation was also observed to be induced at Y643, Y614, and Y476 (Figure 5D). To validate mass spectrometry data, Spry1 was epitope-tagged (C-terminus FLAG tag) and cloned to pMSCVneo. This pMSCVneo-Spry1(FLAG) template was used to prepare VSV-G packaged retroviruses, which were then stably transduced and expressed in derived UT7epo-Spry1(Flag) cells. To confirm (via Western blotting) EPO effects on Y53 phosphorylation, this high-complexity polyclonal cell line was cultured in the absence of hematopoietic cytokines (for 20 hours), challenged with EPO (15 minutes, 3 U/mL), and used to prepare Igepal lysates for anti-FLAG immunoprecipitation. The latter was accomplished by solid-phase immunoprecipitation, stringent washing, and FLAG peptide elution. Anti-PY Western blotting confirmed EPO-dependent tyrosine phosphorylation of Spry1 (supplemental Figure 5A). In addition, ectopic expression of Spry1(flag) limited EPO-dependent UT7epo cell growth, whereas the expression of a point-mutated Spry1-Y53F(flag) construct somewhat enhanced UT7epo expansion (supplemental Figure 6). This work therefore specifically establishes a novel and robust connection between ligated EPOR/Jak2 complexes and Spry1 activation via N-terminal PY site phosphorylation.
Based on the discovery that ligated EPOR/Jak2 complexes activate Spry1, the prospect that Spry1 might affect Jak2 next was tested using primary proerythroblasts from conditional Spry1-deficient versus control mice. Specifically, bone marrow–derived erythroid progenitors were expanded for a limited interval. Lin+ cells then were depleted. After a 5-hour period of hepatocyte growth factor withdrawal, cells were challenged with EPO (1 U/mL) for 0, 8, and 24 minutes. Levels of Jak2, Erk1,2, and Akt activation then were determined by Western blot analyses (using antibodies to PY-1007, 1008- Jak2; PT-202 PY-204 Erk1,2; and PS-473 Akt). Notably, within Spry1-deficient cells, Jak2 was activated by EPO at cumulative levels approximating 400% of control levels (Figure 6A). By direct comparison, no marked differential in activation was observed for Akt. EPO-induced phosphorylation of Erk1,2, however, proved to be sustained. When Stat5 was analyzed, an apparent modest increase in PY phosphorylation was observed because of Spry1 deletion, and a second molecular species of PY-Stat5 also was clearly generated (supplemental Figure 7). Therefore, within primary bone marrow erythroid progenitors, Jak2 is proposed to be subject to suppression by Spry1 (Figure 6B). Decreases in Spry1 expression therefore are suggested to lead to a dysregulation of erythroid progenitor expansion (Figure 6C), especially during anemia. During late-stage erythroblast differentiation, positive roles for Spry1 also may exist.
Figure 6.

In primary erythroid progenitors, conditional deletion of Spry1 derepresses Jak2. (A) Erythroid progenitors from the bone marrow of control (Spry1flox/flox) or Spry1-deleted mice were expanded via short-term culture in SP34ex. Lin+ cells were then depleted (including a second round of Ter119+ cell depletion). Kit+CD71highTer119− stage E1 cells were then isolated and cultured for 5.5 hours in the absence of hepatocyte growth factors. Cells then were exposed to EPO (1 U/mL for 0, 8, or 24 minutes), and lysates were directly prepared. Levels of PY-1007, 1008; Jak2 plus Jak2; P-Ser473-Akt plus Akt; and P-T202, P-Y204 Erk1,2 plus Erk1,2 then were determined by ECL Western blotting. Right panel: fold modulation of Jak2 PY-1007, 1008 phosphorylation by EPO in control versus Spry1-deficient cells is quantitatively analyzed (at 8 plus 24 minutes of EPO exposure). (B-C) Proposed routes for Spry1 regulation of erythropoiesis. (B) A model is outlined via which Spry1 acts to inhibit Jak2 as well as Erk1,2. (C) Routes for Spry1 regulation of proerythroblast expansion are also outlined, including heightened E1 and E2 cell expansion resulting from Spry1 disruption. A potential limiting of late-stage erythroblast (and red cell progeny) formation by Spry1 is also shown.
Discussion
For the presently reported findings, significance resides in 3 prime contexts. First, among 4 Spry genes, Spry1 is selectively highly expressed in developing primary bone marrow (pro)erythroblasts. Second, via a conditional loss-of-function approach, Spry1 is indicated to be functionally important in regulating balanced levels of erythroid progenitors versus differentiating erythroblast progeny, especially during anemia. In particular, deletion of Spry1 predominantly may dysregulate the expansion of proerythroblasts at the apparent expense of their advanced development. Third, a new connection is forged among the EPOR, Jak2, and Spry1 in which Spry1 composes a strong target for EPOR/Jak2-directed PY53 phosphorylation and acts (at least in part) in a negative feedback mode to inhibit Jak2 within activated EPOR complexes. These major aspects are discussed in further detail (later in this section). Possible implications for Spry proteins with hematologic disease contexts also are considered.
With regards to regulators of Spry expression, analyses in nonhematopoietic tissues previously have described strong expression of Spry proteins in FGF-responsive tissues,1–3,36 and FGFR and RTK induction of Spry genes has been described.37 Regulation of Spry gene expression, however, is obviously more complex. In kidney, for example, Spry1, Spry2, and Spry4 are expressed mainly in epithelial cells,36 and within uteric buds Spry1 is predominant.23,36 In contrast, during tooth development, Spry2 and Spry4 expression becomes restricted to epithelium and mesenchyme, respectively.6 Presently, Spry1 is shown to be selectively expressed at high levels in developing bone marrow–derived erythroid progenitors and (pro)erythroblasts. In silico analyses (see NCBI Geo GDS 1245) also suggest roles for Gata1 as a possible regulator in that Gata1 knockdown leads to a several-fold decrease in Spry1 expression within splenic progenitors. In addition, in global transcriptome analyses of EPO response circuits in primary bone marrow, CFUe-like progenitors (R. Verman, A Narayanan, D. Khurt, L. Li, S. Su, R. Asch, E. Jachimowicz, D.M.W., Transcriptome-based analysis of murine bone marrow CRUe to erythroblast development, including EPO-regulated targets and comparisons to human erythroid development, manuscript under revision) reveal modest yet significant EPO induction of Spry1 expression. Taken together, this indicates dynamic regulation of Spry1 expression during erythroid development.
To advance an understanding of functional roles of Spry1 during erythropoiesis and hematopoiesis, we used an Mx1-Cre conditional knockout approach. At steady state, and within peripheral blood, the deletion of Spry1 proved to affect only erythroid cells. Specifically, this involved reticulocytosis together with an activation of splenic erythropoiesis. This reticulocytosis did not lead to elevated hematocrits: Underlying factors might include effects on red cell half-life because of stress erythropoiesis production in the absence of Spry1 and/or possible hemolysis. During stress, Spry proteins can play extended roles.38–40 Spry4 (for example) has recently been characterized as a hypoxia-response factor.41 In keeping with this concept, erythropoietic roles for Spry1 became obvious during anemia. This included impaired red cell formation resulting from Spry1 deletion as observed during both hemolytic anemia and the anemia of short-term bone marrow transplantation. For hemolytic anemia, both severity and duration were affected. Essentially normal hematocrits, however, were ultimately restored. In phenylhydrazine and bone marrow transplant experiments, apparent increases in splenic erythroid progenitor populations among Spry1-null mice did not translate to increased erythrocyte levels. This might be attributable to heightened apoptosis among Spry1-null stage E3 erythroblasts or potential effects of Spry1 deficiency on macrophage and blood island formation. After bone marrow transplantation, recipient mice also recovered when transplanted with Spry1−/− donor marrow (but at markedly delayed rates; data not shown). Results of bone marrow transplant experiments, in addition, indicate erythroid cell- intrinsic roles for Spry1. In future experiments, whether rebound effects might reflect heightened contributions of known stress erythropoiesis mechanisms (eg, glucocorticoids or BMP4),42,43 or possible compensating effects of other Spry proteins17 will be of interest to discover. In the present studies, possible up-modulation of Spry2, Spry3, and/or Spry4 was assessed by RT-PCR of primary erythroid progenitors. No significant skewing, however, was observed (supplemental Figure 8).
From our in vivo and ex vivo analyses, Spry1 might also be suggested to limit the expansion of erythroid progenitor pools and to perhaps also promote late-stage erythroblast development. The prior case derives from an observed hyperexpansion of CFUe-like progenitors as well as proerythroblasts among Spry1-deficient cells in anemia contexts. At steady state, BFU-e and CFU-e levels were not significantly skewed because of Spry1 deletion. In addition, no apparent survival advantage was obvious among Spry1-null progenitors (data not shown). Proerythroblast hyperexpansion therefore is proposed to result from sustained activation of proliferative signals or possibly a differentiation block. Support for a case for hyperexpansion exists at a molecular level based on an observed elevated activation of Jak2 via the EPOR in Spry1-deficient erythroid progenitor cells. Previously, Spry proteins have been demonstrated through gain-of-function and/or loss-of-function studies to modulate Ras/Raf/Mek/Erk,1–3,5,7,9 pTEN/PI3K/Akt,10 Cbl,11,12 and select additional signal transduction routes.1–3 Consistent with the ability of Spry1 to attenuate a Raf/Mek/Erk axis,1–3,5,7,9 sustained activation of Erk1,2 also was presently observed in Spry1-deficient erythroid progenitors. Akt, in contrast, was not markedly affected. Notably, for a knocked-in PY-deficient EPOR allele (EPOR-HM), enhanced coupling selectively to Mek1 and Erk1,2 (but not Akt) also was observed and similarly associated with dysregulated erythroblast development.44 Consistent with this proposed feedback loop, Spry1 was discovered in UT7epo cells to be a major target for EPO/EPOR-mediated Y53 phosphorylation (Figure 5). Whether this is mediated directly by EPOR/Jak2 or possibly downstream PTKs is not yet resolved.
The present findings are novel in that, to our knowledge, Spry proteins have not previously been associated with erythropoiesis, EPO/EPOR action, or Jak kinase activity. With regards to Jak2 regulation, mechanisms engaged by Spry1 are of significant interest to consider based on Jak2 kinase mutations in myeloproliferative disorders, and polycythemia.21,22 Molecular mechanisms for Spry1 suppression of Jak2 presently are speculative. Based on previously reported Spry partners, 2 rational candidates are Cbl and Shp protein tyrosine phosphatases.1–3,11–13 If Spry1 sequestered Cbl, however, one prediction might be a more global activation of EPOR signaling, including Akt. Shp-1 and/or Shp-2 are perhaps attractive candidates in that Shp protein tyrosine phosphatases previously have been shown to inhibit Jak245,46 (and to bind Spry at a conserved cysteine-rich domain).3,13 Other potential routes also exist. Grb2 (for example) binds Spry proteins,3,14 and Grb2 recently has been suggested to be involved in Shp1 (and SOCS1) inhibition of Jak2.47 In a related vein, it is noteworthy that decreases in Spry2 levels have been associated with B-cell lymphomagenesis.20 Here, IL7R-Jak kinase circuits might also merit consideration. Future investigations will seek to specifically define molecular mechanisms of Spry1 inhibition of Jak2.
Supplementary Material
Acknowledgments
The authors thank Cell Signaling Technology Inc for LC-MS/MS analyses of regulated phosphotyrosine events (Tyrosine PhosphoScan), Dr T. Hunter and the Vermont Genetics Network (8P20GM103449 National Institute of General Medical Sciences [NIGMS] grant) for expert efforts in microarray hybridizations and initial analyses, C. Emerson for strong technical support in flow cytometry, Clinical Assessment Services in Histopathology, The Jackson Laboratory for peripheral blood cell analyses, and MMCRI's mouse genetic core for rederivations of Spry1flox/flox mice. Rederivations of Sprylflox/flox mice was supported in part by Maine Medical Center Research Institute's Mouse Transgenic Core (a core facility of MMCRI's COBRE Center in Vascular Biology P20RR015555/8P30GM103392).
This work was supported by the National Institutes of Health (NIH; R01DK089439, D.M.W.; and R01CA059998, J.D.L.) and the Maine Medical Center Research Institute core facilities in Flow Cytometry and Progenitor Cell Analysis and Genomics and Bioinformatics: as supported by the NIH/NIGMS (P20RR18789/8P20GM103465, D.M.W.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: P.S., J.D.L., and A.P. generated and maintained mouse models; P.S., A.D., and M.U. (with assistance from D.M.W.) performed EPO and anemia challenge experiments (and gene profiling experiments); A.P. contributed to flow cytometry, Western blotting, and ex vivo expansions; D.M.W. and J.D.L. provided lead roles in designing and directing experiments; and all authors contributed in substantial ways to data analyses, interpretations, and manuscript construction.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Don M. Wojchowski, Center of Excellence in Stem Cell Biology and Regenerative Medicine, Maine Medical Center Research Institute, 81 Research Dr, Scarborough, ME 04074; e-mail: wojchd@mmc.org.
References
- 1.Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006;16(1):45–54. doi: 10.1016/j.tcb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Cabrita MA, Christofori G. Sprouty proteins, masterminds of receptor tyrosine kinase signaling. Angiogenesis. 2008;11(1):53–62. doi: 10.1007/s10456-008-9089-1. [DOI] [PubMed] [Google Scholar]
- 3.Guy GR, Jackson RA, Yusoff P, Chow SY. Sprouty proteins: modified modulators, matchmakers or missing links? J Endocrinol. 2009;203(2):191–202. doi: 10.1677/JOE-09-0110. [DOI] [PubMed] [Google Scholar]
- 4.Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow MA. Sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell. 1998;92(2):253–263. doi: 10.1016/s0092-8674(00)80919-8. [DOI] [PubMed] [Google Scholar]
- 5.Casci T, Vinos J, Freeman M. Sprouty, an intracellular inhibitor of Ras signaling. Cell. 1999;96(5):655–665. doi: 10.1016/s0092-8674(00)80576-0. [DOI] [PubMed] [Google Scholar]
- 6.Klein OD, Minowada G, Peterkova R, et al. Sprouty genes control diastema tooth development via bidirectional antagonism of epithelial-mesenchymal FGF signaling. Dev Cell. 2006;11(2):181–190. doi: 10.1016/j.devcel.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sasaki A, Taketomi T, Kato R, et al. Mammalian Sprouty4 suppresses Ras-independent ERK activation by binding to Raf1. Nat Cell Biol. 2003;5(5):427–432. doi: 10.1038/ncb978. [DOI] [PubMed] [Google Scholar]
- 8.Akbulut S, Reddi AL, Aggarwal P, et al. Sprouty proteins inhibit receptor-mediated activation of phosphatidylinositol-specific phospholipase C. Mol Biol Cell. 2010;21(19):3487–3496. doi: 10.1091/mbc.E10-02-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanafusa H, Matsumoto K, Nishida E. Regulation of ERK activity duration by Sprouty contributes to dorsoventral patterning. Nat Cell Biol. 2009;11(1):106–109. doi: 10.1038/ncb1820. [DOI] [PubMed] [Google Scholar]
- 10.Edwin F, Singh R, Endersby R, Baker SJ, Patel TB. The tumor suppressor PTEN is necessary for human Sprouty 2-mediated inhibition of cell proliferation. J Biol Chem. 2006;281(8):4816–4822. doi: 10.1074/jbc.M508300200. [DOI] [PubMed] [Google Scholar]
- 11.Pennock S, Wang Z. A tale of two Cbls: interplay of c-Cbl and Cbl-b in epidermal growth factor receptor downregulation. Mol Cell Biol. 2008;28(9):3020–3037. doi: 10.1128/MCB.01809-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun Q, Jackson RA, Ng C, Guy GR, Sivaraman J. Additional serine/threonine phosphorylation reduces binding affinity but preserves interface topography of substrate proteins to the c-Cbl TKB domain. PLoS One. 2010;5(9):e12819. doi: 10.1371/journal.pone.0012819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jarvis LA, Toering SJ, Simon MA, Krasnow MA, Smith-Bolton RK. Sprouty proteins are in vivo targets of Corkscrew/SHP-2 tyrosine phosphatases. Development. 2006;133(6):1133–1142. doi: 10.1242/dev.02255. [DOI] [PubMed] [Google Scholar]
- 14.Lao DH, Chandramouli S, Yusoff P, et al. A Src homology 3-binding sequence on the C terminus of Sprouty2 is necessary for inhibition of the Ras/ERK pathway downstream of fibroblast growth factor receptor stimulation. J Biol Chem. 2006;281(40):29993–30000. doi: 10.1074/jbc.M604044200. [DOI] [PubMed] [Google Scholar]
- 15.Eckfeldt CE, Mendenhall EM, Flynn CM, et al. Functional analysis of human hematopoietic stem cell gene expression using zebrafish. PLoS Biol. 2005;3(8):e254. doi: 10.1371/journal.pbio.0030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckfeldt C, Mendenhall E, Verfaillie C. SPRY1 is a negative regulator of long-term in vivo engraftment and ex vivo expansion of primitive human umbilical cord blood cells [abstract]. Blood (ASH Annual Meeting Abstracts) 2005;106(11) Abstract 1715. [Google Scholar]
- 17.Bundschu K, Walter U, Schuh K. Getting a first clue about SPRED functions. Bioessays. 2007;29(9):897–907. doi: 10.1002/bies.20632. [DOI] [PubMed] [Google Scholar]
- 18.Takatsu K, Nakajima H. IL-5 and eosinophilia. Curr Opin Immunol. 2008;20(3):288–294. doi: 10.1016/j.coi.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Miyamoto K, Miyamoto T, Kato R, Yoshimura A, Motoyama N, Suda T. FoxO3a regulates hematopoietic homeostasis through a negative feedback pathway in conditions of stress or aging. Blood. 2008;112(12):4485–4493. doi: 10.1182/blood-2008-05-159848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frank MJ, Dawson DW, Bensinger SJ, et al. Expression of sprouty2 inhibits B-cell proliferation and is epigenetically silenced in mouse and human B-cell lymphomas. Blood. 2009;113(11):2478–2487. doi: 10.1182/blood-2008-05-156943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tefferi A. JAK2 mutations and clinical practice in myeloproliferative neoplasms. Cancer J. 2007;13(6):366–371. doi: 10.1097/PPO.0b013e318159467b. [DOI] [PubMed] [Google Scholar]
- 22.Marubayashi S, Koppikar P, Taldone T, et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest. 2010;120(10):3578–3593. doi: 10.1172/JCI42442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basson MA, Akbulut S, Watson-Johnson J, et al. Sprouty1 is a critical regulator of GDNF/RET-mediated kidney induction. Dev Cell. 2005;8(2):229–239. doi: 10.1016/j.devcel.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 24.Sathyanarayana P, Houde E, Marshall D, et al. CNTO 530 functions as a potent EPO mimetic via unique sustained effects on bone marrow proerythroblast pools. Blood. 2009;113(20):4955–4962. doi: 10.1182/blood-2008-08-172320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dev A, Fang J, Sathyanarayana P, Pradeep A, Emerson C, Wojchowski DM. During EPO or anemia challenge, erythroid progenitor cells transit through a selectively expandable proerythroblast pool. Blood. 2010;116(24):5334–5346. doi: 10.1182/blood-2009-12-258947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang J, Menon M, Kapelle W, et al. EPO modulation of cell-cycle regulatory genes, and cell division, in primary bone marrow erythroblasts. Blood. 2007;110(7):2361–2370. doi: 10.1182/blood-2006-12-063503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sathyanarayana P, Dev A, Fang J, et al. EPO receptor circuits for primary erythroblast survival. Blood. 2008;111(11):5390–5399. doi: 10.1182/blood-2007-10-119743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Komatsu N, Nakauchi H, Miwa A, et al. Establishment and characterization of a human leukemic cell line with megakaryocytic features: dependency on granulocyte-macrophage colony-stimulating factor, interleukin 3, or erythropoietin for growth and survival. Cancer Res. 1991;51(1):341–348. [PubMed] [Google Scholar]
- 29.Wojchowski DM, Sathyanarayana P, Dev A. Erythropoietin receptor response circuits. Curr Opin Hematol. 2010;17(3):169–176. doi: 10.1097/MOH.0b013e328338008b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muntean AG, Crispino JD. Differential requirements for the activation domain and FOG-interaction surface of GATA-1 in megakaryocyte gene expression and development. Blood. 2005;106(4):1223–1231. doi: 10.1182/blood-2005-02-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keller MA, Addya S, Vadigepalli R, et al. Transcriptional regulatory network analysis of developing human erythroid progenitors reveals patterns of coregulation and potential transcriptional regulators. Physiol Genomics. 2006;28(1):114–128. doi: 10.1152/physiolgenomics.00055.2006. [DOI] [PubMed] [Google Scholar]
- 32.Menon MP, Karur V, Bogacheva O, Bogachev O, Cuetara B, Wojchowski DM. Signals for stress erythropoiesis are integrated via an erythropoietin receptor-phosphotyrosine-343-Stat5 axis. J Clin Invest. 2006;116(3):683–694. doi: 10.1172/JCI25227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mason JM, Morrison DJ, Bassit B, et al. Tyrosine phosphorylation of Sprouty proteins regulates their ability to inhibit growth factor signaling: a dual feedback loop. Mol Biol Cell. 2004;15(5):2176–2188. doi: 10.1091/mbc.E03-07-0503. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.de Haan G, Weersing E, Dontje B, et al. In vitro generation of long-term repopulating hematopoietic stem cells by fibroblast growth factor-1. Dev Cell. 2003;4(2):241–251. doi: 10.1016/s1534-5807(03)00018-2. [DOI] [PubMed] [Google Scholar]
- 35.Yamauchi H, Hotta Y, Konishi M, Miyake A, Kawahara A, Itoh N. Fgf21 is essential for haematopoiesis in zebrafish. EMBO Rep. 2006;7(6):649–654. doi: 10.1038/sj.embor.7400685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang S, Lin Y, Itaranta P, Yagi A, Vainio S. Expression of Sprouty genes 1, 2 and 4 during mouse organogenesis. Mech Dev. 2001;109(2):367–370. doi: 10.1016/s0925-4773(01)00526-3. [DOI] [PubMed] [Google Scholar]
- 37.Ozaki K, Kadomoto R, Asato K, Tanimura S, Itoh N, Kohno M. ERK pathway positively regulates the expression of Sprouty genes. Biochem Biophys Res Commun. 2001;285(5):1084–1088. doi: 10.1006/bbrc.2001.5295. [DOI] [PubMed] [Google Scholar]
- 38.Abou-Khalil R, Brack AS. Muscle stem cells and reversible quiescence: the role of sprouty. Cell Cycle. 2010;9(13):2575–2580. doi: 10.4161/cc.9.13.12149. [DOI] [PubMed] [Google Scholar]
- 39.Taniguchi K, Sasaki K, Watari K, et al. Suppression of Sproutys has a therapeutic effect for a mouse model of ischemia by enhancing angiogenesis. PLoS One. 2009;4(5):e5467. doi: 10.1371/journal.pone.0005467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lito P, Mets BD, Appledorn DM, Maher VM, McCormick JJ. Sprouty 2 regulates DNA damage-induced apoptosis in Ras-transformed human fibroblasts. J Biol Chem. 2009;284(2):848–854. doi: 10.1074/jbc.M808045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haigl B, Mayer CE, Siegwart G, Sutterluty H. Sprouty4 levels are increased under hypoxic conditions by enhanced mRNA stability and transcription. Biol Chem. 2010;391(7):813–821. doi: 10.1515/BC.2010.082. [DOI] [PubMed] [Google Scholar]
- 42.Bauer A, Tronche F, Wessely O, et al. The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev. 1999;13(22):2996–3002. doi: 10.1101/gad.13.22.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu DC, Paulson RF. Hypoxia regulates BMP4 expression in the murine spleen during the recovery from acute anemia. PLoS One. 2010;5(6):e11303. doi: 10.1371/journal.pone.0011303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menon MP, Fang J, Wojchowski DM. Core erythropoietin receptor signals for late erythroblast development. Blood. 2006;107(7):2662–2672. doi: 10.1182/blood-2005-02-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu D, Qu CK. Protein tyrosine phosphatases in the JAK/STAT pathway. Front Biosci. 2008;13:4925–4932. doi: 10.2741/3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han Y, Amin HM, Franko B, Frantz C, Shi X, Lai R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood. 2006;108(8):2796–2803. doi: 10.1182/blood-2006-04-017434. [DOI] [PubMed] [Google Scholar]
- 47.Minoo P, Zadeh MM, Rottapel R, Lebrun JJ, Ali S. A novel SHP-1/Grb2-dependent mechanism of negative regulation of cytokine-receptor signaling: contribution of SHP-1 C-terminal tyrosines in cytokine signaling. Blood. 2004;103(4):1398–1407. doi: 10.1182/blood-2003-07-2617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.