

Abstract
This study was aimed at establishing buffalo embryonic stem cells (ESCs) from in vitro fertilized (IVF), parthenogenetic, and hand-made cloned (HMC) embryos and to check their equivalency in terms of stem cell marker expression, longevity, proliferation, and differentiation pattern. ESCs derived from all three sources were found by immunofluorescence to express the pluripotency markers SSEA-4, TRA-1-60, TRA-1-81, OCT4, and SOX2 and were able to form embryoid bodies containing cells expressing genes specific to endoderm (AFP, HNF4, and GATA4), mesoderm (MSX1, BMP4, and ASA), and ectoderm (cytokeratin 8 and NF68). Reverse transcriptase PCR (RT-PCR) showed cells from all sources to be positive for pluripotency markers OCT4, SOX2, NANOG, STAT3, REX1, FOXD3, NUCLEOSTEMIN, and TELOMERASE. Pluripotency markers OCT4, SOX2, NANOG, and c-MYC were also analyzed by real-time PCR. No significant differences were observed among ESCs from all three sources for all these genes except NANOG, whose expression was higher (p<0.05) in HMC-derived ESCs (6.897±2.3) compared to that in parthenogenesis- and IVF-derived cells (1.603±0.315 and 1±0, respectively). Pluripotent, stable buffalo ESC lines derived from IVF, parthenogenesis, and HMC embryos may be genetically manipulated to provide a powerful tool for studies involving embryonic development, genomic imprinting, gene targeting, cloning, chimera formation, and transgenic animal production.
Introduction
Embryonic stem cells (ESCs) are derived from the inner cell mass (ICM) of the blastocyst-stage embryos. These cells have the unique characteristics of propagating in an undifferentiated state for an indefinite period of time while maintaining their pluripotent status, i.e., the ability to differentiate to any cell type, including gametes. The establishment of ESC lines from laboratory and livestock animals is one of the most important achievements in the last decade and is expected to have a big influence on the genetic modification of livestock, both through genetically engineered production of transgenic animals and through large-scale cloning of embryos. However, one of the major reasons limiting the application of ESC technology to livestock species has been the lack of availability of true ESC lines capable of undergoing germ-line transmission. In addition to a number of laboratory animal species such as rat (Iannaccone et al., 1994; Vassilieva et al., 2000), hamster (Doetschman et al., 1988), and rabbit (Schoonjans et al., 1996), cell lines of ESC-like cells have been isolated from many species of farm animals such as pig (Chen et al., 1999; Li et al., 2004; Notarianni et al., 1990; Wheeler, 1994), cattle (Cibelli et al., 1998; First et al., 1994; Mitalipova et al., 2001; Wang et al., 2005), sheep (Notarianni et al., 1991; Zhu et al., 2007), horse (Saito et al., 2002), goat (Keefer et al., 1996), and buffalo (Anand et al., 2011; Verma et al., 2007).
In addition to embryos produced by in vitro fertilization (IVF), those produced through hand-made cloning (HMC)/nuclear transfer (NT) or parthenogenesis are equally valuable for generation of ESCs, especially in the case of humans where homozygosity is prime. Somatic cells obtained from patients could be used to generate blastocyst-stage embryos through somatic cell nuclear transfer or parthenogenesis. The ESCs derived from such embryos would offer high histocompatibility and could be used for developing patient-specific drug-testing systems. In addition to these, another approach could be to use somatic cells for producing induced pluripotent stem cells (iPSCs) (Okita et al., 2007; Park et al., 2008; Takahashi et al., 2007; Wernig et al., 2007), which would offer advantages similar to those mentioned above in terms of being patient specific (Kim et al., 2009; Nishikawa et al., 2008). However, the equivalence of ESCs obtained through these sources with that of ESCs produced by the conventional approach needs confirmation through further investigations.
Our goal in this study was to investigate the efficiency of ESC derivation from blastocyst-stage embryos generated by IVF, parthenogenetic activation, and HMC in buffalo.
Materials and Methods
All of the chemicals and media were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Disposable plastic wares were from Nunc (Roskilde, Denmark), unless otherwise mentioned.
Production of embryos by in vitro fertilization, parthenogenesis, and hand-made cloning
IVF
In vitro maturation and fertilization of buffalo oocytes was carried out as described earlier (Chauhan et al., 1998) with some modifications. Briefly, usable quality cumulus–oocyte complexes (COCs) obtained from slaughterhouse buffalo ovaries were cultured in In Vitro Maturation (IVM) medium, which consisted of tissue culture medium-199 (TCM)-199, 10% fetal bovine serum (FBS), 5 μg mL−1 porcine follicle-stimulating hormone (pFSH), 1 μg mL−1 estradiol-17β, 0.81 mM sodium pyruvate, 10% buffalo follicular fluid, and 50 μg mL−1 gentamicin sulfate in groups of 15–20 COCs per 100-μL droplet of the IVM medium in 35-mm Petri dishes in a CO2 incubator (5% CO2 in air) at 38.5°C for 21 h after covering them with sterile mineral oil. For IVF, two straws of frozen–thawed ejaculated buffalo semen were washed twice with washing Bracket and Oliphant's (BO) medium (BO medium containing 10 mg mL−1 heparin, 137.0 mg mL−1 sodium pyruvate, and 1.942 mg mL−1 caffeine sodium benzoate). The pellet was resuspended in 0.5 mL of the washing BO medium. Matured COCs were washed three times with washing BO medium and transferred to 50-μL droplets (15–20 oocytes/droplet) of the capacitation and fertilization BO medium [washing BO medium containing 10 mg mL−1 fatty acid–free bovine serum albumin (BSA-FAF). The spermatozoa in 50 μL of the capacitation and fertilization BO medium (≈3 million spermatozoa mL−1) were then added to the droplets containing the oocytes, covered with sterile mineral oil, and placed in a CO2 incubator (5% CO2 in air) at 38.5°C for 16–18 h. The spermatozoa used for IVF throughout the study had been tested for IVF efficiency earlier.
The cumulus cells were removed from the oocytes by gentle pipetting at the end of sperm–oocyte incubation. The oocytes were then washed several times with modified Charles Rosenkrans medium with amino acids (mCR2aa) containing 0.8% BSA and cultured in this medium for 48 h postinsemination. After this, the embryos were shifted to the IVC medium (mCR2aa, 0.8% BSA, 10% FBS) and cultured in 100-μL droplets of this medium on original beds of granulosa cells for up to 8 days postinsemination in a humidified CO2 incubator (5% CO2 in air) at 38.5°C. The medium was replaced with 50% of fresh IVC medium every 48 h.
HMC
The donor cells were prepared for HMC as described earlier (Shah et al., 2008). Briefly, ear skin biopsies collected from an adult, a more than 6-year-old Murrah buffalo in sterile Dulbecco's phosphate-buffered saline (DPBS), were cut into small pieces after the removal of skin tissue. These were cultured in Dulbecco's modified Eagle's medium (DMEM) and 20% FBS until the cultures reached 60–70% confluence. The cells were then subcultured by partial trypsinization for up to 10 passages. A confluent monolayer of cells between passages 5 and 10 was cultured for an additional 3 days to enable the cells to achieve overconfluence so that a higher proportion of cells could be available in G1 phase of cell cycle. Just before use as nuclear donors, the cells were trypsinized and washed with T20 buffer (TCM-199, 20% FBS).
The preparation of recipient cytoplasts was carried out as reported earlier (Shah et al., 2008). Briefly, cumulus investment was removed from in vitro matured COCs with expanded cumulus mass by treatment with hyaluronidase (0.5 mg mL−1 in T2). The zona pellucida was removed by pronase (2.0 mg mL−1 in T10, i.e., TCM-199, 10% FBS) treatment. Zona-free oocytes were incubated in T20 at 38.5°C for 10–15 min or until a prominent protrusion cone became easily visible. Protrusion cone–guided bisection was carried out using a microblade (MicroBlades, MTB-05; Micromanipulator Microscope Company, Inc., Carson City, NV, USA) in 4 mL of T20 containing 2.5 μg mL−1 cytochalasin B. The enucleated demicytoplasts were incubated in T20 for 10–15 min at 38.5°C so that they could regain the spherical shape. These were treated with phytohemagglutinin (0.5 mg mL−1 in T2) for 3–4 sec and were then transferred to T2 containing the donor cells. Each demicytoplast was gently rolled over a single, rounded, medium-sized donor cell so that it would attach to the donor cell, after which the demicytoplast–donor cell pairs were transferred to fusion medium which consisted of 0.3 M D-mannitol, 1 mg mL−1 polyvinyl alcohol, 0.1 mM MgCl2, and 0.05 mM CaCl2 for equilibration.
The demicytoplast–donor cell pair was aligned by subjecting it to an A.C. pulse (4 V) using a BTX Electrocell Manipulator 200 (BTX, San Diego, CA, USA) in such a manner that the somatic cell faced the negative electrode. Immediately, another demicytoplast was introduced into the fusion chamber (BTX microslide 0.5-mm gap, model 450; BTX, San Diego, CA, USA) close to the somatic cell. A single D.C. pulse (3.36 kV cm−1 for 4 μsec) was applied for electrofusion immediately after the somatic cell was sandwiched between the demicytoplasts. For rounding up, the triplets were incubated in T20 for 6 h at 38.5°C. For activation, the reconstructed oocytes were incubated in T20 containing 2 μM calcimycin A23187 for 5 min at 38.5°C, after which these were washed three times with T20. The reconstructed oocytes were incubated individually in a CO2 incubator in 5-μL droplets of T20 (containing 2 mM 6-dimethylamino purine) at 38.5°C for 4 h after covering them with mineral oil. The activated embryos were washed four times with RVCL medium containing 1% BSA-FAF and were cultured in 400 μL of this medium in four-well dishes (10–15 embryos per well), after covering them with mineral oil. The dishes were kept undisturbed in a CO2 incubator for 8 days.
Parthenogenesis
Parthenogenetic activation and culture of zona-free oocytes were carried out similarly at 24 h postmaturation, as described above for reconstructed oocytes.
Assessment of blastocyst quality
For examining the health of the embryos, the total cell number of trophectoderm (TE) and ICM of day-8 blastocysts was determined by differential staining protocol, as described previously (Thouas et al., 2001) with certain modifications. Briefly, the blastocysts were washed with DPBS for 10–15 sec and were then immediately transferred to 500 μL of solution I (5 μg mL−1 of Hoechst 33342) and incubated for 40 min at 37°C. The embryos were washed with DPBS and then transferred to solution II (0.04% Triton X-100) for 1 min. The embryos were again washed with DPBS, followed by incubation in 25 μg mL−1 propidium iodide for 40 sec. The embryos were observed under an epifluorescence inverted microscope (Nikon Diaphot) fitted with an ultraviolet (UV) lamp and excitation filters (excitation wavelength, 330–380 nm; barrier filter, 420 nm). ICM cells stained blue, whereas TE cells stained red.
Buffalo ESC culture
ICMs obtained from day-8 blastocyst-stage embryos generated by IVF, parthenogenesis, and HMC were isolated mechanically using a microblade (MicroBlades, MTB-05; Micromanipulator Microscope Company, Inc., Carson City, NV, USA) and cultured in a CO2 incubator (5% CO2 in air) at 37°C on mitomycin C– (10 μg mL−1) treated buffalo fetal fibroblast (bFF) feeder layer, which was prepared by a method described earlier (Verma et al., 2007). The cells were cultured in ESC culture medium consisting of knockout-Dulbecco's modified Eagle's medium (KO-DMEM) supplemented with 15% knockout serum replacement (KSR) (Invitrogen, Carlsbad, CA, USA), 2 mM nonessential amino acids (NEAAs), 2 mM L-glutamine, 50 μg mL−1 gentamicin, 0.1 mM β-mercaptoethanol, 1000 U mL−1 leukemia inhibitory factor (LIF), and 5 ng mL−1 basic fibroblast growth factor-2 (bFGF-2). After the formation of ICM outgrowths (primary colonies), cells were dissociated mechanically using a microblade and placed on fresh feeders. After the first few passages, colonies with ESC-like morphology were selected for further propagation, characterization, and in vitro differentiation. Medium was changed every other day, and the cells were passaged mechanically every 5 or 6 days in a split ratio of 1:2 to 1:4, depending on the colony size. ESCs successfully maintained an undifferentiated morphology for >70 passages, >60 passages, and >40 passages for IVF, parthenogenesis, and HMC-derived ESCs, respectively.
Alkaline phosphatase staining
For alkaline phosphatase (AP) staining, the ESC colonies were washed twice with DPBS and then stained using an AP staining kit (Sigma Chemical Co., USA, Catalog No. 86C) as per the manufacturer's protocol.
Immunocytochemical analysis (ICC)
Buffalo ESCs were characterized at regular intervals by examining specific extracellular/surface markers like SSEA-4, TRA-1-60, and TRA-1-81 and intracellular markers like OCT4 and SOX2 by immunocytochemistry, as described earlier (Anand et al., 2011). The primary antibodies against these antigens, used at a dilution of 1:10 or 1:20, were purchased from Chemicon International (Temecula, CA, USA). Secondary antibodies, which included goat anti-mouse immunoglobulin M–fluorescein isothiocyanate (IgM-FITC) conjugate and goat anti-mouse IgG-FITC conjugate, were purchased from Sigma Chemical Co., whereas goat anti-rat IgM-FITC conjugate was from Pierce Biotechnology Inc. (Rockford, IL, USA). These were used at a dilution of 1:200. In the respective controls, the addition of the primary antibody was omitted.
Reverse transcription polymerase chain reaction (RT-PCR) and sequencing analysis
Total RNA was extracted from buffalo ESCs using TRIzol® (Invitrogen) and subjected to DNase I treatment using a DNA-free™ Kit (Ambion; Cat#AM1906) as per protocol. First-strand cDNAs were synthesized using Superscript III (Invitrogen), according to the manufacturer's instructions, and cDNAs were amplified with GoTaq® Flexi DNA Polymerase (Promega, Cat#M8295). Primer sequences are listed in Supplementary Tables 1, 2, and 3. (Supplementary Data are available at www.liebertonline/cell/.) (PCR reactions were conducted using the following parameters: Denaturation at 95°C for 3 min followed by 35 amplification cycles including denaturation at 95°C for 30 sec, primer-specific annealing temperature for 30 sec, and 72°C for a 30-sec extension. The cycles were followed by a final extension at 72°C for 10 min. Annealing temperatures are listed in Supplementary Tables 1, 2 and 3. Samples without reverse transcriptase, at the reverse transcription step, were used as the control for examining genomic DNA contamination. PCR products were separated by electrophoresis through a 1.5% Tris–acetate–EDTA agarose gel stained with ethidium bromide. Gels were imaged with a Chemidoc XRS system (BioRad Inc., Hercules, CA, USA). To retrieve amplified PCR products, DNA bands were excised from the gel and extracted with Qiagen gel extraction kit (Qiagen, Valencia, CA). The amplified products were custom sequenced.
In vitro differentiation
Embryoid body (EB) outgrowths were prepared from ESCs derived from the three sources (IVF, parthenogenesis, and HMC). For this preparation, the ESC colonies were removed from the feeders and were disintegrated into small clumps with the help of a microblade. The clumps (600–800 cells each) were suspended from the lid surface in hanging drops of 20–30 μL each in ESC culture medium without growth factors. The medium was changed every other day by tilting the plate and allowing the cells to settle. After 4 days in suspension culture, some EBs were transferred to gelatin-coated four-well plates for spontaneous differentiation. The EBs differentiated to several types of cells. The expression of the markers for the three germ layers was examined by reverse transcriptase PCR (RT-PCR).
Quantitative real-time PCR
RNA extractions and cDNA synthesis were performed as described above. Relative levels of OCT4, SOX2, NANOG, and cMYC in the ESCs were determined by quantitative real-time PCR. Gene transcripts from ESCs were quantified using the CFX 96 thermocycler (BioRad Inc.) and detected using Maxima™ SYBR Green/ROX qPCR Master Mix (2×) (Fermentas; Cat#K0221). All reactions were run in triplicate, and three biological replicates were carried. Relative levels of expression was determined using 2−ΔΔCt method, where ΔCt=Ct (target gene) − Ct (internal reference), and ΔΔCt=ΔCt sample − ΔCt calibrator. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as internal reference gene. cDNA from IVF ESCs for each study served as the calibrator. Relative mRNA expression was expressed as n-fold mRNA expression relative to the calibrator. The specificity and integrity of PCR products was ensured through the melt curve analysis. No PCR products were obtained when reverse transcriptase was omitted from cDNA synthesis or when DNA templates were omitted from the PCR reaction.
Statistical analysis
The data was analyzed using SYSTAT 7.0 (SPSS Inc., Chicago, IL, USA) after arc-sine transformation. Differences among mean percentages were analyzed by one-way analysis of variance (ANOVA), followed by Fisher's least significant differences (LSD) test. Significance was determined at p<0.05.
Results
Comparative embryo production in three sources
Oocytes retrieved from slaughterhouse buffalo ovaries and matured in vitro, were subjected to IVF, parthenogenesis, and HMC to produce blastocysts, which served as the primary source for ESC generation. A total of 1250, 622, and 296 oocytes/reconstructed embryos, respectively, were examined for development to blastocyst stage (Table 1). The cleavage rate observed was significantly (p<0.05) higher in embryos derived through parthenogenesis (81.09±2.34%) and HMC (86.94±3.90%) compared to those produced by IVF (70.23±3.34%). Also, blastocyst production varied significantly (p<0.05) among IVF (12.13±1.62%), parthenogenesis (24.60±2.29%) and HMC (48.18±3.63%).
Table 1.
Developmental Competence and Quality Assessment of Buffalo Embryos Produced by In Vitro Fertilization, Parthenogenesis, and Hand-Made Cloning
| Source | No. of oocytes/reconstructed embryos (n) | Cleavage rate n (%) | Blastocyst rate n (%) | Total cell number | ICM cell number | ICM/TE ratio |
|---|---|---|---|---|---|---|
| IVF | 1250 | 898 (70.23±3.34)a | 112 (12.13±1.62)a | 158.50±6.80a | 26.50±2.26a | 0.20±0.02a |
| Parthenogenesis | 622 | 545 (81.09±2.34)b | 153 (24.60±2.29)b | 169.83±11.99a | 27.66±4.05a | 0.19±0.02a |
| HMC | 296 | 238 (86.94±3.90)b | 142 (48.18±3.63)c | 231.33±15.72b | 43.50±1.98b | 0.24±0.03a |
Values with different superscripts within the same column differ significantly (p<0.05).
ICM, inner cell mass; TE, trophectoderm; IVF, in vitro fertilization; HMC, hand-made cloning.
In the present study, quality assessment of embryos was done using modified differential staining (Fig. 1). Total cell number and ICM cell number were significantly higher (p<0.05) in HMC embryos (231.33±15.72 and 43.50±1.98, respectively) compared to embryos produced by parthenogenesis (169.83±11.99 and 27.66±4.05, respectively) and IVF (158.50±6.80 and 26.50±2.26, respectively). Furthermore, when the viability of donor cells used for HMC was checked by flow cytometry, 97.44% cells of gated population were found to be viable.
FIG. 1.

Differential staining of blastocysts derived from in vitro fertilization (I), parthenogenesis (P), and hand-made cloning (C) showing inner cell mass (ICM) cells and trophectodermal cells.
Primary colony formation rate
Comparative evaluation of the primary colony formation rates (PCFRs) in this present study revealed that IVF- and parthenogenesis-derived blastocysts exhibited significantly higher (p<0.05) PCFRs (52.44±2.55 and 45.63±3.12%, respectively) when compared with HMC-derived blastocysts (33.4±5.52%) (Table 2, Fig. 2).
Table 2.
Comparative Primary Colony Formation Rate Following Seeding of ICMs Obtained from Blastocysts Produced by In Vitro Fertilization, Parthenogenesis, and Hand-Made Cloning
| Source | Maximum passage number | ICMs seeded (n) | Time taken for primary colony formation (days) | Primary colony formation rate (%) |
|---|---|---|---|---|
| IVF | 78 | 86 | 8–12 | 52.44±2.55a |
| Parthenogenesis | 61 | 98 | 10–15 | 45.63±3.12a |
| HMC | 45 | 96 | 10–15 | 33.34±5.52b |
Values with different superscripts within the same column differ significantly (p<0.05).
ICM, inner cell mass; IVF, in vitro fertilization; HMC, hand-made cloning.
FIG. 2.
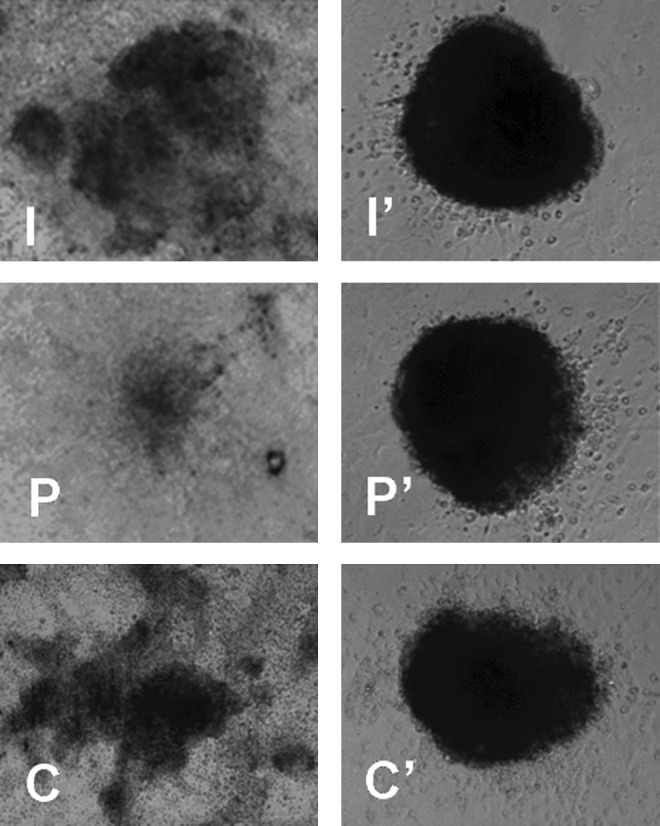
Stem cell colonies derived inner cell mass (ICM) cells of blastocysts derived from in vitro fertilization (I), parthenogenesis (P), and hand-made cloning (C). I, P, and C are primary colonies, whereas I′, P′, and C′ are established colonies at passages 53, 42, and 35 respectively.
Comparative primary colony formation rate at different concentrations of LIF
The PCFR of ESCs was determined with respect to the source of ICM cells (IVF, parthenogenesis, and HMC). To optimize LIF concentration to best suit the culture system, ESCs from three sources were cultured in the presence of varying concentrations of LIF (0, 1000, 2000, and 4000 U mL−1), and the PCFR among different groups was compared. The PCFR obtained with LIF concentrations of 0, 1000, and 2000 U mL−1 was significantly higher (p<0.05) than that with 4000 U mL−1 LIF for all the three sources of ICMs examined (Table 3). For ICMs derived from IVF and parthenogenetic blastocysts, PCFR at LIF concentrations of 0 U mL−1 (29.98±2.33 and 33.575±2.49%, respectively), 1000 U mL−1 (32.16±1.44 and 35.79±2.76%, respectively) and 2000 U mL−1 (24.15±2.30 and 26.59±2.65%, respectively) did not differ significantly (p<0.05) amongst themselves but were significantly higher (p<0.05) than the PCFR at LIF concentrations of 4000 U mL−1 (12.36±2.69 and 9.26±1.67%, respectively). In the case of ICMs from HMC-derived blastocysts, the PCFRs observed at LIF concentrations of 0 U mL−1 (28.51±1.57%) and 1000 U mL−1 (30.64±1.24%) were statistically comparable but were significantly higher (P<0.05) than the PCFR observed at LIF concentrations of 2000 (20.68±1.57%) and 4000 U mL−1 (8.83±1.78%).
Table 3.
Effect of LIF on the Primary Colony Formation Rate Following Seeding of ICMs Obtained from Blastocysts Produced by In Vitro Fertilization, Parthenogenesis, and Hand-Made Cloning
| |
Source |
||
|---|---|---|---|
| |
Primary colony formation rate (%) |
||
| LIF concentration (U mL−1) | IVF | Parthenogenesis | HMC |
| 0 | 29.98±2.33a | 33.575±2.49a | 28.51±1.57a |
| 1000 | 32.16±1.44a | 35.79±2.76a | 30.64±1.24a |
| 2000 | 24.15±2.30a | 26.59±2.65a | 20.68±1.57a* |
| 4000 | 12.36±2.69a* | 9.26±1.67a* | 8.835±1.78a** |
Values with different superscripts within the same row differ significantly (p<0.05).
Indicates significant difference within the same columns (p<0.05).
LIF, leukemia inhibitory factor; ICM, inner cell mass; IVF, in vitro fertilization; HMC, hand-made cloning.
Characterization of ESCs
The characterization of the ESCs with the help of pluripotent markers was carried out at different passages to confirm that the ESCs remained in an undifferentiated state during the course of culture. SSEA-4, TRA-1-60, TRA-1-81, OCT4, and SOX2 protein expression was visualized by ICC (Fig. 3). At least 20 colonies were tested for each marker at one time. The results of ICC revealed that all of these markers were expressed by ESCs derived from IVF, parthenogenesis, or HMC, confirming their pluripotent nature. The expression of these markers diminished when the ESCs differentiated and lost pluripotency. Buffalo ESCs exposed to the respective secondary antibody without prior exposure to the respective primary antibodies served as negative controls, and none of the negative controls exhibited any fluorescence.
FIG. 3.

Immunofluorescence analysis of pluripotency markers in embryonic stem cells (ESCs) derived from blastocysts produced by in vitro fertilization (I), hand-made cloning (C), and parthenogenesis (P). FITC, fluorescein isothiocyanate.
In addition to this, buffalo ESC colonies from all the three sources exhibited high levels of AP activity (Fig. 4). RT-PCR analysis for expression of OCT4, SOX2, NANOG, cMYC, FOXD3, STAT3, REX1, NUCLEOSTEMIN, and TELOMERASE revealed that these genes were expressed by buffalo ESCs derived from all the three sources, thus confirming their pluripotent nature (Fig. 5).
FIG. 4.

Alkaline phosphate expression in embronic stem cell (ESC) colonies derived from blastocysts produced by in vitro fertilization (I), Parthenogenesis (P), and hand-made cloning (C). I, P, and C are colonies before staining, whereas I′, P′, and C′ are respective colonies after staining.
FIG. 5.
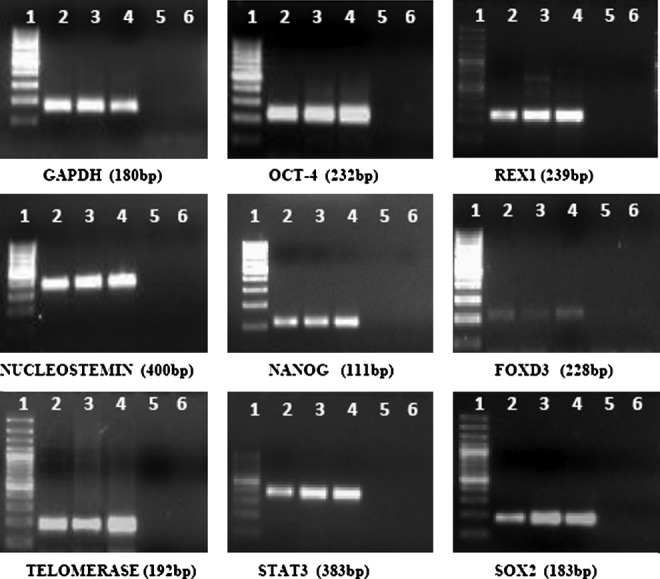
Expression of pluripotency markers in embryonic stem cells (ESCs) derived from blastocysts produced by in vitro fertilization (IVF), parthenogenesis, or hand-made cloning (HMC). Lane 1, 100-bp ladder; lane 2, IVF; lane 3, parthenogenesis; lane 4, HMC; lane 5, PCR −ve); lane 6, reverse transcriptase −ve.
To test the differentiation potential of the ESCs, colonies were dissected mechanically into small clumps of 400–1000 cells and cultured in 20- to 30-μL hanging drops of DMEM and 15% KSR in bacterial dishes in the absence of the growth factors and feeder cells for EB formation. This led to differentiation of the ESCs to form three-dimensional round aggregates. Compact EBs were formed within 2–4 days (Fig. 6) and developed to cystic EBs, when these cultures were maintained for 1–2 weeks (Fig. 7). When the culture period was extended, the size and number of cysts in cystic EBs increased. Harvested cells from EBs expressed markers for ectoderm—viz. NF-68 and cytokeratin 8, mesoderm, msh homeobox 1 (MSX-1), bone morphogenetic protein-4 (BMP-4), and α-skeletal actin (ASA) and endoderm α-fetoprotein (AFP), GATA-4, and hepatocyte nuclear factor-4 (HNF-4). ESCs from all the three sources were positive for these genes (Fig. 8).
FIG. 6.

Compact embryoid bodies produced from embryonic stem cells (ESCs) derived from blastocysts produced by in vitro fertilization (I), Parthenogenesis (P), and hand-made cloning (C). Scale bar, 200 μm.
FIG. 7.

Cystic embryoid bodies produced from embryonic stem cells (ESCs) derived from blastocysts produced by in vitro fertilization (I), parthenogenesis (P), and hand-made cloning (C). Scale bar, 200 μm.
FIG. 8.

Expression of differentiation markers in embryoid bodies produced from embryonic stem cells (ESCs ) derived from blastocysts produced by in vitro fertilization (IVF), parthenogenesis, or hand-made cloning (HMC). Endodermal markers: AFP, HNF4, and GATA4. Mesodermal markers: MSX-1, BMP-4, and ASA. Ectodermal markers: Cytokeratin and NF-68. Lane 1, 100-bp ladder; lane 2, IVF; lane 3, parthenogenetic; lane 4, HMC; lane 5, PCR −ve; lane 6, reverse transcriptase −ve.
Furthermore, 5- to 6-day-old compact EBs became attached when cultured on gelatin-coated tissue culture dishes. The morphology of the cells at the periphery of the EBs changed indicating that these cells had differentiated to different types of cells (Fig. 9).
FIG. 9.

Different cell types produced by spontaneous differentiation from embryonic stem cells (ESCs) derived from blastocysts produced by in vitro fertilization (I), parthenogenesis (P), and hand-made cloning (C).
The occurrence of a stable karyotype, as indicated by the presence of a normal diploid set of chromosomes, was examined for ESCs from the three sources. The buffalo ESCs of all the groups revealed a normal diploid chromosome number (Fig. 10).
FIG. 10.
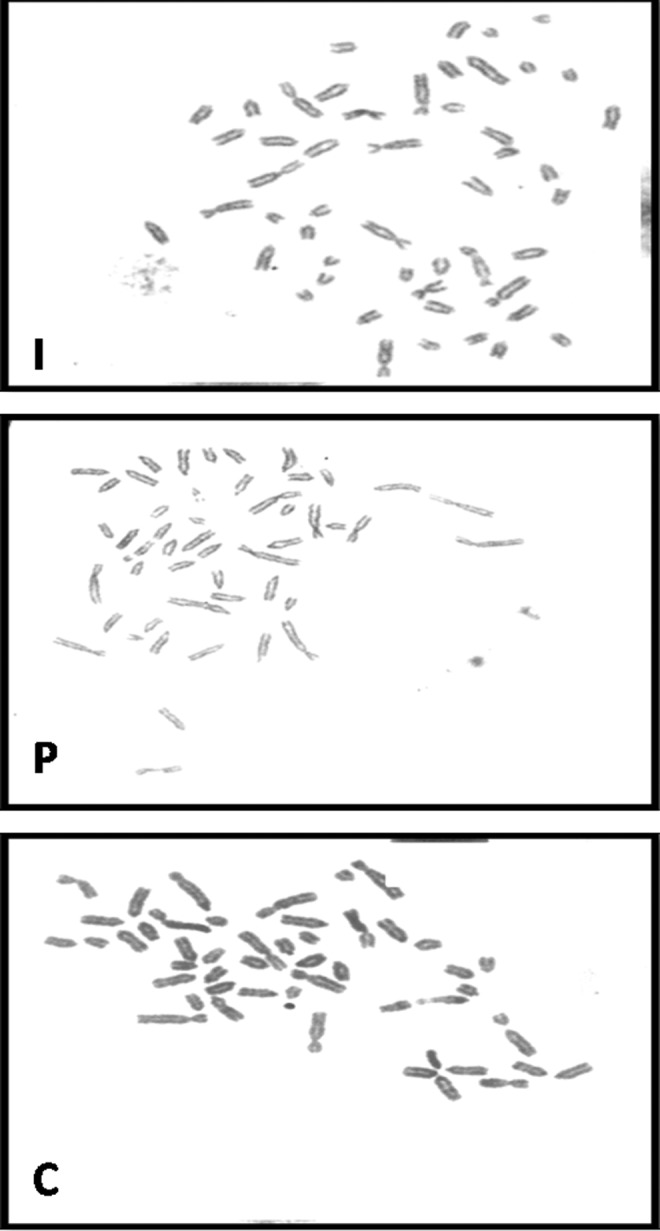
A representative metaphase spread of buffalo embryonic stem cells (ESCs) and ESCs derived from blastocysts produced by in vitro fertilization (I), parthenogenesis (P), and hand-made cloning (C) at passages 46, 42, and 39, respectively. (Magnification, 100×.)
Comparative real-time analysis of pluripotency genes in ESCs from three sources
ICM explants obtained from IVF-derived blastocysts were cultured on inactivated feeders, and the resulting colonies were evaluated for the relative mRNA abundance of the pluripotency genes OCT4, SOX2, NANOG, and cMYC. ESCs from IVF, parthenogenetic, and HMC embryos were established simultaneously to minimize the batch-to-batch variation during culture. The same batch of feeder layer, medium, and passage time was used in all the groups. Except for the higher expression of NANOG in the HMC group, the expression of all other transcription factors was found to be similar in ESCs from all the three sources (Fig. 11).
FIG. 11.
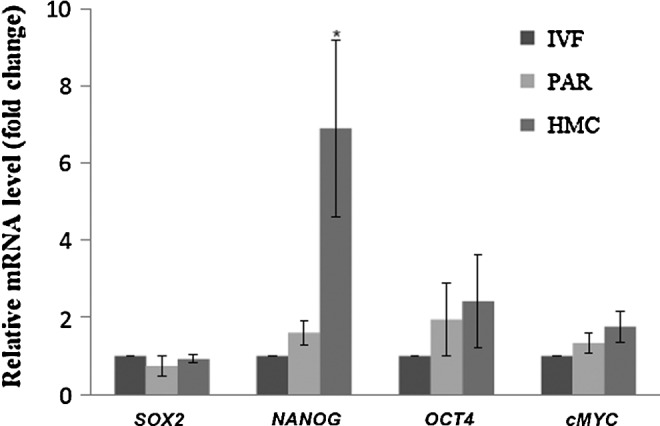
Relative expression level of pluripotency-related genes in embryonic stem cells (ESCs) derived from blastocysts produced by in vitro fertilization (IVF), parthenogenesis (PAR), and hand-made cloning (HMC).
Discussion
This study was aimed at evaluating the feasibility of establishing buffalo ESCs from blastocysts produced by parthenogenesis and HMC, in addition to those produced by IVF. We successfully generated buffalo ESCs from all the three sources. No such comparative study in buffalo ESCs had been carried out earlier.
In this study, the cleavage rate and subsequent embryo development to the blastocyst stage was found to be higher following chemical activation, used in the HMC and parthenogenetic groups, than following fertilization done in the IVF group. This is in accordance with earlier reports (Mishra et al., 2008; Shah et al., 2008). The quality of the frozen semen used and improper capacitation, besides some yet unknown factors, may be the possible reasons for low blastocyst rate in the IVF group. The low blastocyst rate during in vitro embryo production of buffalo embryos (Chauhan et al., 1998; Nandi et al., 2001) has been attributed to low oocyte yield, poor quality of oocytes obtained from the slaughterhouse, and to suboptimal culture conditions (Anand et al., 2008). The quality of blastocysts was judged on the basis of total cell number and ICM/TE ratio determined by differential staining. The cell number of embryos generally correlates with their quality and developmental competence and has been regarded as an important criterion for quality assessment. Also, fast-cleaving embryos show less fragmentation (Tanghe et al., 2004) and apoptosis (Lonergan et al., 1999) and have a higher blastocyst rate (Vandaele et al., 2006), total cell number, ICM/TE ratio, and fetal development (McKiernan and Bavister, 1994; Racowsky et al., 2000; Vajta et al., 2003). Our results suggest that HMC embryos have a higher ability to develop to blastocyst stage with a well-defined ICM and acceptable cell number than those produced by IVF. The higher embryo production rate by HMC might be due to pairing of somatic cell with two demioocytes, thereby increasing the cytoplasmic volume and cell number (Ribeiro et al., 2009).
ICM, when seeded on feeder layer, resulted in outgrowths or primary colonies. In our case, buffalo ESCs from all the three sources, i.e., IVF, parthenogenesis and HMC, formed large, round, and compact colonies with distinct boundaries as observed in the case of bovines (Wang et al., 2005), whereas some of previous studies reported formation of a monolayer sheet (Cibelli et al., 1998; Mitalipova et al., 2001; Stice et al., 1996). The rate of primary colony formation has been found to differ among the ESCs produced from three sources and also across different species (Anand et al., 2011; Gong et al., 2010; Hwang et al., 2005; Intawicha et al., 2009). In all cases, ICMs were found to be attached to the feeder layer by day 3. No comparable study has been reported in the literature; however, the observed time of attachment was lower than 4–7 days reported when whole blastocysts were seeded in case of bovines (Cao et al., 2009).
Additionally we compared the efficiency of colony formation between the ESCs derived from IVF, parthenogenetic, and HMC embryos. Mechanical isolation was preferred for dissociation of cells because this method was found to be more effective than enzymatic treatment, as indicated by the capability of the ICM to survive for a longer period without undergoing differentiation (Anand et al., 2011; Verma et al., 2007). A wide variation in PCFR has been reported across different species. The PCFR of 52% obtained in the present study is lower than that of 73.1% (Verma et al., 2007), 61% (Huang et al., 2010), and 75% (Anand et al., 2011) reported for putative buffalo ESCs derived from blastocysts produced by IVF. Gong et al. (2010) reported a PCFR of 41.2% for bovine ESCs derived from blastocysts produced by IVF, in the presence of LIF, which is comparable to our results. In goat, a PCFR of 66.6% has been reported for IVF-derived ESCs (Pawar et al., 2009). Panasophonkul et al. (2010) failed to establish ESC primary colonies in pig, whereas a PCFR of 24% has been observed for rabbit IVF-derived ESCs, which increased to 57% by addition of hLIF (1000 U mL−1) to cultures (Intawicha et al., 2009). Pashaiasl et al. (2010) observed a PCFR of 53.33% in parthenogenetically derived bovine ESCs that is slightly higher than that obtained in the present study. The discrepancies observed in the PCFRs among the three sources may be attributed partly to variations in sample size, because higher PCFRs could be expected from a small sample size and to the different culture conditions employed by different workers. Lower PCFRs obtained with HMC blastocysts, in spite of their highest cell number, may be due to their lower developmental competence.
While optimizing exogenous LIF concentration in ESC culture, we found that the primary colonies formed less efficiently when seeded in cultures with LIF at a concentration of 4000 U mL−1. Most of the time, the ICMs seeded at this concentration of LIF did not attach well to feeder cells, remained suspended in the medium, and showed signs of differentiation or cell death after 2–3 days. This could be due to inhibitory effects of high levels of LIF on ICM attachment and or proliferation. To our knowledge, this is the first study of LIF requirements in buffalo ESC culture. In the case of bovine ESCs, the primary colony formation rate has been observed to be higher (41.2%) in the presence of 1000 U mL−1 hLIF compared to that of 36.9% in the absence of hLIF (Gong et al., 2010). It is further conceived that extrapolation of such studies within or across the species warrants a critical consideration of the species involved as well as the source of LIF.
Characterization of the newly generated cell lines demonstrated that at molecular and chromosomal levels the examined buffalo ESCs were similar to regular ESCs. Also buffalo cells in our colonies displayed positive AP staining throughout the culture period. ESCs from IVF, parthenogenesis, and HMC were found to be positive for SSEA-4, TRA-1-60, TRA-1-81, OCT4, and SOX2, markers when examined by immunocytochemistry. Positive marker expression of transcriptional markers, viz. OCT4, SOX2, NANOG, FOXD3, REX1, NUCLEOSTEMIN, and TELOMERASE, and that of surface markers such as SSEA-4, TRA-1-60, and TRA-1-81 in buffalo ESCs that are characteristic for ESCs, resembled human and mouse ESCs (Chambers et al., 2003; Boyer et al., 2005).
Huang et al. (2010) reported the expression of SSEA-1, SSEA-3, and SSEA-4, whereas Anand et al. (2011) reported positive expression of SSEA-4 only but not SSEA-1 and SSEA-3 in IVF-derived buffalo ESCs. Parthenogenetically derived buffalo ESCs exhibited SSEA-1, SSEA-4, TRA-1-60, and TRA-1-81, but not SSEA-3 (Sritanaudomchai et al., 2007) and NT-ESCs (HMC) were positive for OCT-4, SOX-2, NANOG, NUCLEOSTEMIN, STAT-3, and FOXD-3 as detected by RT-PCR and SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81, but not SSEA-1 by ICC (George et al., 2011). The reason for these variations is still not clear, but more conclusive inferences can be drawn if bubaline-specific antibodies can be developed and used for this purpose. Expression of these markers in ESCs from all sources in the present study confirmed their equivalency.
It is possible that the buffalo ESCs derived from IVF, parthenogenesis, and HMC employ self-renewal and pluripotency pathways similar to those of human and mouse ESCs. The possibility of the interchangeability of use of ESCs from the three sources examined in the present study also exists. Furthermore, our relative mRNA abundance results from real-time PCR are complemented by protein expression by ICC, which further authenticated our results. Importance of characterization has been realized because the undifferentiated cells can be easily distinguished and/or separated from the differentiated ones. Buffalo ESC characterization at regular intervals will guarantee their pluripotent undifferentiated state as well as in-depth knowledge for unrevealing mechanisms involved in differentiation.
In differentiation studies, buffalo ESCs from all the three sources were found to be capable of forming both compact and cystic EBs, which were able to undergo spontaneous differentiation to different cell types. It is considered that ESCs that are able to form only compact EBs have restricted pluripotency (Sukoyan et al., 2002). The formation of EBs, which was reported for the first time following suspension culture of mouse ESCs (Doetschman et al., 1985), was later reported in many other species like buffalo (Anand et al., 2011; Chauhan et al., 2006; Verma et al., 2007), cattle (Yadav et al., 2005), sheep (Dattena et al., 2006), canine (Hatoya et al., 2006; Wilcox et al., 2009), and human (Reubinoff et al., 2000).
The cells from harvested EBs from IVF-, parthenogenesis-, and HMC-derived buffalo ESCs were analyzed for various differentiation markers by RT-PCR. Parthenogenesis-derived buffalo ESCs easily formed EBs in suspension culture. We identified derivatives of all three embryonic germ layers in these EBs. Parthenogenesis-derived ESCs from M. fascicularis are reported to have normal in vitro differentiation potential (Cibelli et al., 2002). Yu et al. (2011) have reported establishment of ESC lines from parthenogenesis-derived mouse embryos, which could be differentiated to various types of cells from three germ layers in vitro, as confirmed by analysis of EBs by immunohistochemistry and RT-PCR.
NT-ESCs have been established in a number of species, such as bovine (Cibelli et al., 1998; Munoz et al., 2008; Roach et al., 2006; Saito et al., 2003; Stice et al., 1996; Strelchenko, 1996; Wang et al., 2005) and mouse (Kawase et al., 2000; Munsie et al., 2000; Wakayama et al., 2003; Wakayama, 2005), and were capable of differentiating to EBs containing cells from all the three germ layers. Mouse ESCs have been characterized and differentiated via EB formation (Kobolak et al., 2010). It was observed in the present study that the EB-derived cells from all the three sources of ESCs expressed genes for ectoderm, mesoderm, and endoderm indicative of their pluripotent status. The pluripotent status of the ESCs was further confirmed by their ability to spontaneously differentiate to at least two different cell types.
The buffalo ESCs produced in the present study showed normal euploidy. ESCs having more than 50% of cells with normal chromosome numbers were suggested to be efficient for germ-line transmission due to successful segregation of chromosomes throughout meiosis (Longo et al., 1997; Suzuki et al., 1997). During the in vitro culture, the buffalo ESCs derived from IVF, parthenogenesis, and HMC were able to maintain their undifferentiated status along with their normal chromosomal compliment.
Real-time comparison among the buffalo ESCs from three sources revealed that, except for the higher expression of NANOG in HMC group, the expression of all other transcription factors was similar in ESCs from the three sources. Overexpression of NANOG in the HMC-derived ESCs can be attributed partly to epigenetic differences among ESCs derived from the three sources, in addition to NANOG being one of the important genes involved in nuclear reprogramming (Zhang et al., 2011). No previous report exists on comparative pluripotent gene expression from three sources. Further studies are warranted to confirm the findings.
Conclusion
We have successfully established buffalo ESCs lines from blastocysts derived from IVF, parthenogenesis, and nuclear transfer (HMC). By using current protocols, ESCs could be isolated efficiently and subjected to long-term cultures on buffalo fetal fibroblast feeders in medium containing murine (m) LIF and bFGF-2. Characteristics such as the pluripotent stem cell maker profile of buffalo ESCs make them appear similar to human ESC lines, yet on the other hand, their LIF-dependence resembles to that found in mouse ESCs. The differences in the profile of pluripotency markers among different species are an important component of the species differences in ESCs. It is suggested that ESCs derived from blastocysts produced by parthenogenesis or HMC could be possible alternatives to the ones derived from blastocysts produced by IVF because their equivalent nature was established by immunocytochemistry, expression of pluripotency genes, and differentiation potential. Pluripotent, stable buffalo ESC lines derived from embryos produced by IVF, parthenogenesis, and HMC could be genetically manipulated and would provide a powerful tool for the studies of early embryonic development, genomic imprinting, gene targeting, cloning, chimera formation, and transgenic animal production.
Supplementary Material
Acknowledgments
This research was funded by National Agriculture Innovative Project (NAIP) Grant to S.K.S. (C 2-1-(5)/2007) and M.S.C. (C-2067 and 075).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
- Anand T. Kumar D. Chauhan M.S., et al. Cysteamine supplementation of in vitro maturation medium, in vitro culture medium or both media promotes in vitro development of buffalo (Bubalus bubalis) embryos. Reprod. Fertil. Dev. 2008;20:253–257. doi: 10.1071/rd07167. [DOI] [PubMed] [Google Scholar]
- Anand T. Kumar D. Singh M.K., et al. Buffalo (Bubalus bubalis) embryonic stem cell-like cells and preimplantation embryos exhibit comparable expression of pluripotency-related antigens. Reprod. Domest. Anim. 2011;46:50–58. doi: 10.1111/j.1439-0531.2009.01564.x. [DOI] [PubMed] [Google Scholar]
- Boyer L.A. Lee T.I. Cole M.F., et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S. Wang F. Chen Z., et al. Isolation and culture of primary bovine embryonic stem cell colonies by a novel method. J. Exp. Zool. A Ecol. Genet. Physiol. 2009;311:368–376. doi: 10.1002/jez.535. [DOI] [PubMed] [Google Scholar]
- Chambers I. Colby D. Robertson M., et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- Chauhan M.S. Singla S.K. Palta P., et al. In vitro maturation and fertilization, and subsequent development of buffalo (Bubalus bubalis) embryos, effects of oocyte quality and type of serum. Reprod. Fertil. Dev. 1998;10:173–177. doi: 10.1071/r97080. [DOI] [PubMed] [Google Scholar]
- Chauhan M.S. Verma V. Manik R.S., et al. Development of inner cell mass and formation of embryoid bodies on a gelatin coated dish and on the feeder layer in buffalo (Bubalus bubalis) Reprod. Fertil. Dev. 2006;18:205–206. [Google Scholar]
- Chen L.R. Shiue Y.L. Bertolini L., et al. Establishment of pluripotent cell lines from porcine preimplantation embryos. Theriogenology. 1999;52:195–212. doi: 10.1016/S0093-691X(99)00122-3. [DOI] [PubMed] [Google Scholar]
- Cibelli J.B. Stice S.L. Golueke P.J., et al. Transgenic bovine chimeric offspring produced from somatic cell-derived stem-like cells. Nat. Biotechnol. 1998;16:642–646. doi: 10.1038/nbt0798-642. [DOI] [PubMed] [Google Scholar]
- Cibelli J.B. Grant K.A. Chapman K. B., et al. Parthenogenetic stem cells in nonhuman primates. Science. 2002;295:819. doi: 10.1126/science.1065637. [DOI] [PubMed] [Google Scholar]
- Dattena M. Chessa B. Lacerenza D., et al. Isolation, culture and charecterization of embryonic cell lines from vitrified sheep blastocysts. Mol. Reprod. Dev. 2006;31:31–39. doi: 10.1002/mrd.20378. [DOI] [PubMed] [Google Scholar]
- Doetschman T. Williams P. Maeda N. Establishment of hamster blastocyst-derived embryonic stem (ES) cells. Dev Biol. 1988;127:224–227. doi: 10.1016/0012-1606(88)90204-7. [DOI] [PubMed] [Google Scholar]
- Doetschman T.C. Eistetter H. Katz M., et al. The in vitro development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J. Embryol. Exp. Morphol. 1985;87:27–45. [PubMed] [Google Scholar]
- First N.L. Sims M.M. Park S.P. Kent-First M.J. Systems for production of calves from cultured bovine embryonic cells. Reprod. Fertil. Dev. 1994;6:553–562. doi: 10.1071/rd9940553. [DOI] [PubMed] [Google Scholar]
- George A. Sharma R. Singh K.P., et al. Production of cloned and transgenic embryos using Buffalo (Bubalus bubalis) embryonic stem cell-like cells isolated from in vitro fertilized and cloned blastocysts. Cell. Reprogram. 2011;13:263–272. doi: 10.1089/cell.2010.0094. [DOI] [PubMed] [Google Scholar]
- Gong G. Roach M.L. Jiang L., et al. Culture conditions and enzymatic passaging of bovine ESC-like cells. Cell. Reprogram. 2010;12:151–160. doi: 10.1089/cell.2009.0049. [DOI] [PubMed] [Google Scholar]
- Hatoya S. Torii R. Kondo Y., et al. Isolation and characterization of embryonic stem-like cells from canine blastocysts. Mol. Reprod. Dev. 2006;73:298–305. doi: 10.1002/mrd.20392. [DOI] [PubMed] [Google Scholar]
- Huang B. Li T. Wang X.L., et al. Generation and characterization of embryonic stem-like cell lines derived from in vitro fertilization Buffalo (Bubalus bubalis) embryos. Reprod. Domest. Anim. 2010;45:122–128. doi: 10.1111/j.1439-0531.2008.01268.x. [DOI] [PubMed] [Google Scholar]
- Hwang W.S. Roh S.I. Lee B.C., et al. Patient-specific embryonic stem cells derived from human SCNT blastocysts. Science. 2005;308:1777–1783. doi: 10.1126/science.1112286. [DOI] [PubMed] [Google Scholar]
- Iannaccone P.M. Taborn G.U. Garton R.L., et al. Pluripotent embryonic stem cells from the rat are capable of producing chimeras. Develop. Biol. 1994;163:288–292. doi: 10.1006/dbio.1994.1146. [DOI] [PubMed] [Google Scholar]
- Intawicha P. Ou Y.W. Lo N.W., et al. Characterization of embryonic stem cell lines derived from New Zealand white rabbit embryos. Cloning Stem Cells. 2009;11:27–38. doi: 10.1089/clo.2008.0040. [DOI] [PubMed] [Google Scholar]
- Kawase E. Yamazaki Y. Yagi T., et al. Mouse embryonic stem (ES) cell lines established from neuronal cell-derived cloned blastocysts. Genesis. 2000;28:156–163. [PubMed] [Google Scholar]
- Keefer C.L. Karatzas C.N. Lazaris-Karatzas A., et al. Isolation and maintanance of putative embryonic stem cells derived from Nigerian dwarf goat embryos. Biol. Reprod. 1996;54:462. [Google Scholar]
- Kim D. Kim C.H. Moon J.I., et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobolak J. Bodo S. Rungsiwiwut R., et al. Generation of mouse embryonic stem cell lines from zona-free nuclear transfer embryos. Cell. Reprogram. 2010;12:105–113. doi: 10.1089/cell.2009.0040. [DOI] [PubMed] [Google Scholar]
- Li M. Li Y.H. Hou Y., et al. Isolation and culture of pluripotent cells from in vitro produced porcine embryos. Zygote. 2004;12:43–48. doi: 10.1017/s0967199404002679. [DOI] [PubMed] [Google Scholar]
- Lonergan P. Khatir H. Piumi F., et al. Effect of time interval from insemination to first cleavage on the developmental characteristics, sex ratio and pregnancy rate after transfer of bovine embryos. J. Reprod. Fertil. 1999;117:159–167. doi: 10.1530/jrf.0.1170159. [DOI] [PubMed] [Google Scholar]
- Longo L. Bygrave A. Grosveld F.G., et al. The chromosome make-up of mouse embryonic stem cells is predictive of somatic and germ cell chimaerism. Transgenic Res. 1997;6:321–328. doi: 10.1023/a:1018418914106. [DOI] [PubMed] [Google Scholar]
- McKiernan S.H. Bavister B.D. Timing of development is a critical parameter for predicting successful embryogenesis. Hum. Reprod. 1994;9:2123–2129. doi: 10.1093/oxfordjournals.humrep.a138403. [DOI] [PubMed] [Google Scholar]
- Mishra V. Misra A.K. Sharma R. A comparative study of parthenogenic activation and in vitro fertilization of bubaline oocytes. Anim. Reprod. Sci. 2008;103:249–259. doi: 10.1016/j.anireprosci.2006.12.019. [DOI] [PubMed] [Google Scholar]
- Mitalipova M. Beyhan Z. First N.L. Pluripotency of bovine embryonic cell line derived from precompacting embryos. Cloning. 2001;3:59–67. doi: 10.1089/15204550152475563. [DOI] [PubMed] [Google Scholar]
- Munoz M. Diez C. Caamano J. N., et al. Embryonic stem cells in cattle. Reprod. Domest. Anim. 2008;43:32–37. doi: 10.1111/j.1439-0531.2008.01229.x. [DOI] [PubMed] [Google Scholar]
- Munsie M. J. Michalska A. E. O'Brien , et al. Isolation of pluripotent embryonic stem cells from reprogrammed adult mouse somatic cell nuclei. Curr. Biol. 2000;10:989–992. doi: 10.1016/s0960-9822(00)00648-5. [DOI] [PubMed] [Google Scholar]
- Nandi S. Chauhan M.S. Palta P. Effect of environmental temperature on quality and developmental competence in vitro of buffalo oocytes. Vet. Rec. 2001;148:278–279. doi: 10.1136/vr.148.9.278. [DOI] [PubMed] [Google Scholar]
- Nishikawa S. Goldstein R.A. Nierras C.R. The promise of human induced pluripotent stem cells for research and therapy. Nat. Rev. Mol. Cell Biol. 2008;9:725–729. doi: 10.1038/nrm2466. [DOI] [PubMed] [Google Scholar]
- Notarianni E. Laurie S. Moor R.M., et al. Maintenance and differentiation in culture of pluripotential embryonic cell lines from pig blastocysts. J. Reprod. Fertil. 1990;41:51–56. [PubMed] [Google Scholar]
- Notarianni E. Galli C. Laurie S., et al. Derivation of pluripotent, embryonic cell lines from the pig and sheep. J. Reprod. Fertil. 1991;43:255–260. [PubMed] [Google Scholar]
- Okita K. Ichisaka T. Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Panasophonkul S. Tharasanit T. Techakumphu1 M. Establishment of porcine embryonic stem-like cells from parthenogenetic and in vivo derived embryos. Thai J. Vet. Med. 2010;40:273–280. [Google Scholar]
- Park I.H. Zhao R. West J.A., et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Pashaiasl M. Khodadadi K. Holland M.K., et al. The efficient generation of cell lines from bovine parthenotes. Cell. Reprogram. 2010;12:571–579. doi: 10.1089/cell.2009.0118. [DOI] [PubMed] [Google Scholar]
- Pawar S.S. Malakar D. De A.K., et al. Stem cell-like outgrowths from in vitro fertilized goat blastocysts. Indian J. Exp. Biol. 2009;47:635–642. [PubMed] [Google Scholar]
- Racowsky C. Jackson K.V. Cekleniak N.A., et al. The number of eight-cell embryos is a key determinant for selecting day 3 or day 5 transfer. Fertil. Steril. 2000;73:558–564. doi: 10.1016/s0015-0282(99)00565-8. [DOI] [PubMed] [Google Scholar]
- Reubinoff B. E. Pera M. F. Fong C. Y., et al. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat. Biotechnol. 2000;18:399–404. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]
- Ribeiro Ede S. Gerger R.P. Ohlweiler L.U., et al. Developmental potential of bovine handmade clone embryos reconstructed by aggregation or fusion with distinct cytoplasmic volumes. Cloning Stem Cells. 2009;11:377–386. doi: 10.1089/clo.2009.0022. [DOI] [PubMed] [Google Scholar]
- Roach M. Wang L. Yang X., et al. Bovine embryonic stem cells. Methods Enzymol. 2006;418:21–37. doi: 10.1016/S0076-6879(06)18002-7. [DOI] [PubMed] [Google Scholar]
- Saito S. Ugai H. Sawai K., et al. Isolation of embryonic stem-like cells from equine blastocysts and their differentiation in vitro. FEBS Lett. 2002;531:389–396. doi: 10.1016/s0014-5793(02)03550-0. [DOI] [PubMed] [Google Scholar]
- Saito S. Sawai K. Ugai H., et al. Generation of cloned calves and transgenic chimeric embryos from bovine embryonic stem-like cells. Biochem. Biophys. Res. Commun. 2003;309:104–113. doi: 10.1016/s0006-291x(03)01536-5. [DOI] [PubMed] [Google Scholar]
- Schoonjans L. Albright G.M. Li J.L., et al. Pluripotential rabbit embryonic stem (ES) cells are capable of forming overt coat color chimeras following injection into blastocysts. Mol. Reprod. Develop. 1996;45:439–443. doi: 10.1002/(SICI)1098-2795(199612)45:4<439::AID-MRD5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Shah R.A. George A. Singh M.K., et al. Handmade cloned buffalo (Bubalus bubalis) embryos: comparison of different media and culture systems. Cloning Stem Cells. 2008;10:435–442. doi: 10.1089/clo.2008.0033. [DOI] [PubMed] [Google Scholar]
- Sritanaudomchai H. Pavasuthipaisit K. Kitiyanant Y., et al. Characterization and multilineage differentiation of embryonic stem cells derived from a buffalo parthenogenetic embryo. Mol. Reprod. Dev. 2007;74:1295–1302. doi: 10.1002/mrd.20592. [DOI] [PubMed] [Google Scholar]
- Stice S.L. Strelchenko N.S. Keefer C.L., et al. Pluripotent bovine embryonic cell lines direct embryonic development following nuclear transfer. Biol Reprod. 1996;54:100–110. doi: 10.1095/biolreprod54.1.100. [DOI] [PubMed] [Google Scholar]
- Strelchenko N.S. Bovine pluripotent stem cells. Theriogenology. 1996;45:131–140. [Google Scholar]
- Sukoyan M.A. Kerkis A.Y. Mello M.R., et al. Establishment of new murine embryonic stem cell lines for the generation of mouse models of human genetic diseases. Braz. J. Med. Biol. Res. 2002;35:535–542. doi: 10.1590/s0100-879x2002000500004. [DOI] [PubMed] [Google Scholar]
- Suzuki H. Kamada N. Ueda O., et al. Germ-line contribution of embryonic stem cells in chimeric mice: Influence of karyotype and in vitro differentiation ability. Exp. Anim. 1997;46:17–23. doi: 10.1538/expanim.46.17. [DOI] [PubMed] [Google Scholar]
- Takahashi K. Tanabe K. Ohnuki M., et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tanghe S. Van Soom A. Duchateau L., et al. Inhibition of bovine sperm-oocyte fusion by the p-aminophenyl derivative of D-mannose. Mol Reprod Dev. 2004;67:224–232. doi: 10.1002/mrd.10387. [DOI] [PubMed] [Google Scholar]
- Thouas G.A. Korfiatis N.A. French A.J., et al. Simplified technique for differential staining of inner cell mass and trophectoderm cells of mouse and bovine blastocysts. Reprod. Biomed. Online. 2001;3:25–29. doi: 10.1016/s1472-6483(10)61960-8. [DOI] [PubMed] [Google Scholar]
- Vajta G. Lewis I.M. Trounson A.O., et al. Handmade somatic cell cloning in cattle: analysis of factors contributing to high efficiency in vitro. Biol Reprod. 2003;68:571–578. doi: 10.1095/biolreprod.102.008771. [DOI] [PubMed] [Google Scholar]
- Vandaele L. Mateusen B. Maes D., et al. Is apoptosis in ovine in vitro produced embryos related to early developmental kinetics and in vivo bull fertility? Theriogenology. 2006;65:1691–1703. doi: 10.1016/j.theriogenology.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Vassilieva S. Guan K. Pich U., et al. Establishment of SSEA-1- and Oct-4-expressing rat embryonic stem-like cell lines and effects of cytokines of the IL-6 family on clonal growth. Exp. Cell Res. 2000;258:361–373. doi: 10.1006/excr.2000.4940. [DOI] [PubMed] [Google Scholar]
- Verma V. Gautam S.K. Singh B., et al. Isolation and characterization of embryonic stem cell-like cells from in vitro-produced buffalo (Bubalus bubalis) embryos. Mol. Reprod. Dev. 2007;74:520–529. doi: 10.1002/mrd.20645. [DOI] [PubMed] [Google Scholar]
- Wakayama S. Ohta H. Kishigami S., et al. Establishment of male and female nuclear transfer embryonic stem cell lines from different mouse strains and tissues. Biol. Reprod. 2005;72:932–936. doi: 10.1095/biolreprod.104.035105. [DOI] [PubMed] [Google Scholar]
- Wakayama T. Cloned mice and embryonic stem cell lines generated from adult somatic cells by nuclear transfer. Oncol. Res. 2003;13:309–314. doi: 10.3727/096504003108748500. [DOI] [PubMed] [Google Scholar]
- Wang L. Duan E. Sung L.Y., et al. Generation and characterization of pluripotent stem cells from cloned bovine embryos. Biol. Reprod. 2005;73:149–155. doi: 10.1095/biolreprod.104.037150. [DOI] [PubMed] [Google Scholar]
- Wernig M. Meissner A. Foreman R., et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- Wheeler M.B. Development and validation of swine embryonic stem cells: A review. Reprod. Fertil. Develop. 1994;6:563–568. doi: 10.1071/rd9940563. [DOI] [PubMed] [Google Scholar]
- Wilcox J.T. Semple E. Gartley C., et al. Characterization of canine embryonic stem cell lines derived from different niche microenvironments. Stem Cells Dev. 2009;18:1167–1178. doi: 10.1089/scd.2008.0336. [DOI] [PubMed] [Google Scholar]
- Yadav P.S. Kues W.A. Herrmann D., et al. Bovine ICM derived cells express the Oct4 ortholog. Mol. Reprod. Dev. 2005;72:182–190. doi: 10.1002/mrd.20343. [DOI] [PubMed] [Google Scholar]
- Yu S.M. Yan X.R. Chen D.M., et al. Isolation and characterization of parthenogenetic embryonic stem (pES) cells containing genetic background of the Kunming mouse strain. Asian-Aust. J. Anim. Sci. 2011;24:37–44. [Google Scholar]
- Zhang L. Luo Y.B. Bou G., et al. Overexpression Nanog activates pluripotent genes in porcine fetal fibroblasts and nuclear transfer embryos. Anat Rec (Hoboken). 2011;294:1809–1817. doi: 10.1002/ar.21457. [DOI] [PubMed] [Google Scholar]
- Zhu S.X. Sun Z. Zhang J.P. Ovine (Ovis aries) blastula from an in vitro production system and isolation of primary embryonic stem cells. Zygote. 2007;15:35–41. doi: 10.1017/S0967199406003959. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.