

Abstract
The capacity of cutaneous, including trigeminal endings, to detect chemicals is known as chemesthesis or cutaneous chemosensation. This sensory function involves the activation of nociceptor and thermoreceptor endings and has a protective or defensive function, as many of these substances are irritants or poisonous. However, humans have also developed a liking for the distinct sharpness or pungency of many foods, beverages, and spices following activation of the same sensory afferents. Our understanding of the cellular and molecular mechanisms of chemosensation in the trigeminal system has experienced enormous progress in the past decade, following the cloning and functional characterization of several ion channels activated by physical and chemical stimuli. This brief review attempts to summarize our current knowledge in this field, including a functional description of various sensory channels, especially TRP channels, involved in trigeminal chemosensitivy. Finally, some of these new findings are discussed in the context of the pathophysiology of trigeminal chemosensation, including pain, pruritus, migraine, cough, airway inflammation, and ophthalmic diseases.
Keywords: Chemestesis, pungency, spices, capsaicin, menthol, TRP channel, TRPV1, TRPV2, TRPV3, TRPA1, TRPM8, KCNK, pain, cough, asthma, dry eye
A large variety of plant-derived natural products and other chemical agents evoke sensory responses with an infinite shade of perceptual qualities. The perception of chemical stimuli by sensory means is referred to as chemosensation or chemoreception. In humans, the olfactory and the gustatory systems are the principal chemosensory systems and the substrates for the senses of smell and taste, respectively (1). The perception of chemical agents, such as a perfume or the smell of a fruit, is strongly linked to memory and can evoke pleasant feelings. They are also important in nutrition and food selection. However, detection of many chemicals can also have a protective or defensive function, as many are irritants or are poisonous. Their presence in plants is precisely to act as deterrents against animals searching for food.
Albeit less well recognized, the trigeminal somatosensory system also plays a fundamental role in chemosensation and the overall “flavor” of foods. Sensory endings of the trigeminal (V cranial) nerve innervate the skin covering the face, the mucous membranes of the nasal and oral cavities, and the cornea and conjunctiva of the eye (Figure 1). These endings can be activated by physical stimuli (mechanical forces and temperature) and by a huge array of chemical agents (2), and evoke sensations of touch, temperature, and pain. The capacity of cutaneous, including trigeminal endings, to detect chemicals is known as chemesthesis or cutaneous chemosensation (3). Chemosensitive glossopharyngeal (IX cranial) and vagal (X cranial) nerve fibers innervating the oropharynx and respiratory tract fall in the same category. Oral chemesthesis explains the pungent or sharp feel of many different foods and spices such as chili peppers, horseradish, wasabi roots, and Szechuan pepper, the coolness of peppermint, the tingle of carbonated drinks, and the irritation produced by substances such as nicotine or raw garlic extracts (Table 1). Protective responses evoked by trigeminal stimulation include salivation, tearing, coughing, respiratory depression, and sneezing. Moreover, pungent irritation or burning pain is a very common side effect of drugs applied topically to the skin.
Figure 1.
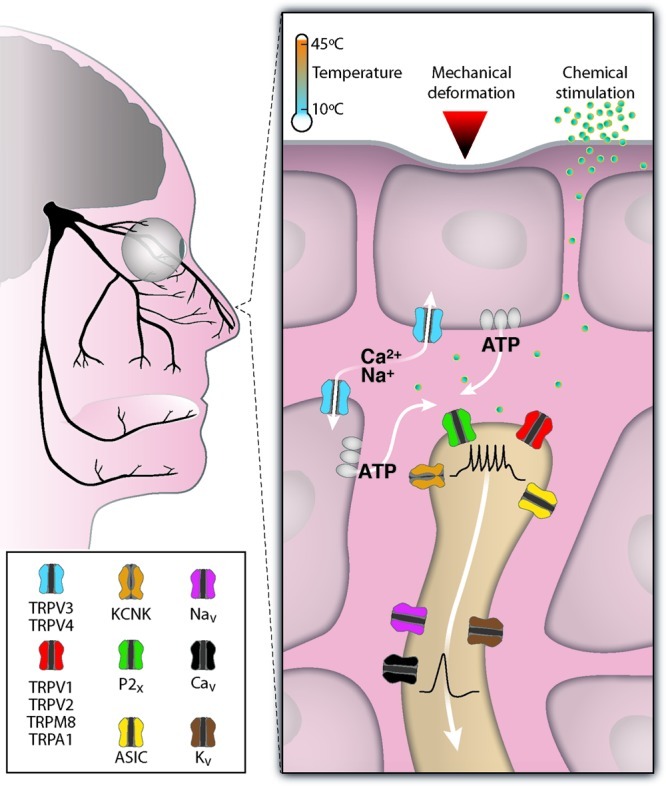
Molecular determinants of chemosensation in trigeminal nerve terminals. Branches of the human trigeminal nerve innervating the face, the eye, and the nasal and oral cavities. Temperature and many chemical agents can stimulate chemosensitive channels directly. These channels are expressed in sensory nerve terminals and mucosal epithelial cells or skin keratinocytes. The opening of cationic channels (TRPs, ASICs) or the closing of potassium-selective channels (KCNK) generate a graded transduction current, depolarization, and the firing of action potentials. Voltage gated sodium (NaV), calcium (CaV), and potassium (KV) channels participate in action potential electrogenesis and propagation of the nerve impulse to the brainstem. Mechanical deformation of the skin can trigger the release of ATP from keratinocytes and the activation of purinergic (P2X) receptors from sensory nerve terminals.
Table 1. Plant-Derived Agonists of Chemosensitive Ion Channelsa.
| chemical agonist | natural source | receptor | ref |
|---|---|---|---|
| capsaicin | chili peppers (Capsicum sp.) | V1 | (13), (17) |
| resiniferatoxin | dried latex of the cactus (Euphorbia resinifera) | V1 | (13), (72) |
| piperine | black pepper (Piper nigrum) | V1 > A1 | (24),161, (162) |
| Δ9-tetrahydrocannabinol | marijuana plant (Cannabis sativa) | A1, V2 | (83), (84), (109) |
| cannabidiol | marijuana plant (Cannabis sativa) | V1, V2 | (83), (84) |
| camphor | camphor laurel tree (Cinnamomum camphora) | V3, V1 | (59), (85), (87) |
| incensole acetate | incense (Boswellia sp.) | V3 | (89) |
| thymol | thyme (Thymus vulgaris) | V3, A1 | (88), (100), (115), |
| carvacrol | oregano (Origanum vulgare) | V3, A1 | (88), (100) |
| eugenol | clove oil (Eugenia charophyllata, Syzygium aromaticum) | V3, V1, A1 | (96), (100) |
| allyl isothiocyanate | mustard oil (Brassica nigra), horseradish (Armoracia rusticana) wasabi (Wasabia japonica) | A1 > V1 | (96), (97), (107), (109) |
| cinnamaldehyde | cinnamon oil (Cinnamomum cassia) | A1 | (96), (103), (110), |
| allicin | garlic (Allium sativum) | A1 > V1 | (98), (99), (163) |
| diallyl disulfide | garlic (Allium sativum) | A1 > V1 | (98), (99), (164) |
| 1′-acetoxychavicol acetate | galangal (Alpinia galanga, Kaempferia galanga) | A1 | (165) |
| methyl salicylate | wintergreen oil (Betula lenta, Spiraea sp., Gaultheria sp.) | A1, V1 | (96),107 |
| gingerols | ginger (Zingiber officinale) | A1, V1 | (96), (166) |
| menthol | peppermint oil (Mentha piperita) | M8, A1 > V3 | (16), (59), (115), (117), (123) |
| eucalyptol | eucalyptus oil (Eucalyptus polybractea) | M8 | (16), (123) |
| linalool | coriander (Coriandrum sativum), laurel (Laurus nobilis), Szechuan pepper (Zanthoxylum piperitum) | M8, A1 | (123), (129) |
| citral | lemongrass (Cymbopogon citratus) | M8 > V1 > A1 > V3 | (86)b |
| nicotine | tobacco plant (Nicotiana tabacum) | A1 | (114) |
| hydroxy-α-sanshool | Szechuan pepper (Zanthoxylum piperitum) | KCNK, A1, V1 | (14), (129) |
The references cited focus on data obtained from recombinant channels expressed in heterologous systems. In many of the same references, responses to the same agonists were also observed in trigeminal or DRG sensory neurons. The symbol > denotes that the receptor on the left shows a higher affinity, or responds stronger, to the agonist. Note that in most cases the natural source of a given agonist includes other species belonging to the same or other genera.
Reference (123) could not demonstrate effects of citral on mouse TRPM8.
The sensitivity of somatosensory terminals to chemical irritants was first recognized by Parker in 1912 while studying aversive responses in fish, coining the term “common chemical sense” to describe this property (2,4,5). Some early researchers, including Parker, described this sense as distinct from taste, odor, touch and pain, while others defended the view that pain and the common chemical sensitivity were mediated by the same nerve endings (6). Most modern authors agree that cutaneous chemosensation is determined primarily by the chemical activation of nociceptors and thermoreceptors (7,8).
Chemical signaling of taste and smell involves a metabotropic mechanism; binding of the chemical agent to the receptor activates a heterotrimeric G protein that initiates a transduction cascade (1). In contrast, activation of somatosensory afferents by chemicals involves the direct gating of an ion channel by the chemical stimulus and is called ionotropic transduction. In either case, the chemical signal leads to a change in ionic permeability and the depolarization of the sensory receptor. The sensations evoked by chemical agents applied to the skin or mucous membranes vary in their quality (e.g., burning, stinging, itch, tingling, cold), temporal profile, and intensity. Even sensations evoked by individual chemicals are complex. An explanation for this complexity is provided by the overlapping sensitivity of individual ion channels to chemical agonists (Table 1), which results in the combinatorial activation of different classes of endings by a single chemical (9,10).
In recent years, a tremendous progress has been achieved in our understanding of how different chemicals are detected by trigeminal endings (8,11,12). A landmark finding was the use of capsaicin to clone the vanilloid receptor and the identification of transient receptor potential (TRP) channels as transducers of physical (e.g., temperature) and chemical signals in mammalian sensory fibers (13) (Figure 2). In addition to TRP channels, other ion channels expressed in trigeminal neurons appear to play important roles in chemosensation (14,15). Other natural products (e.g., menthol) were also fundamental in the identification and functional characterization of other sensory channels (16). This brief review attempts to summarize our current knowledge of the cellular and molecular mechanisms of chemosensation in the trigeminal system. Some of these new findings are discussed in the context of the pathophysiology of trigeminal chemosensation. Other authors have addressed some aspects of this subject in recent years (17−19).
Figure 2.

Chemosensitive responses of cultured mice trigeminal sensory neurons. Fura-2 calcium imaging of agonist-evoked responses of individual sensory neurons to capsaicin (100 nM) and warm temperature (A), menthol (100 μM) and cold temperature (B), nifedipine (10 μM) and mustard oil (100 μM) (C), clotrimazole (10 μM), capsaicin (100 μM), menthol (100 μM), and mustard oil (100 μM) (D), nicotine (1 mM) and mustard oil (100 μM) (E), ATP (1 mM) (F). Responses were obtained with different imaging systems, and absolute amplitude cannot be compared. (A) by R. Madrid (unpublished), (B) from Madrid et al. (159), (C) from Fajardo et al. (113), (D) from Meseguer et al. (160), (E) by V. Meseguer (unpublished), and (F) by E. de la Pena (unpublished). The record in (D) was obtained from a Trpv1−/− mouse.
Pungency
Pungency refers to the sharp or biting quality of sensation evoked by some foods, spices, or volatile compounds in contact with trigeminal endings of the nasal or oral epithelium. The response threshold of trigeminal fibers to chemical stimuli is generally 1−2 log units higher than olfactory thresholds for the same compounds (2,20). This is not true for the corneal endings in the eye that can be extremely sensitive to chemical irritants. To reach the nerve endings and evoke a sensation, the agonists first need to pass though the lipid phase of the epithelial barrier or through tight junctions. It is somewhat puzzling why some potent agonists of nociceptive transduction channels lack pungency when applied to the skin or mucosal surfaces. A classical study by Szolcsanyi and Jancso-Gabor showed that some capsaicin analogues produced rapid desensitization of the sensory terminals but lacked pungency characteristics (21). Olvanil (N-9-Z-octadecenoyl-vanillamide) is another agonist of TRPV1 channels lacking the pungency of capsaicin. Also, SDZ249-665, a potent agonist of TRPV1 channels, shows little irritancy when instilled in the nose or eye of guinea pigs (22). This is in contrast with the pungent irritation produced by capsacin, which is followed by a prolonged desensitization, a feature exploited clinically to achieve antinociception. Dermal application of anandamide, an endogeneous cannabinoid, produces skin microvessel dilation via a TRPV1-dependent mechanism (23). Interestingly, anandamide application on the skin does not result in pain.
The mechanisms explaining this lack of pungency of some agonists of TRPV1 are not clear. In the case of anandamide, a nonselective agonist of TRPV1, it is possible that well-known additional effects, such as inhibition of sodium and calcium channels, and activation of potassium channels could prevent the electrical propagation of the afferent message. Another possibility is that binding to the receptor by nonpungent agonists is slow, producing a progressive depolarization and inactivation of the receptor potential at the nerve terminals. Recently, Ursu et al. evaluated the pungency of several TRPV1 agonists covering a large range of potencies and found a strong correlation between the kinetics of calcium flux, pungency (estimated by the increment in eye-wipes produced by the compound), and lipophilicity (24). The authors evaluated the effect of agonists on membrane potential and action potential firing, with current-clamp recordings of DRG neurons, and found that the onset of the depolarization induced by olvanil (i.e., nonpungent agonist) was much slower, and irreversible, compared to the effects induced by capsaicin. This slow depolarization, albeit of similar magnitude to that produced by capsaicin, translated into a low probability of evoking action potentials. A molecular understanding of the mechanisms involved in long lasting TRPV1 desensitization by capsaicin agonists is critical for its potential application in therapies targeting the TRPV1 receptor, including treatment of chronic pain and overactive bladder syndrome.
Perceptual Interaction between Temperature and Chemical Sensitivity
In the trigeminal system, the sensations evoked by many chemical agents show clear cross-interactions with temperature (25). Many chemical agents (e.g., capsaicin) sensitize the perception of temperature. In addition, temperature and touch introduce additional perceptual qualities to the sensory experience evoked by chemicals in contact with the trigeminal system. Menthol potentiates cold sensations (26). In agreement with these psychophysical findings, menthol sensitizes responses of trigeminal endings to cold temperature (27).
These interactions are easily explained by the allosteric gating of TRP channels by chemical and thermal stimuli (see below) (13,28,29). TRP channels with a marked sensitivity to temperature are referred to as thermoTRPs (30).
Chemosensory Signaling between Skin Cells and Nerve Fibers
A special type of chemosensory communication is provided by the signaling between cells in the skin (e.g., keratinocytes, mast cells) and neighboring nerve endings (31,32). Chemosensing by non-neuronal cells is also important in tissues normally exposed to food- or blood-borne substances, such as the gastrointestinal epithelium.
It has become apparent that certain thermoTRP channels are functionally expressed in numerous non-neuronal cell types of the skin. It has been postulated that traumatic or thermal activation of skin cells can release diffusible molecules that activate neighboring nerve endings (Figure 1). In a co-culture system, mechanical activation of keratynocytes results in a propagated Ca2+ wave and the activation of adjacent DRG neurons by a mechanism involving released ATP (33). A recent elegant study provided strong evidence for the thermal activation of TRPV3 channels on keratinocytes and the subsequent release of ATP to the extracellular space (34). ATP is an accepted transmitter or co-transmitter in different sensory nerves (35,36). Indeed, sensory neurons placed in close proximity to activated keratinocytes responded with a depolarizing inward current (34).
The Sensation of Itch
Itch, formally known as pruritus, is defined as an unpleasant sensation that triggers an overwhelming urge to scratch. Severe pruritus is a frequent medical problem with poor treatment. A number of chemical agents behave as mediators of pruritus (i.e., pruritogens), including endogenous substances such as histamine, neuropeptides, prostaglandin E2, proteases, and cytokines. Drugs such as chloroquine, clonidine, and morphine can also induce pruritus.
Itch and pain are closely related sensations, with important similarities (e.g., sensitization), but nevertheless are distinct in their behavioral manifestations. There is a strong overlap between mediators of itch and pain, as well as neuronal pathways involved in their expression (37). However, recent studies provide strong support for the concept of a specific sensory labeled line for itch separate from the one reporting pain. Thus, selective ablation of a specific class of interneurons in the dorsal horn of the spinal cord can abolish itch while pain sensations remain completely normal (38). In contrast, the loss of a different population of inhibitory interneurons in the superficial dorsal horn potentiates itch responses with no change in nociception (39). Also in this line, selective removal of the glutamate transporter VGlut2 from sensory afferents expressing TRPV1 results in a strong potentiation of spontaneous scratching behaviors in mice (40). Finally, recent work implicates altered function of TRPV3 channels in causing allergic and pruritic dermatitis in rodents (41).
Chemical Sensitivity to Protons
Extracellular acidification is typical of tissues under conditions of ischemia or inflammation. Acidic solutions applied to the skin or mucosae induce pain (reviewed in ref (42)). These solutions activate and sensitize trigeminal nerve terminals of polymodal nociceptors (43,44) and induce inward currents in small diameter trigeminal sensory neurons (45)(46). Currents in individual neurons have different kinetic properties, pharmacology, and pH sensitivity, suggesting that they are mediated by more than one receptor type. Several ion channels expressed in trigeminal nociceptors are sensitive to low pH, including TRPV1, ASICs, and TASK channels.
The acid-sensing ion channels (ASICs), members of the DEG/ENaC superfamily, are activated by extracellular protons and are overexpressed during inflammation. Several ASICs are expressed in TG neurons (e.g., ASIC-α, ASIC-β, DRASIC). Amiloride reduces or eliminates proton-gated currents in many TG neurons, suggestion and important contribution of ASICs (46). Moreover, psychophysical and pharmacological data in humans suggest that ASICs have an important role in the sensation of cutaneous acid-induced pain.
Extracellular protons also activate TRPV1 channels and strongly sensitize the response to vanilloid agonists (13). Many studies have identified a pH-sensitive current blocked by specific TRPV1 antagonists (e.g., capsazepine) in TG neurons (13,46,47). Unlike ASIC-mediated currents, these currents are generally noninactivating.
Among the 2P-domain K+ channels, TASK subunits (TASK-1, TASK-2, TASK-3) represent background outward rectifiers that are constitutively active at resting membrane potential and are inhibited by extracellular acidic pH values (48). Closure of these channels will depolarize the neuron. TASK-1 and TASK-3 channels have been found in some sensory neurons with nociceptive properties (47). Moreover, the pH-sensitive current in these neurons is only partially sensitive to capsazepine.
Chemical Sensitivity to CO2
Carbonated drinks, containing dissolved CO2, elicit a very distinct sensation that combines a stinging and pungent quality. This complex sensation is certainly responsible for the popularity of carbonated beverages among consumers. Until recently, the neural basis of this sensation remained somewhat obscure; some authors favored a mechanical hypothesis (i.e., the bursting of tiny CO2 bubbles would excite mechanoreceptors) over a chemosensitive mechanism. It is well established that CO2 activates trigeminal nociceptors in the cornea and nasal epithelium (49,50). Although many CO2-sensitive fibers are capsaicin-sensitive, the mechanism of action appears to be independent of TRPV1 because the activation is not blocked by capsazepine and is only partially desensitized by capsaicin (51). In agreement with the neurophysiological data, the sensations evoked by CO2 solutions to the tongue are only partially desensitized by capsaicin (52). Some studies postulated the conversion of CO2 into carbonic acid, a reaction catalyzed by carbonic anhydrase, and the stimulation of polymodal nociceptors by protons. However, a large fraction of lingual nerve fibers sensitive to carbonated water are not activated by acidic solution, indicating that changes in mucosal pH are not the direct mediators of the response to CO253. The mechanism was clarified recently in an elegant study (see below) and appears to be mediated by intracellular acidification of trigeminal nerve terminals and activation of TRPA1 channels (54). It is interesting that cooling enhances the pungent and tingling sensation of carbonated water (55). Whether this is due to physicochemical factors (i.e., the effect of temperature on CO2 solubility) or to potentiation of CO2-mediated responses in TRPA1 nociceptors by low temperature is unknown (56).
TRP Channels as Polymodal Transducers
The transient receptor potential (TRP) ion channel superfamily, which was first described in the fruit fly Drosophila melanogaster, forms a large group of cation-permeable ion channels conserved in yeast, worms, insect, fish, and mammals (57). These receptors share structural similarities with voltage-gated K+ channels and are classified according to their primary amino acid sequence (58). They are probably tetrameric, and each subunit is predicted to have six transmembrane (TM) spanning domains with a short pore-forming hydrophobic loop between TM domains 5 and 6.
A distinct feature of many TRP channels is their polymodal activation by physical (e.g., temperature, mechanical forces) and chemical stimuli (9,59). This characteristic is ideal to serve as a detector of environmental stimuli, acting as a molecular interface between the external world and the nervous system. Many TRP channels are expressed in sensory neurons (60) and skin cells, and can be activated by a variety of physical and chemical stimuli to function as signal integrators (61). Importantly, the different stimuli show allosteric interactions in the gating of the channels: exposure to one stimulus sensitizes the response to another (29). The prototype for this characteristic is the TRPV1 receptor that integrates noxious heat, tissue acidosis, and chemical stimuli which are all known to cause pain (13,62).
Sensory Ion Channels and Chemosensation
Next, I will briefly address the chemical sensitivity of individual ion channels expressed in trigeminal sensory neurons (Figure 1). Emphasis is placed on the functional relevance of the different channels to the detection of external stimuli. The reader is referred to a number of recent reviews which provide an extensive account of the pharmacological properties and chemical sensitivity of some of these transduction channels (19,63−66).
TRPV1
TRPV1, also known as the “capsaicin or vanilloid receptor”, is a Ca2+-permeable, nonselective cationic channel that belongs to the TRP family of ion channels. The identification and functional characterization of this protein as a polymodal transducing molecule for physical and chemical stimuli in the mammalian somatosensory system represents a milestone in molecular studies of pain and trigeminal chemosensation (13). TRPV1 is activated by capsaicin (13,67), the pungent component of “hot” chilli peppers long since known as a selective activator of polymodal nociceptor endings (reviewed in refs (68 and 69)). Topical application of capsaicin to the skin or mucosae elicits a sensation of burning pain (70). Fittingly, the channel is also activated by noxious heat temperatures (threshold ∼ 43 °C) and by extracellular protons (reviewed in ref (71)). The early response to capsaicin application includes an augmented sensitivity to noxious and innocuous stimuli.
Other vanilloids also activate the TRPV1 receptor (17,66). Among them, resiniferotoxin, an alkaloid extracted from some species of Euphorbia cactuslike plants, is one of the most potent chemical agonists of TRPV1 known (72). Various fatty acid derivatives, referred to as endovanilloids, are activators of TRPV1 channels (66). Recently, some animal toxins were identified as TRPV1 agonists (73).
TRPV1 is preferentially expressed in small size, unmyelinated primary sensory neurons of the trigeminal and DRG ganglia, the classical polymodal nociceptors. Therefore, TRPV1 was proposed as the molecular substrate for polymodality in these neurons, providing them with the ability to respond to heat and to potentially injurious chemical stimuli causing pain (13,62) (Figure 2A). More recently, TRPV1 has been detected in other tissues, including keratinocytes, urothelium, endothelial cells, and brain nuclei, expanding its biological role.
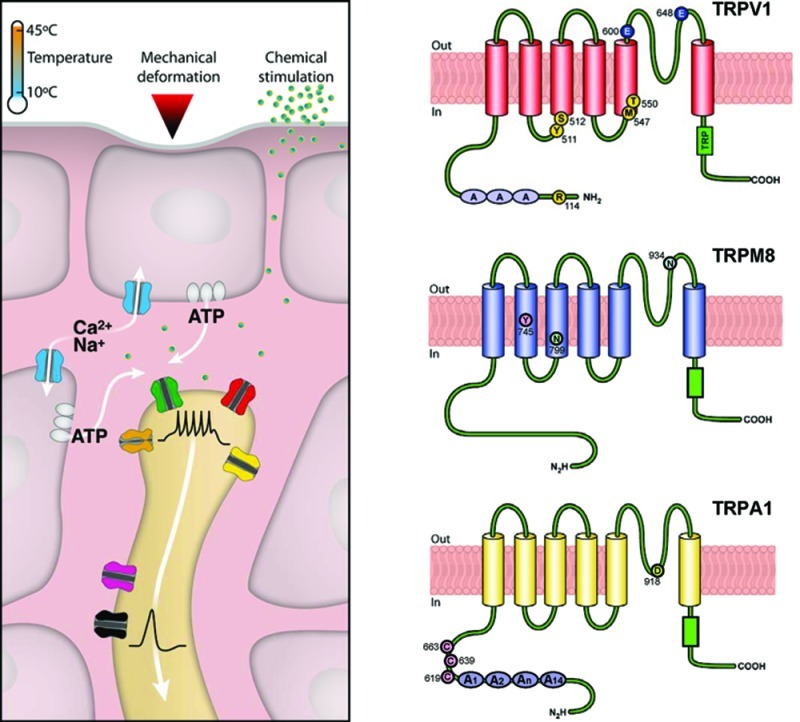
Bird orthologues of TRPV1 are insensitive to capsaicin. This characteristic was exploited by Jordt and Julius, making chimeric proteins between mammalian and avian TRPV1 receptors, to identify structural elements in TRPV1 important for capsaicin sensitivity (67). The authors found that that Tyr 511 and Ser 512, located in the region between the second intracellular loop and the third TM domain, are critical for the action of vanilloid ligands. Further studies have established the importance of additional residues in capsaicin sensitivity (reviewed in ref (71)). Other studies have established the molecular determinants of heat and proton sensitivity in TRPV1. The acidic residue Glu 600, located within the extracellular loop close to the channel pore, is involved in proton potentiation of TRPV1 activity, whereas Glu 648 is critical for proton-evoked activation of TRPV1 (74).
An important functional characteristic of TRPV1 channels is their high permeability to Ca2+ ions (13). In addition, prolonged exposure of TRPV1 to vanilloid agonists results in a modification of the ionic pore properties (i.e., “pore dilation”) (75) with a threefold increase in the relative permeability to Ca2+ (as compared to sodium). This results in a Ca2+ overload of capsaicin-sensitive nerve terminals, the retraction of epidermal nerve fibers, and the long-lasting functional desensitization of nociceptive sensory endings (76). These structural and functional changes are the basis for the therapeutic use of skin patches with high capsaicin content for the treatment of peripheral neuropathic conditions, including postherpetic neuralgia, diabetic neuropathy, and sensory neuropathy associated to HIV infection (77,78). This functional pore dilation of TRPV1 by agonists is also the basis of a novel delivery method of the membrane-impermeant local anesthetic QX-314 to TRPV1-expressing sensory nerve terminals, leading to their selective block (79,80).
TRPV2
TRPV2 is an ion channel with an amino acid sequence ∼50% identical to TRPV1 (81). It is expressed in sensory neurons of medium and large diameter. The channel is insensitive to capsaicin and low external pH. TRPV2 is activated by noxious heat, with an activation threshold (∼52 °C) higher than that of TRPV1 (81) and to changes in osmolarity (82). Very few natural chemical agonists of TRPV2 are known, and none are considered selective. Δ9-Tetrahydrocannabinol and cannabidiol of the marijuana plant activate the channel (83,84).
TRPV3
TRPV3 is an ion channel structurally analogous to TRPV1 that is strongly expressed in skin keratinocytes. Expression of TRPV3 in peripheral sensory neurons is a controversial subject (85,86). When transfected into mammalian cells, this channel responds to temperature with a threshold around 33 °C but not to capsaicin or pH changes (reviewed in ref (30)). TRPV3 null mice have strong deficits in responses to innocuous and noxious heat, indicating that it participates in thermosensation (85).
TRPV3 is activated by a number of natural chemical compounds, including camphor (87) and carvacrol, a monoterpenoid phenol present at high concentrations in the essential oil of oregano. Other monoterpenes such as 6-tert-butyl-m-cresol, eugenol (derived from cloves), dihydrocarveol, thymol (from thyme), carveol, and (+)-borneol also activate TRPV3 (87,88). Incensole acetate, a compound in the resin obtained from Boswellia trees, also activates TRPV3 (89). Farnesyl pyrophosphate (FPP), an intermediate metabolite in the mevalonate pathway, is a specific activator of TRPV3. The cutaneous sensations produced by heating and cooling can be enhanced by camphor coapplication (90).
TRPA1
TRPA1 is a nonselective cationic TRP channel (1119 amino acids in humans) phylogenetically distant to other mammalian TRP proteins. Like other TRPs, TRPA1 channels have six putative transmembrane-spanning domains, with cytoplasmic N- and C-terminal ends (91,92). Activation of this channel is responsible for the pungent feel of many foods and condiments, including isothiocyanates occurring naturally in fruits and plants such as mustard greens and capers (Table 1). Most notably, the chemical sensitivity of this receptor has been preserved during evolution, being similar in humans and flies (93).
Chemical activators of TRPA1 have been reviewed recently in several publications (59,91,92,94,95). The list of TRPA1 agonists keeps growing daily and includes many natural and synthetic irritants such as allyl isothiocyanate (in mustard oil) (96,97) (Figure 2C), cinnamaldehyde (in cinnamon oil) (96), allicin and diallyl disulfide (in garlic extract) (98,99), methyl salicylate (in wintergreen oil) (96), ginger (96), carvacrol (in oregano) (100), formalin (101), natural fungal deterrents like isovelleral (102), unsaturated aldehydes like acrolein (103), isocyanates (104), and oxidizing agents like hypochlorite (OCl−) and hydrogen peroxide (H2O2) (64,105). Several odorants (α-terpineol, amyl acetate, benzaldehyde, toluene) known to activate trigeminal fibers also activate TRPA1 channels (106). The unique chemical sensitivity of this ion channel for some of these irritants was clarified in TRPA1 knockout mice (103). These animals were completely insensitive to mustard oil and allicin (but see ref (107)). In addition, these animals had pronounced deficits in bradykinin-evoked nociceptor excitation, highlighting a fundamental role for TRPA1 in neurogenic inflammatory pain (96,97,103,108).
Mechanisms of TRPA1 Activation
An old puzzle in the pain literature concerned the diverse physicochemical properties of chemical sensory irritants. Recent work has unveiled the unique molecular mechanism of activation of TRPA1 channels, explaining its capacity to sense many structurally unrelated chemicals. Unlike traditional chemoreceptors (e.g., olfactory receptors) that can discriminate between closely related chemical structures, TRPA1 is activated by structurally different molecules with high chemical reactivity. These highly electrophilic compounds interact with thiol groups in side chains of intracellular cysteines in the intracellular N-terminal region of the TRPA1 molecule, forming covalent bonds (109,110). These covalent modifications produce long lasting channel openings that can be reversed by reducing agents such as dithiothreitol (110). Mutational studies have identified several cysteine and lysine residues involved in channel activation (109,110). In human TRPA1, crucial residues for channel activation by allyl isothiocyanate include a cluster of cysteines (Cys619, Cys639, and Cys663) and Lys708 (109). Actually, activation of TRPA1 by electrophilic compounds is more complex than outlined here (for further details, consult refs (91, 95, and 111). For example, certain Cys-reacting compounds show species-specific responses in TRPA1 channels (112).
Other agonists of TRPA1 appear to act by conventional reversible ligand−receptor interactions. They include, among others, icilin, nifedipine (113), nicotine (114) (Figure 2E), menthol (115) (Figure 2D), eugenol, carvacrol, and Δ9-tetrahydrocannabinol. Mutating the critical cysteines to nonreactive serines does not prevent the agonist action of some of these compounds (109,110).
Dissolved CO2 activates TRPA1-expressing trigeminal neurons. This activation is abrogated in neurons obtained from TRPA1−/− animals. Furthermore, intracellular protons activate TRPA1 channels in excised patches in a dose dependent manner, suggesting that the mechanism of activation in intact neurons involves diffusion of CO2 across the plasma membrane and intracellular acidification by the re-equilibration of CO2 and H2O in bicarbonate (HCO3−) and a free proton (H+) (54).
TRPM8
The transient receptor potential melastatin 8 channel (TRPM8) is a nonselective cation channel activated by mild cold temperatures (threshold around 25 °C in heterologous systems) and cooling compounds such as menthol. The TRPM8 protein is 1104 amino acids long (16,116,117) and is found in a subpopulation (10−15%) of small-diameter, cold-sensitive peripheral sensory neurons (16,117). The same neurons are activated by cooling compounds (Figure 2B). Thermosensitive nerve endings of these sensory neurons innervate the skin and mucosae (cornea, oral cavity) (118−120) where TRPM8 plays a clear physiological role in the detection of low temperature ambient signals (reviewed in refs (121 and 122)).
In addition to cold, TRPM8 can be activated by natural and synthetic cooling mimetic agents such as icilin (AG-3-5), eucalyptol, menthol, and an abundance of menthol analogues (123−125), with WS-12 being the most potent one described to date in the scientific literature. Other natural weak agonists of TRM8 include hydroxy-citronellal, geraniol, and linalool (123). Chemicals modulators of TRPM8 and their mechanisms of channel gating have been reviewed recently (63).
KCNK Channels
Alkylamides are a unique class of compounds that elicit a distinct tingling sensation when applied to the tongue (126). Natural plant-derived alkylamides, such as α-hydroxy-sanshool, are found in Szechuan pepper (Zanthoxylum piperitum) and are heavily used in oriental cooking. At the cellular level, α-hydroxy-sanshool activates mechanoreceptors and thermosensitive trigeminal neurons (127,128). The sensation evoked is clearly distinct from the pungent sensation evoked by capsaicin or isothiocyanates, specific activators of TRPV1 and TRPA1, respectively. This suggests a distinct molecular mechanism of action. Recently, Bautista et al. found that natural alkylamides inhibit a particular class of background or leak potassium channels with two pore domains (KCNK3, KCNK9, and KCNK18) (14). Closure of these potassium-selective channels leads to depolarization and firing. In vitro recordings from sensory fibers established that sanshool has a potent excitatory effect on D-hair cells, an ultrasensitive light-touch receptor, and Aβ mechanoreceptors. Sanshool also activated a subpopulation of very slowly conducting C fibers (128). The firing evoked by sanshool was not suppressed by TRPA1 and TRPV1 antagonists. However, other studies have established excitatory effects of sanshool on TRPV1 and TRPA1 channels (129).
Purinergic Receptors
Application of ATP to skin blisters is painful. Normally, ATP can be coreleased with noradrenaline from sympathetic nerve terminals or from skin cell in response to mechanical deformation (e.g., shear stress) (36). Application of ATP or α,β-methylene ATP excites capsaicin-sensitive trigeminal lingual afferents (130) (Figure 2F). Primary sensory neurons and nerve terminals express all subtypes of ionotropic purinergic receptors (P2X). The P2X3 subunit is selectively expressed in IB4(+) nociceptors, forming heteromeric assemblies with P2X2 subunits (15). In addition, a high percentage of sensory neurons express metabotropic P2Y receptors.
Patophysiology of Trigeminal Chemosensitivity
Trigeminal irritant stimuli applied repeatedly at short interstimulus intervals produce a sensitization of the response (3). Also, during inflammation, trigeminal nociceptor terminals can become sensitized, increasing their excitability and responding inadequately to innocuous stimuli (allodynia) and augmenting their response to noxious stimuli (hyperalgesia) (131).
The molecular mechanisms of nociceptor sensitization following injury are complex (131,132). One mechanism involves the sustained activation, posttranslational modifications, and membrane translocation of TRPV1 channels by a variety of endogenous agents released after injury or inflammation, including bradykinin and endogenous lipids, leading to the sensitizing/excitatory action of polymodal nociceptor neurons during these pathological states (133−138). TRPV1 channels are critical in inflammatory thermal hyperalgesia (139).
The expression of TRPV1 and TRPA1 channels in vagal chemosensory unmyelynated afferents innervating the lung and airways (140,141) suggests that activation of these receptors could be involved in the sympthomathology and pathogenesis of different airway diseases. Indeed, TRP channels are emerging as important mediators of pathologies such as asthma, chemical hypersensitivity, chronic cough, and chronic obstructive pulmonary disease (reviewed in refs (95 and 142−144)). Many TRPA1 agonists are volatile organic compounds, present in cigarette smoke and smog, which trigger eye irritation, sneezing, cough, bronchoconstriction, respiratory depression, mucous secretion, and neurogenic inflammation of the airways, reflexes aimed at limiting exposure of the affected tissues to potentially harmful chemicals.
Capsaicin is a powerful tussive agent, and TRPV1 is upregulated during airway inflammation (144). TRPA1 agonists, including acrolein and crotonaldehide, two α,β-unsaturated aldehydes present in cigarette smoke (145), can also trigger cough reflexes, a response inhibited by HC-030031, a selective TRPA1 antagonist (146,147).
Endogenous unsaturated aldehydes, such as 4-hydroxynonenal and 4-oxo-nonenal, also activate TRPA1 (105,148) and vagal bronchopulmonary fibers (149). These α,β-unsaturated aldehydes are produced during oxidation of membrane phospholipids in response to tissue injury, inflammation, and oxidative stress. Inhibition of TRPA1 with HC-030031 prevents neuropeptide-mediated constriction of bronchial rings produced by exposure to cigarette smoke or α,β-unsaturated aldehydes (145). Other endogenous products of oxidative stress also activate TRPA1 including hydrogen peroxide (H2O2) and the cyclopentenone prostaglandin, 15-deoxy-Δ(12,14)-prostaglandin J2, an electrophilic compund (105,150,151). Hydrogen sulfide (H2S) is a gaseous molecule formed endogenously during inflammation. It activates mouse and human TRPA1 in recombinant systems and induces bladder detrussor overactivity, suggesting a role for this inflammatory mediator in symptoms secondary to lower urinary tract infection (152). Nitroleic acid, a byproduct of nitrative stress also activates TRPA1 via covalent modification (153). Hypochlorite, the oxidazing mediator of chlorine, activates recombinant TRPA1 channels and TRPA1-expressing neurons (64).
A study in an animal model suggests that TRPA1 channels are involved in the etiology of the airway inflammation and hyperreactivity characteristic of asthma (154). Following an ovoalbumin challenge in sensitized mice, the inflammatory and allergic response, characterized by an increase in leukocytes (primarily eosiniphils) and Th2 interleukins and TNF-α, was abrogated by genetic ablation or pharmacological inhibition of TRPA1 (154). Moreover, airway hyperreactivity to acetylcholine was absent in the TRPA1 ko mice or after specific TRPA1 block.
Exposures to many volatile chemical irritants, such as formaldehyde, toluene, or acrolein, can trigger headache. A recent study by Kunkler et al. showed that activation of TRPA1 channels and release of CGRP are critical for the meningeal vasodilatation induced by environmental irritants (155). These findings suggest that TRPA1 participates in the activation of the trigeminovascular system following exposure to volatile irritants, unveiling a novel potential therapeutic target for the treatment of migraine headache.
Basal tearing is essential for maintaining ocular surface wetness. In a recent study, Parra and colleagues found a reduced basal tearing rate in TRPM8 ko mice (120). These animals showed a complete absence of spontaneous and cold-evoked activity in trigeminal corneal thermoreceptor endings, and the authors hypothesize that a reduced reflex input of cold- and menthol-activated corneal sensory afferents to lacrimal glands reduces basal tearing. Consistent with this hypothesis, increasing the surface temperature of the cornea in human volunteers, a maneuver that is expected to silence corneal cold thermoreceptors, also decreased basal tearing. These findings have important implications for the pathophysiology and potential treatment of dry eye disease and other pathologies of mucosal surfaces such as burning mouth syndrome and vaginal dryness.
Mice lacking the Trpv3 gene have skin alterations and curly whiskers (156). In keratinocytes, the channel forms a signaling complex with TGF-α/EGFR. Activation of EGFR leads to increased TRPV3 channel activity and Ca2+ influx, which in turn stimulates TGF-α release, an important effector in hair morphogenesis and epidermal development (156). A recent study by Yoshioka et al. suggests an important role of TRPV3 in the development of allergic and pruritic dermatitis (41). Mice with a gain-of-function mutation (G573S) of TRPV3 (157) expressed higher TRPV3 levels in epidermal keratinocytes, were hairless, and developed typical signs of dermatitis, including erythema, edema, dry skin, skin erosions, and excoriations (41). These mice also exhibited significantly increased scratching behavior compared with wildtype, suggesting the spontaneous development of pruritus. These findings make TRPV3 channels promising candidates for the development of new therapies for the treatment of itch (158).
Concluding Remarks
Trigeminal chemosensitivity is an important aspect of human sensory physiology, relevant to food enjoyment, nutrition, peripheral mechanisms of itch and pain, thermoregulation, skin physiology, environmental toxicology, respiratory diseases, and eye pathologies. Work over the past decade has expanded tremendously our understanding of the cellular and molecular aspects of trigeminal chemosensation. The identification of specific ion channels, especially TRP channels, involved in different pathophysiological mechanisms including pain, pruritus, headache, chronic cough, and asthma, opens new opportunities for their treatment. Unfortunately, the number of specific antagonists targeting TRP channels is still very limited and their characterization using in vivo models of disease is very fragmentary. However, the investment in drug discovery programs by major pharmaceutical companies in this area is high. There is significant hope that the pace of progress in this field will speed up, and significant developments are expected in the upcoming years, hopefully resulting in novel therapeutics for some of these diseases.
Acknowledgments
The author is grateful to current and former members of the Sensory Transduction and Nociception Group for original recordings and discussions. Stuart Ingham is credited for help with Figure 1.
The author acknowledges funding from the Spanish MICINN Projects BFU2007-61855, CONSOLIDER-INGENIO 2010 CSD2007-0002, and Generalitat Valenciana project PROMETEO/2010/046.
References
- Fain G. L. (2003) Sensory Transduction, Sinauer Associates Inc., Sunderland, MA. [Google Scholar]
- Bryant B. P. and Silver W. L. (2000) in The Neurobiology of Taste and Smell (Finger T. E. and Silver W. L., Eds.), pp 73−100, Wiley-Liss, New York. [Google Scholar]
- Green B. G. (1996) Trends Food Sci. Technol. 7, 415–420. [Google Scholar]
- Parker G. H. (1912) J. Acad. Nat. Sci. 15, 221–234. [Google Scholar]
- Keele C. A. (1962) Arch. Int. Pharmacodyn. Ther. 139, 547–557. [PubMed] [Google Scholar]
- Jones M. H. (1954) Am. J. Psychol. 67, 696–699. [PubMed] [Google Scholar]
- Wood J. N.; Docherty R. (1997) Annu. Rev. Physiol. 59, 457–482. [DOI] [PubMed] [Google Scholar]
- Lee Y.; Lee C. H.; Oh U. (2005) Mol. Cells 20, 315–324. [PubMed] [Google Scholar]
- Belmonte C.; Viana F. (2008) Mol. Pain 4, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Erickson R. P.; Simon S. A. (1993) J. Gen. Physiol. 101, 843–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patapoutian A.; Tate S.; Woolf C. J. (2009) Nat. Rev. Drug Discovery 8, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B.; Voets T. (2004) Novartis Found. Symp. 258, 140–149. [PubMed] [Google Scholar]
- Caterina M. J.; Schumacher M. A.; Tominaga M.; Rosen T. A.; Levine J. D.; Julius D. (1997) Nature 389, 816–824. [DOI] [PubMed] [Google Scholar]
- Bautista D. M.; Sigal Y. M.; Milstein A. D.; Garrison J. L.; Zorn J. A.; Tsuruda P. R.; Nicoll R. A.; Julius D. (2008) Nat. Neurosci. 11, 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. C.; Akopian A. N.; Sivilotti L.; Colquhoun D.; Burnstock G.; Wood J. N. (1995) Nature 377, 428–431. [DOI] [PubMed] [Google Scholar]
- McKemy D. D.; Neuhausser W. M.; Julius D. (2002) Nature 416, 52–58. [DOI] [PubMed] [Google Scholar]
- Calixto J. B.; Kassuya C. A.; Andre E.; Ferreira J. (2005) Pharmacol. Ther. 106, 179–208. [DOI] [PubMed] [Google Scholar]
- Gerhold K. A.; Bautista D. M. (2009) Ann. N.Y. Acad. Sci. 1170, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriens J.; Nilius B.; Vennekens R. (2008) Curr. Neuropharmacol. 6, 79–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker D. (1963) J. Gen. Physiol 46, 453–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolcsanyi J.; Jancso-Gabor A. (1975) Arzneim. Forsch. 25, 1877–1881. [PubMed] [Google Scholar]
- Urban L.; Campbell E. A.; Panesar M.; Patel S.; Chaudhry N.; Kane S.; Buchheit K.; Sandells B.; James I. F. (2000) Pain 89, 65–74. [DOI] [PubMed] [Google Scholar]
- Movahed P.; Evilevitch V.; Andersson T. L.; Jonsson B. A.; Wollmer P.; Zygmunt P. M.; Hogestatt E. D. (2005) Br. J. Pharmacol. 146, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursu D.; Knopp K.; Beattie R. E.; Liu B.; Sher E. (2010) Eur. J. Pharmacol. 641, 114–122. [DOI] [PubMed] [Google Scholar]
- Green B. G. (2004) J. Neurobiol. 61, 13–29. [DOI] [PubMed] [Google Scholar]
- Green B. G. (1985) Physiol. Behav. 35, 427–434. [DOI] [PubMed] [Google Scholar]
- Schafer K.; Braun H. A.; Isenberg C. (1986) J. Gen. Physiol. 88, 757–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkia A.; Madrid R.; Meseguer V.; de la P. E.; Valero M.; Belmonte C.; Viana F. (2007) J. Physiol. 581, 155–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T.; Droogmans G.; Wissenbach U.; Janssens A.; Flockerzi V.; Nilius B. (2004) Nature 430, 748–754. [DOI] [PubMed] [Google Scholar]
- Patapoutian A.; Peier A. M.; Story G. M.; Viswanath V. (2003) Nat. Rev. Neurosci. 4, 529–539. [DOI] [PubMed] [Google Scholar]
- Dussor G.; Koerber H. R.; Oaklander A. L.; Rice F. L.; Molliver D. C. (2009) Brain Res. Rev. 60, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Caterina M. J. (2005) Pflugers Arch. 451, 160–167. [DOI] [PubMed] [Google Scholar]
- Koizumi S.; Fujishita K.; Inoue K.; Shigemoto-Mogami Y.; Tsuda M.; Inoue K. (2004) Biochem. J. 380, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandadi S.; Sokabe T.; Shibasaki K.; Katanosaka K.; Mizuno A.; Moqrich A.; Patapoutian A.; Fukumi-Tominaga T.; Mizumura K.; Tominaga M. (2009) Pflugers Arch. 458, 1093–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn P. M.; Zhong Y.; Burnstock G. (2001) Prog. Neurobiol. 65, 107–134. [DOI] [PubMed] [Google Scholar]
- Burnstock G. (2007) Physiol Rev. 87, 659–797. [DOI] [PubMed] [Google Scholar]
- Ikoma A.; Steinhoff M.; Stander S.; Yosipovitch G.; Schmelz M. (2006) Nat. Rev. Neurosci. 7, 535–547. [DOI] [PubMed] [Google Scholar]
- Sun Y. G.; Zhao Z. Q.; Meng X. L.; Yin J.; Liu X. Y.; Chen Z. F. (2009) Science 325, 1531–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross S. E.; Mardinly A. R.; McCord A. E.; Zurawski J.; Cohen S.; Jung C.; Hu L.; Mok S. I.; Shah A.; Savner E. M.; Tolias C.; Corfas R.; Chen S.; Inquimbert P.; Xu Y.; McInnes R. R.; Rice F. L.; Corfas G.; Ma Q.; Woolf C. J.; Greenberg M. E. (2010) Neuron 65, 886–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerstrom M. C.; Rogoz K.; Abrahamsen B.; Persson E.; Reinius B.; Nordenankar K.; Olund C.; Smith C.; Mendez J. A.; Chen Z. F.; Wood J. N.; Wallen-Mackenzie A.; Kullander K. (2010) Neuron 68, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka T.; Imura K.; Asakawa M.; Suzuki M.; Oshima I.; Hirasawa T.; Sakata T.; Horikawa T.; Arimura A. (2009) J. Invest. Dermatol. 129, 714–722. [DOI] [PubMed] [Google Scholar]
- Reeh P. W.; Steen K. H. (1996) Prog. Brain Res. 113, 143–151. [DOI] [PubMed] [Google Scholar]
- Belmonte C.; Gallar J.; Pozo M. A.; Rebollo I. (1991) J. Physiol. 437, 709–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant B. P.; Moore P. A. (1995) Am. J. Physiol. 268, R58–R65. [DOI] [PubMed] [Google Scholar]
- Krishtal O. A.; Pidoplichko V. I. (1981) Neuroscience 6, 2599–2601. [DOI] [PubMed] [Google Scholar]
- Liu L.; Simon S. A. (2000) Physiol. Behav. 69, 363–378. [DOI] [PubMed] [Google Scholar]
- Baumann T. K.; Chaudhary P.; Martenson M. E. (2004) Eur. J. Neurosci. 19, 1343–1351. [DOI] [PubMed] [Google Scholar]
- Talley E. M.; Sirois J. E.; Lei Q.; Bayliss D. A. (2003) Neuroscientist 9, 46–56. [DOI] [PubMed] [Google Scholar]
- Chen X.; Gallar J.; Pozo M. A.; Baeza M.; Belmonte C. (1995) Eur. J. Neurosci. 7, 1154–1163. [DOI] [PubMed] [Google Scholar]
- Cain W. S.; Murphy C. L. (1980) Nature 284, 255–257. [DOI] [PubMed] [Google Scholar]
- Chen X.; Belmonte C.; Rang H. P. (1997) Pain 70, 23–29. [DOI] [PubMed] [Google Scholar]
- Dessirier J. M.; Simons C. T.; Carstens M. I.; O’Mahony M.; Carstens E. (2000) Chem. Senses 25, 277–284. [DOI] [PubMed] [Google Scholar]
- Komai M.; Bryant B. P. (1993) Brain Res. 612, 122–129. [DOI] [PubMed] [Google Scholar]
- Wang Y. Y.; Chang R. B.; Liman E. R. (2010) J. Neurosci. 30, 12958–12963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green B. G. (1992) Chem. Senses 17, 435–450. [Google Scholar]
- Karashima Y.; Talavera K.; Everaerts W.; Janssens A.; Kwan K. Y.; Vennekens R.; Nilius B.; Voets T. (2009) Proc. Natl. Acad. Sci. U. S. A 106, 1273–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell C. (2005) Sci. Signaling 2005, re3. [Google Scholar]
- Clapham D. E.; Julius D.; Montell C.; Schultz G. (2005) Pharmacol. Rev. 57, 427–450. [DOI] [PubMed] [Google Scholar]
- Macpherson L. J., Hwang S. W., Miyamoto T., Dubin A. E., Patapoutian A., and Story G. M. (2006) Mol. Cell Neurosci. 32, 335−343. [DOI] [PubMed] [Google Scholar]
- Eid S. R.; Cortright D. N. (2009) Handb. Exp Pharmacol. 261–281. [DOI] [PubMed] [Google Scholar]
- Clapham D. E. (2003) Nature 426, 517–524. [DOI] [PubMed] [Google Scholar]
- Tominaga M.; Caterina M. J.; Malmberg A. B.; Rosen T. A.; Gilbert H.; Skinner K.; Raumann B. E.; Basbaum A. I.; Julius D. (1998) Neuron 21, 531–543. [DOI] [PubMed] [Google Scholar]
- Malkia A., Morenilla-Palao C., and Viana F. (2010) Curr. Pharm. Biotechnol. Nov 8 [Epub ahead of print] PMID: 20932258. [DOI] [PubMed] [Google Scholar]
- Bessac B. F.; Sivula M.; von Hehn C. A.; Escalera J.; Cohn L.; Jordt S. E. (2008) J. Clin. Invest. 118, 1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingle S. C.; Matta J. A.; Ahern G. P. (2007) Handb. Exp. Pharmacol. 155–171. [DOI] [PubMed] [Google Scholar]
- Vriens J.; Appendino G.; Nilius B. (2009) Mol. Pharmacol. 75, 1262–1279. [DOI] [PubMed] [Google Scholar]
- Jordt S. E.; Julius D. (2002) Cell 108, 421–430. [DOI] [PubMed] [Google Scholar]
- Szolcsanyi J. (2004) Neuropeptides 38, 377–384. [DOI] [PubMed] [Google Scholar]
- Szolcsanyi J. (1977) J. Physiol. (Paris) 73, 251–259. [PubMed] [Google Scholar]
- Rentmeister-Bryant H.; Green B. G. (1997) Chem. Senses 22, 257–266. [DOI] [PubMed] [Google Scholar]
- Tominaga M. and Tominaga T. (2005) Pflugers Arch. 451, 143−150. [DOI] [PubMed] [Google Scholar]
- Szallasi A.; Blumberg P. M. (1989) Neuroscience 30, 515–520. [DOI] [PubMed] [Google Scholar]
- Bohlen C. J.; Priel A.; Zhou S.; King D.; Siemens J.; Julius D. (2010) Cell 141, 834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt S. E.; Tominaga M.; Julius D. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 8134–8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung M. K.; Guler A. D.; Caterina M. J. (2008) Nat. Neurosci. 11, 555–564. [DOI] [PubMed] [Google Scholar]
- Kennedy W. R.; Vanhove G. F.; Lu S. P.; Tobias J.; Bley K. R.; Walk D.; Wendelschafer-Crabb G.; Simone D. A.; Selim M. M. (2010) J. Pain 11, 579–587. [DOI] [PubMed] [Google Scholar]
- Backonja M.; Wallace M. S.; Blonsky E. R.; Cutler B. J.; Malan P. Jr.; Rauck R.; Tobias J. (2008) Lancet Neurol. 7, 1106–1112. [DOI] [PubMed] [Google Scholar]
- Noto C.; Pappagallo M.; Szallasi A. (2009) Curr. Opin. Invest. Drugs 10, 702–710. [PubMed] [Google Scholar]
- Binshtok A. M.; Bean B. P.; Woolf C. J. (2007) Nature 449, 607–610. [DOI] [PubMed] [Google Scholar]
- Kim H. Y.; Kim K.; Li H. Y.; Chung G.; Park C. K.; Kim J. S.; Jung S. J.; Lee M. K.; Ahn D. K.; Hwang S. J.; Kang Y.; Binshtok A. M.; Bean B. P.; Woolf C. J.; Oh S. B. (2010) Pain 150, 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina M. J.; Rosen T. A.; Tominaga M.; Brake A. J.; Julius D. (1999) Nature 398, 436–441. [DOI] [PubMed] [Google Scholar]
- Muraki K.; Iwata Y.; Katanosaka Y.; Ito T.; Ohya S.; Shigekawa M.; Imaizumi Y. (2003) Circ. Res. 93, 829–838. [DOI] [PubMed] [Google Scholar]
- Bisogno T.; Hanus L.; De Petrocellis L.; Tchilibon S.; Ponde D. E.; Brandi I.; Moriello A. S.; Davis J. B.; Mechoulam R.; Di M., V (2001) Br. J. Pharmacol. 134, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N.; Neeper M. P.; Liu Y.; Hutchinson T. L.; Lubin M. L.; Flores C. M. (2008) J. Neurosci. 28, 6231–6238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moqrich A.; Hwang S. W.; Earley T. J.; Petrus M. J.; Murray A. N.; Spencer K. S.; Andahazy M.; Story G. M.; Patapoutian A. (2005) Science 307, 1468–1472. [DOI] [PubMed] [Google Scholar]
- Stotz S. C.; Vriens J.; Martyn D.; Clardy J.; Clapham D. E. (2008) PLoS One 3, e2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Blair N. T.; Clapham D. E. (2005) J. Neurosci. 25, 8924–8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt-Eisele A. K.; Weber K.; Sherkheli M. A.; Vielhaber G.; Panten J.; Gisselmann G.; Hatt H. (2007) Br. J. Pharmacol. 151, 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussaieff A.; Rimmerman N.; Bregman T.; Straiker A.; Felder C. C.; Shoham S.; Kashman Y.; Huang S. M.; Lee H.; Shohami E.; Mackie K.; Caterina M. J.; Walker J. M.; Fride E.; Mechoulam R. (2008) FASEB J. 22, 3024–3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green B. G. (1990) J. Invest Dermatol. 94, 662–666. [DOI] [PubMed] [Google Scholar]
- Viana F.; Ferrer-Montiel A. (2009) Expert. Opin. Ther. Pat 19, 1787–1799. [DOI] [PubMed] [Google Scholar]
- Garcia-Anoveros J.; Nagata K. (2007) Handb. Exp. Pharmacol. 347–362. [DOI] [PubMed] [Google Scholar]
- Kang K.; Pulver S. R.; Panzano V. C.; Chang E. C.; Griffith L. C.; Theobald D. L.; Garrity P. A. (2010) Nature 464, 597–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterlin Z.; Chesler A.; Firestein S. (2007) Neuron 53, 635–638. [DOI] [PubMed] [Google Scholar]
- Bessac B. F.; Jordt S. E. (2008) Physiology (Bethesda) 23, 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandell M.; Story G. M.; Hwang S. W.; Viswanath V.; Eid S. R.; Petrus M. J.; Earley T. J.; Patapoutian A. (2004) Neuron 41, 849–857. [DOI] [PubMed] [Google Scholar]
- Jordt S. E.; Bautista D. M.; Chuang H. H.; McKemy D. D.; Zygmunt P. M.; Hogestatt E. D.; Meng I. D.; Julius D. (2004) Nature 427, 260–265. [DOI] [PubMed] [Google Scholar]
- Macpherson L. J.; Geierstanger B. H.; Viswanath V.; Bandell M.; Eid S. R.; Hwang S.; Patapoutian A. (2005) Curr. Biol. 15, 929–934. [DOI] [PubMed] [Google Scholar]
- Bautista D. M.; Movahed P.; Hinman A.; Axelsson H. E.; Sterner O.; Hogestatt E. D.; Julius D.; Jordt S. E.; Zygmunt P. M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 12248–12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Delling M.; Jun J. C.; Clapham D. E. (2006) Nat. Neurosci. 9, 628–635. [DOI] [PubMed] [Google Scholar]
- McNamara C. R.; Mandel-Brehm J.; Bautista D. M.; Siemens J.; Deranian K. L.; Zhao M.; Hayward N. J.; Chong J. A.; Julius D.; Moran M. M.; Fanger C. M. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 13525–13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalera J.; von Hehn C. A.; Bessac B. F.; Sivula M.; Jordt S. E. (2008) J. Biol. Chem. 283, 24136–24144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista D. M.; Jordt S. E.; Nikai T.; Tsuruda P. R.; Read A. J.; Poblete J.; Yamoah E. N.; Basbaum A. I.; Julius D. (2006) Cell 124, 1269–1282. [DOI] [PubMed] [Google Scholar]
- Bessac B. F.; Sivula M.; von Hehn C. A.; Caceres A. I.; Escalera J.; Jordt S. E. (2009) FASEB J. 23, 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson D. A.; Gentry C.; Moss S.; Bevan S. (2008) J. Neurosci. 28, 2485–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards P.; Johnson E. C.; Silver W. (2010) Chemosens. Perception 3, 190–199. [Google Scholar]
- Ohta T.; Imagawa T.; Ito S. (2009) Mol. Pharmacol. 75, 307–317. [DOI] [PubMed] [Google Scholar]
- Kwan K. Y.; Allchorne A. J.; Vollrath M. A.; Christensen A. P.; Zhang D. S.; Woolf C. J.; Corey D. P. (2006) Neuron 50, 277–289. [DOI] [PubMed] [Google Scholar]
- Hinman A.; Chuang H. H.; Bautista D. M.; Julius D. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 19564–19568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson L. J.; Dubin A. E.; Evans M. J.; Marr F.; Schultz P. G.; Cravatt B. F.; Patapoutian A. (2007) Nature 445, 541–545. [DOI] [PubMed] [Google Scholar]
- Nilius B., Prenen J., and Owsianik G. (2010) J. Physiol. Nov 15 [Epub ahead of print]. [Google Scholar]
- Chen J.; Zhang X. F.; Kort M. E.; Huth J. R.; Sun C.; Miesbauer L. J.; Cassar S. C.; Neelands T.; Scott V. E.; Moreland R. B.; Reilly R. M.; Hajduk P. J.; Kym P. R.; Hutchins C. W.; Faltynek C. R. (2008) J. Neurosci. 28, 5063–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo O.; Meseguer V.; Belmonte C.; Viana F. (2008) Channels (Austin) 2, 429–438. [DOI] [PubMed] [Google Scholar]
- Talavera K.; Gees M.; Karashima Y.; Meseguer V. M.; Vanoirbeek J. A.; Damann N.; Everaerts W.; Benoit M.; Janssens A.; Vennekens R.; Viana F.; Nemery B.; Nilius B.; Voets T. (2009) Nat. Neurosci. 12, 1293–1299. [DOI] [PubMed] [Google Scholar]
- Karashima Y.; Damann N.; Prenen J.; Talavera K.; Segal A.; Voets T.; Nilius B. (2007) J. Neurosci. 27, 9874–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsavaler L.; Shapero M. H.; Morkowski S.; Laus R. (2001) Cancer Res. 61, 3760–3769. [PubMed] [Google Scholar]
- Peier A. M.; Moqrich A.; Hergarden A. C.; Reeve A. J.; Andersson D. A.; Story G. M.; Earley T. J.; Dragoni I.; McIntyre P.; Bevan S.; Patapoutian A. (2002) Cell 108, 705–715. [DOI] [PubMed] [Google Scholar]
- Dhaka A.; Earley T. J.; Watson J.; Patapoutian A. (2008) J. Neurosci. 28, 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y.; Daniels R. L.; Knowlton W.; Teng J.; Liman E. R.; McKemy D. D. (2007) J. Neurosci. 27, 14147–14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra A.; Madrid R.; Echevarria D.; Del Olmo S.; Morenilla-Palao C.; Acosta M. C.; Gallar J.; Dhaka A.; Viana F.; Belmonte C. (2010) Nat. Med. 16, 1396–1399. [DOI] [PubMed] [Google Scholar]
- Reid G. (2005) Pflugers Arch. 451, 250–263. [DOI] [PubMed] [Google Scholar]
- Daniels R. L.; McKemy D. D. (2007) Mol. Pain 3, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrendt H. J.; Germann T.; Gillen C.; Hatt H.; Jostock R. (2004) Br. J. Pharmacol. 141, 737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodding M.; Wissenbach U.; Flockerzi V. (2007) Cell Calcium 42, 618–628. [DOI] [PubMed] [Google Scholar]
- Beck B.; Bidaux G.; Bavencoffe A.; Lemonnier L.; Thebault S.; Shuba Y.; Barrit G.; Skryma R.; Prevarskaya N. (2007) Cell Calcium 41, 285–294. [DOI] [PubMed] [Google Scholar]
- Albin K. C.; Simons C. T. (2010) PLoS One 5, e9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant B. P.; Mezine I. (1999) Brain Res. 842, 452–460. [DOI] [PubMed] [Google Scholar]
- Lennertz R. C.; Tsunozaki M.; Bautista D. M.; Stucky C. L. (2010) J. Neurosci. 30, 4353–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riera C. E.; Menozzi-Smarrito C.; Affolter M.; Michlig S.; Munari C.; Robert F.; Vogel H.; Simon S. A.; le Coutre J. (2009) Br. J. Pharmacol. 157, 1398–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong W.; Burnstock G.; Spyer K. M. (2000) J. Physiol. 524 Pt 3, 891–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold M. S. and Gebhart G. F. (2010) Nat. Med. 16, 1248−1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum A. I.; Bautista D. M.; Scherrer G.; Julius D. (2009) Cell 139, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planells-Cases R.; Garcia-Sanz N.; Morenilla-Palao C.; Ferrer-Montiel A. (2005) Pflugers Arch. 451, 151–159. [DOI] [PubMed] [Google Scholar]
- Di M., V; Blumberg P. M.; Szallasi A. (2002) Curr. Opin. Neurobiol. 12, 372–379. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Huang J.; McNaughton P. A. (2005) EMBO J. 24, 4211–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt P. M.; Petersson J.; Andersson D. A.; Chuang H.; Sorgard M.; Di M., V; Julius D.; Hogestatt E. D. (1999) Nature 400, 452–457. [DOI] [PubMed] [Google Scholar]
- Bhave G.; Gereau R. W. (2004) J. Neurobiol. 61, 88–106. [DOI] [PubMed] [Google Scholar]
- Szallasi A.; Cortright D. N.; Blum C. A.; Eid S. R. (2007) Nat. Rev. Drug Discovery 6, 357–372. [DOI] [PubMed] [Google Scholar]
- Caterina M. J.; Leffler A.; Malmberg A. B.; Martin W. J.; Trafton J.; Petersen-Zeitz K. R.; Koltzenburg M.; Basbaum A. I.; Julius D. (2000) Science 288, 306–313. [DOI] [PubMed] [Google Scholar]
- Fajardo O.; Meseguer V.; Belmonte C.; Viana F. (2008) J. Neurosci. 28, 7863–7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassenstein C.; Kwong K.; Taylor-Clark T.; Kollarik M.; Macglashan D. M.; Braun A.; Undem B. J. (2008) J. Physiol. 586, 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colsoul B., Nilius B., and Vennekens R. (2009) Clin. Exp. Allergy 39, 1456−1466. [DOI] [PubMed] [Google Scholar]
- Taylor-Clark T. E.; Nassenstein C.; McAlexander M. A.; Undem B. J. (2009) Pulm. Pharmacol. Ther. 22, 71–74. [DOI] [PubMed] [Google Scholar]
- Geppetti P.; Patacchini R.; Nassini R.; Materazzi S. (2010) Lung 188(Suppl 1), S63–S68. [DOI] [PubMed] [Google Scholar]
- Andre E.; Campi B.; Materazzi S.; Trevisani M.; Amadesi S.; Massi D.; Creminon C.; Vaksman N.; Nassini R.; Civelli M.; Baraldi P. G.; Poole D. P.; Bunnett N. W.; Geppetti P.; Patacchini R. (2008) J. Clin. Invest. 118, 2574–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andre E.; Gatti R.; Trevisani M.; Preti D.; Baraldi P. G.; Patacchini R.; Geppetti P. (2009) Br. J. Pharmacol. 158, 1621–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell M. A.; Belvisi M. G.; Grace M.; Sadofsky L.; Faruqi S.; Hele D. J.; Maher S. A.; Freund-Michel V.; Morice A. H. (2009) Am. J. Respir. Crit. Care Med. 180, 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisani M.; Siemens J.; Materazzi S.; Bautista D. M.; Nassini R.; Campi B.; Imamachi N.; Andre E.; Patacchini R.; Cottrell G. S.; Gatti R.; Basbaum A. I.; Bunnett N. W.; Julius D.; Geppetti P. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 13519–13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor-Clark T. E.; McAlexander M. A.; Nassenstein C.; Sheardown S. A.; Wilson S.; Thornton J.; Carr M. J.; Undem B. J. (2008) J. Physiol. 586, 3447–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Orengo L.; Dhaka A.; Heuermann R. J.; Young T. J.; Montana M. C.; Cavanaugh E. J.; Kim D.; Story G. M. (2008) Mol. Pain 4, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor-Clark T. E.; Undem B. J.; MacGlashan D. W. Jr.; Ghatta S.; Carr M. J.; McAlexander M. A. (2008) Mol. Pharmacol. 73, 274–281. [DOI] [PubMed] [Google Scholar]
- Streng T.; Axelsson H. E.; Hedlund P.; Andersson D. A.; Jordt S. E.; Bevan S.; Andersson K. E.; Hogestatt E. D.; Zygmunt P. M. (2008) Eur. Urol. 53, 391–399. [DOI] [PubMed] [Google Scholar]
- Taylor-Clark T. E.; Ghatta S.; Bettner W.; Undem B. J. (2009) Mol. Pharmacol. 75, 820–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres A. I.; Brackmann M.; Elia M. D.; Bessac B. F.; del Camino D.; D’Amours M.; Witek J. S.; Fanger C. M.; Chong J. A.; Hayward N. J.; Homer R. J.; Cohn L.; Huang X.; Moran M. M.; Jordt S. E. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 9099–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkler P. E., Ballard C. J., Oxford G. S., and Hurley J. H. (2010) Pain Nov 12 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Jin J.; Hu L.; Shen D.; Dong X. P.; Samie M. A.; Knoff J.; Eisinger B.; Liu M. L.; Huang S. M.; Caterina M. J.; Dempsey P.; Michael L. E.; Dlugosz A. A.; Andrews N. C.; Clapham D. E.; Xu H. (2010) Cell 141, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakawa M.; Yoshioka T.; Matsutani T.; Hikita I.; Suzuki M.; Oshima I.; Tsukahara K.; Arimura A.; Horikawa T.; Hirasawa T.; Sakata T. (2006) J. Invest. Dermatol. 126, 2664–2672. [DOI] [PubMed] [Google Scholar]
- Steinhoff M.; Biro T. (2009) J. Invest. Dermatol. 129, 531–535. [DOI] [PubMed] [Google Scholar]
- Madrid R.; Donovan-Rodriguez T.; Meseguer V.; Acosta M. C.; Belmonte C.; Viana F. (2006) J. Neurosci. 26, 12512–12525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meseguer V.; Karashima Y.; Talavera K.; D’Hoedt D.; Donovan-Rodriguez T.; Viana F.; Nilius B.; Voets T. (2008) J. Neurosci. 28, 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara F. N.; Randall A.; Gunthorpe M. J. (2005) Br. J. Pharmacol. 144, 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okumura Y.; Narukawa M.; Iwasaki Y.; Ishikawa A.; Matsuda H.; Yoshikawa M.; Watanabe T. (2010) Biosci. Biotechnol. Biochem. 74, 1068–1072. [DOI] [PubMed] [Google Scholar]
- Salazar H.; Llorente I.; Jara-Oseguera A.; Garcia-Villegas R.; Munari M.; Gordon S. E.; Islas L. D.; Rosenbaum T. (2008) Nat. Neurosci. 11, 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi K.; Iwasaki Y.; Narukawa M.; Iitsuka Y.; Fukao T.; Seki T.; Ariga T.; Watanabe T. (2009) Biochem. Biophys. Res. Commun. 382, 545–548. [DOI] [PubMed] [Google Scholar]
- Narukawa M.; Koizumi K.; Iwasaki Y.; Kubota K.; Watanabe T. (2010) Biosci. Biotechnol. Biochem. 74, 1694–1696. [DOI] [PubMed] [Google Scholar]
- Dedov V. N.; Tran V. H.; Duke C. C.; Connor M.; Christie M. J.; Mandadi S.; Roufogalis B. D. (2002) Br. J. Pharmacol. 137, 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]