

Abstract
The kainic acid (KA) receptors belong to the class of glutamate (Glu) receptors in the brain and constitute a promising target for the treatment of neurological and/or psychiatric diseases such as schizophrenia, major depression, and epilepsy. Five KA subtypes have been identified and named GluK1−5. In this article, we present the discovery of (2S,3R)-3-(3-carboxyphenyl)-pyrrolidine-2-carboxylic acid (1) based on a rational design process. Target compound 1 was synthesized by a stereoselective strategy in 10 steps from commercially available starting materials. Binding affinities of 1 at native ionotropic Glu receptors were determined to be in the micromolar range (AMPA, 51 μM; KA, 22 μM; NMDA 6 μM), with the highest affinity for cloned homomeric KA receptor subtypes GluK1,3 (3.0 and 8.1 μM, respectively). Functional characterization of 1 by two electrode voltage clamp (TEVC) electrophysiology at a nondesensitizing mutant of GluK1 showed full competitive antagonistic behavior with a Kb of 11.4 μM.
Keywords: Glutamate receptors, kainic acid receptors, antagonist, rational design
Introduction
In the mammalian central nervous system (CNS), (S)-glutamate (Glu) functions as the major excitatory neurotransmitter. Once released from the presynaptic neuron into the glutamatergic synapse, Glu activates a number of pre- and postsynaptic glutamate receptors. On the basis of pharmacological profile and ligand selectivity studies, the Glu receptors have been grouped into two main classes: the fast-acting ionotropic receptors (iGluRs) (1,2) and the G-protein coupled metabotropic receptors (mGluRs), which produce a much slower signal transduction through second messenger systems (3). Within the class of iGluRs, three subgroups have been established on the basis of ligand affinity studies: the AMPA (subunits GluA1−4), kainic acid (KA) (subunits GluK1−5), and NMDA receptors (subunits GluN1, GluN2A-D, and GluN3A,B). Likewise for the mGluRs, three subgroups have been defined and named group I (subunits mGluR1,5), group II (subunits mGluR2,3) and group III (subunits mGluR4,6−8). The uptake of Glu is carried out by the action of the excitatory amino acid transporters (EAATs) and plays an important role in the termination of the neuronal signaling process and in keeping the concentration of Glu below neurotoxic levels.
In order to understand the physiological role of the different Glu receptor subunits, the development of subtype selective ligands is a key strategy. Such pharmacological tools, agonists, antagonists, and positive/negative modulators may be used for the functional studies of isolated receptors, neuronal tissue, and in vivo. With a focus on the KA receptors, they have been suggested to be involved in modulating synaptic transmission and plasticity (4,5), particularly in the hippocampal region (6,7). Thus KA receptors may be promising targets in the treatment of schizophrenia, major depression, epilepsy, and neurodegeneration (8). However, despite an extensive effort only truly subtype selective antagonists for the GluK1 subtype have been reported (9). In this article, we present the discovery of 1 as the lead structure for a new class of selective iGluR antagonists, which due to its vast substitution options holds potential for future development into new subtype selective KA receptor antagonists.
Results and Discussion
From a drug design point of view, it is of the essence to distinguish between competitive and noncompetitive antagonists. While the chemical scaffold and binding site of noncompetitive antagonists may vary extensively, the chemical diversity of competitive antagonists is constrained by the nature of the orthosteric binding pocket. An analysis of reported competitive AMPA/KA receptor antagonists which comprise an α-amino acid moiety (Table 1) shows that such ligands are characterized by a longer distance between the α-carbon and the distal acidic functional group, as compared to reported agonists (10). This common structural feature prevents the bringing together of domain 1 (D1) and domain 2 (D2) of the ligand binding domain (LBD) (Figure 1), a biostructural reorganization which is essential for AMPA/KA receptor activation (calcium channel opening) (11−13).
Table 1. Selected Examples of Competitive iGluR Antagonists Which Comprise an α-Amino Acid Moiety and the Distance from the α-Carbon to the Carbon of the Distal Carboxylic Acid in [Å].

| dist [Å] | subtype, PDB code | ligand class | |
|---|---|---|---|
| Glu | 3.15 | GluK1, 1TFX | agonist |
| KA | 3.35 | GluK2, 1TT1 | agonist |
| ATPO | 5.31 | GluK1, 1VSO | antagonist |
| UBP310 | 7.55 | GluK1, 2F34 | antagonist |
| LY466195 | 7.31 | GluK1, 2QS4 | antagonist |
| 1a | 5.53 |
Low energy conformation located by performing a stochastic conformational search (for details, see Methods).
Figure 1.
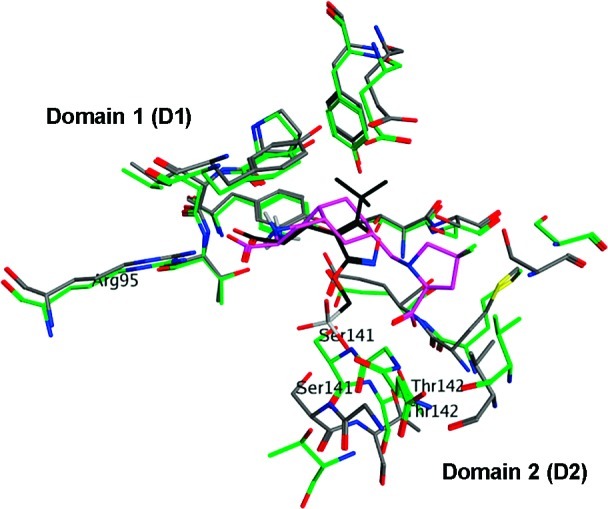
Superimposition of X-ray structures of the LBD of GluK1 cocrystallized with ATPO (black; 1VSO in type code) and with LY466195 (purple, 2QS4 in green) with an indication of the two binding domains D1 and D2. The two antagonists occupy distinct spatial positions with respect to their distal functional groups. Furthermore, the two residues important for the binding of the distal acid group, Ser141 and Thr142, are being forced out by ATPO by 2.85 Å and 1.55 Å, respectively.
Depending on the chemical nature of the antagonist, the KA receptor may be captured in diverse open states. For example, a superimposition study of X-ray structures of ATPO and LY466195 in GluK1 shows distinct spatial organization of the backbone and orientation of side chains of the D2 pocket (Figure 1). However, it remains to be understood if the distinct binding affinities of ATPO and LY466195 is a result thereof or simply due to disfavored ligand−receptor interactions.
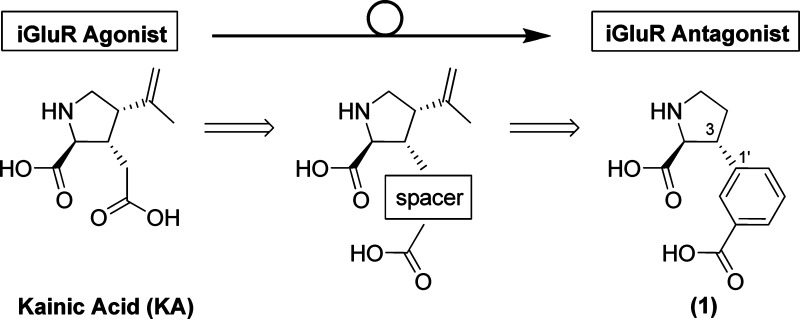
In our search for new lead structures for the development of KA antagonists which display a subtype selectivity profile, we looked toward the nonselective KA receptor agonist KA (Figure 2). We believed this compound served as an attractive starting point for the rational design of novel KA antagonists as it holds several possibilities for the introduction of substituents. By incorporation of a chemical spacer at the 3-position of the proline ring, the distance between the α-amino acid moiety and the distal carboxylic acid could be extended as desired. Several chemical spacers would do this; however, spatial orientation of the distal carboxylic acid group in the molecule’s low-energy conformation, the possibility for future incorporation of substituents, and synthetic tractability pointed toward a meta-substituted phenyl ring as in 1 (Figure 2) as a first attempt. In this compound, the distance between the α-amino acid moiety and the distal carboxylic acid was calculated to be 5.53 Å.
Figure 2.

Chemical structures of iGluR agonist kainic acid (KA) and KA with the incorporation of a chemical spacer (KA-CS) leading to the rational design of potential iGluR antagonist 1.
A superimposition study of the low-energy conformation of 1 with the binding conformations of the KA receptor antagonists ATPO, UBP310, and LY466195 obtained from their respective KA receptor X-ray crystal structure was carried out (Figure 3). It shows that the distal carboxylic acid of 1 finds itself in between the distal functional groups of ATPO and UBP310/LY466195, being able to make essential interactions with Ser141 and Thr142 (GluK1 numbering). From the in silico study, it is also evident that 1 provides several options for the introduction of substituents into unexplored areas of the receptor which may be advantageous in subsequent work to improve potency and/or the subtype selectivity profile.
Figure 3.
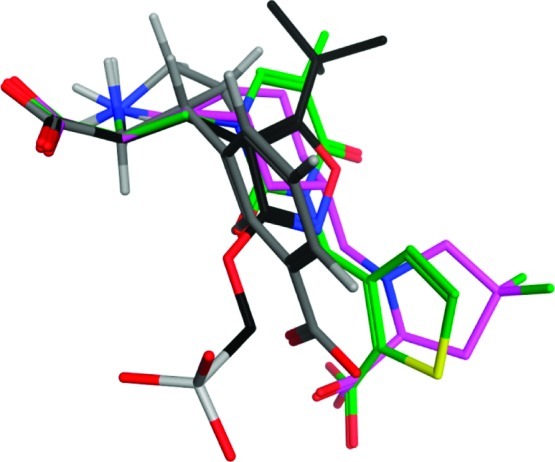
Superimposition of the low-energy conformation of 1 (type code) with the binding conformations of ATPO (black), UBP310 (green), and LY466195 (purple) obtained from X-ray crystal structures with PDB codes 1VSO, 2QS4, and 2F34, respectively.
A retro synthetic analysis of 1 suggests (S)-pyroglutaminol (2) (Scheme 1) as the source of chirality for the α-amino acid. This stereogenic center may furthermore be used to direct the asymmetric introduction of the phenyl ring in the 3-position. It is well established that cuprate addition to enone 4 proceeds selectively with 2,3-anti stereo selectivity and that high yields may be obtained by the use of a method previously developed in our laboratories.
Scheme 1. Synthetic Pathway toward 1.

Reagents and conditions: (a) TBSCl, Et3N, CH2Cl2; (b) BOC2O, Et3N, DMAP, CH2Cl2 (87% two steps); (c) LHMDS, THF, −78 °C, then PhSeCl; (d) H2O2, EtOAc, 0 °C to RT (65% two steps); (e) 5, t-BuLi, CuCN, TMSCl, THF, −50 °C, then 4 (90%); (f) BH3·THF, reflux, then NaOH, H2O2, 0 °C to rt (71%); (g) TBAF, THF, rt (94%); (h) RuCl3, NaIO4, H2O, MeCN, EtOAc (75%); (i) HCl/dioxane, rt, then recrystallization from AcOH (44%); (j) TBSCl, imidazole, DMF (96%).
The synthesis commenced with O-TBS and N-BOC protection of 2 under standard conditions followed by the introduction of the double bond by α-selenation and oxidative elimination to give 4(14). The cyano-Gilman cuprate of O-TBS-protected m-bromobenzylalcohol 5 was generated according to previously described methods (15,16) and reacted with 4 to give the conjugated addition product 6, in a stereoselective fashion as expected. Compound 6 was reduced using borane under reflux to give 7. Removal of the TBS protecting groups by treatment with tetrabutylammonium fluoride (TBAF) gave free diol 8. This deprotection step was performed after the reduction of the endocarbonyl group in order to prevent undesired migration of the BOC group (17). Diol 8 was oxidized using Ru(III) in catalytic amount and periodate as the co-oxidant to give 9, according to the modified Sharpless procedure (18). Finally, N-Boc deprotection of 9 using HCl in dioxane, followed by recrystallization from acetic acid gave the target compound 1, as the HCl salt.
Pharmacological characterization of 1 commenced with binding studies at native AMPA, KA, and NMDA receptors from rat synaptosomes (Table 2). At AMPA and KA (predominantly the high KA affinity subtypes GluK4,5) receptors, 1 displayed medium-range micromolar affinity (51 and 22 μM, respectively), whereas it showed a 3−9-fold higher affinity for the NMDA receptors (6.0 μM). To fully investigate its pharmacological profile at the KA receptors, 1 underwent binding affinity studies at cloned homomeric KA subtypes, GluK1−3 (Table 2). Here, 1 showed a clear selectivity profile for GluK1 and GluK3 over GluK2 (3 and 8 vs >100 μM, respectively). In the same assay, the binding affinity of 1 was furthermore determined for AMPA receptor subtype GluA2, and the affinity was in agreement with the data obtained for native AMPA receptors (67 vs 51 μM, respectively).
Table 2. Binding Affinities of 1 at Native AMPA, KA, and NMDA Receptors (Rat Synaptosomes) and at Cloned Homomeric Rat GluA2 and GluK1-3 Receptorsa.
| AMPA | KA | NMDA | GluA2 | GluK1 | GluK2 | GluK3 | |
|---|---|---|---|---|---|---|---|
| IC50 | IC50 | Ki | Ki | Ki | Ki | Ki | |
| 1 | 51 ± 10b | 22 ± 2b | 6.0 ± 0.5b | 67 ± 16c | 4.3 ± 0.4c | >100c | 8.1 ± 0.6c |
| ATPO | 16f | >100f | >100f | 60 ± 5c | 2.6 ± 0.4c | >100c | >1000 |
| UBP310d | -- | -- | -- | >100 | 0.010e | >100 | 0.023 |
| LY466195e | -- | -- | -- | 270 | 0.05 | -- | 8.9 |
All values are in μM.
Mean values ± SEM of three individual experiments. Radioligands used: AMPA, [3H]AMPA; KA, [3H]KA; NMDA, [3H]CGP 39653.
Mean values ± SEM of at least three experiments, conducted in triplicate at 12−16 drug concentrations. Radioligands used: GluA2, [3H]AMPA; GluK1−3, [3H](2S,4R)-4-methyl Glu (SYM2081). Hill coefficients were not different from unity.
Kb values for the inhibition of 100 μM glutamate-induced Ca2+ influx in HEK 293 cells expressing human homomeric AMPA or KA receptor subtypes (from refs (19) and (20)).
IC50 value for the inhibition of glutamate-evoked currents in HEK 293 cells expressing homomeric KA receptors.
From ref (21).
From ref (22). --: no data available.
To determine if 1 was capable of fully antagonizing KA receptor subtypes GluK1 and GluK3, two electrode voltage clamp (TEVC) electrophysiology was employed using homomeric nondesensitizing mutants of GluK1−3 (23). In this assay, 1 was shown to be a competitive full antagonist (efficacy = 0) at GluK1 and GluK3 with Kb values of 11.4 and 275 μM, respectively (Table 3 and Figure 4). The Kb value of 1 at GluK1 was within the expected range based on the binding affinity data, whereas Kb for GluK2 was too high to determine, which was anticipated from the radioligand binding data. The unexpected lower potency (34-fold) of 1 at the nondesensitizing GluK3 mutant compared to the binding affinity at wild-type GluK3 is not readily explained from a molecular or structural perspective. A reasonable explanation could be that the nondesensitizing GluK3 mutant is not well representative of the wild-type receptor, because the disulfide linkage which prevents conformational change of the receptor into the desensitized state could also interfere with D1−D2 domain movements associated with ligand binding. As such, 1 may not have the same LBD contacts in the mutant receptor as it has in the wild-type receptor, leading to a possible loss of binding affinity. Notably, the competitive antagonist UBP310 also exhibited a 17-fold higher Kb value at the nondesensitizing GluK3 receptor (Table 3) compared to the Kb value at wild-type GluK3 (Table 2). Finally, the wild-type GluK3 subtype has been shown to exhibit atypical function as compared to that of wild-type GluK1,2 (24).
Table 3. Functional Characterization of 1 Using TEVC Electrophysiology at Non-Desensitizing Mutant Homomeric GluK1-3a.
| GluK1 Kb | GluK2 Kb | GluK3 Kb | |
|---|---|---|---|
| 1b | 11.4 ± 2.8 (5) | >300 (5) | 275 ± 12 (6) |
| (S)-ATPOc | 24 | >300 | -- |
| UBP310b | 0.019 ± 0.003 (6) | 207 ± 34 (5) | 0.394 ± 0.035 (9) |
| LY466195d | 0.024 | -- | -- |
All values are in μM.
Shown are the mean ± SEM values from concentration−response curves conducted in duplicate at 7−9 antagonist concentrations. The number of experiments is indicated in parentheses. No agonistic activity was detected at 1 mM compound.
From ref (22). --: no data available.
Figure 4.

Inhibition of Glu responses by 1 at the nondesensitizing mutants of GluK1 and GluK3, expressed in X. laevis oocytes and measured by TEVC electrophysiology. Shown are the pooled data normalized to the control response in the absence of antagonist from 5 to 6 experiments conducted in duplicate. GluK1, Glu = 100 μM, IC50 = 18.6 μM; GluK3, Glu = 5 mM, IC50 = 323 μM. Inset: traces from one oocyte expressing the GluK3 mutant. Stimulations in duplicate at increasing concentrations of 1 (in μM): 0, 5, 10, 20, 50, 100, 200, 300, 1000; (Vh = −90 mV). Scale bars: 50 nA and 1 min.
On the comparison of 1 with KA antagonists ATPO, UBP310, and LY466195, it is clear that the pharmacological profile of 1 resembles mostly the profile observed for ATPO (Table 3). This finding is not surprising given the information obtained from the initial molecular modeling study. While UBP310 and LY466195 occupy very similar GluK1 LBD spaces and are both highly potent selective GluK1 antagonists, ATPO deviates in spatial orientation, domain closure, and side chain/backbone organization as well as the pharmacological profile (Figure 1). With 1 estimated to exhibit medicinal chemistry properties in between ATPO and UBP310/LY466195, it is not unexpected that 1 could turn out to be ATPO-like rather than UBP310/LY466195-like in its pharmacological profile.
Conclusions
We have discovered a new class of iGluR antagonists by the rational design and synthesis of 1. The compound was synthesized by a stereoselective approach in 10 steps from commercially available starting materials with the overall yield of 11%. The binding affinity profile reveals that 1 is fully selective for iGluRs, with no affinity for the mGluRs (>1000 μM) (26). In more detail, 1 displayed a 3−9-fold higher affinity for native NMDA receptors over that of AMPA and KA receptors. Furthermore, affinity studies at cloned homomeric AMPA and KA subtypes revealed that 1 has low micromolar affinity for GluK1 with a >20 fold selectivity over that of GluK2,3. Functional characterization of 1 in a TEVC electrophysiological assay using nondesensitizing homomeric mutants of GluK1−3 showed that 1 is a competitive full antagonist at GluK1 and GluK3. In all, 1 serves as a promising lead structure for the discovery of iGluR antagonists with novel subtype selectivity profiles. This is due to its broad but selective binding affinity profile for the iGluRs and its chemical scaffold, which allows for the introduction of substituents in diverse positions.
Methods
Chemistry
All reactions involving air- and moisture-sensitive reagents were performed in flame-dried glassware and under a nitrogen atmosphere using syringe−septum cap techniques. All reagents and solvents were purchased from Sigma-Aldrich and used without further purification unless otherwise specified. THF was distilled from Na/benzophenone under a nitrogen atmosphere prior to use. Et2O was dried with and stored over Na-thread. DMF, CH2Cl2, and dioxane were dried by storing over molecular sieves (4 Å). Analytical thin-layer chromatography (TLC) was carried out using Merck Silica gel 60 F254 aluminum sheets. Compounds were detected as single spots on TLC plates and visualized using UV light (254 or 366 nm) and KMnO4 or ninhydrin. Merck silica gel (35−70 mesh) was used for flash chromatography. Melting points were recorded on an SRS Optimelt apparatus and are uncorrected. 1H NMR spectra were recorded on a 300 MHz Varian Mercury 300BB with a 5 mm 1H probe. 13C NMR spectra were recorded on a 75 MHz Varian Gemini 2000BB spectrometer with a 5 mm 31P, 13C, 1H, and 19F probe. NMR samples were prepared as CDCl3 solutions using solvent peaks as a reference unless otherwise noted. The purity of tested compounds was determined by elementary analysis to be >95%.
(5S)-N-tert-Butoxycarbonyl-5-(tert-butyldimethylsilyloxymethyl)pyrrolidin-2-one (3) (14)
DMAP and Et3N were used instead of imidazole. CH2Cl2 was used as the solvent instead of DMF. Yield 87%; 1H NMR δ 4.17 (m, 1H), 3.92 (dd, 1H, J = 10 and 4 Hz), 3.69 (dd, 1H, J = 10 and 2 Hz), 2.72 (dt, J = 17 and 10 Hz), 2.38 (qd, J = 17, 9, and 2 Hz), 2.08 (m, 2H), 1.55 (s, 9H), 0.89 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
(5S)-N-tert-Butoxycarbonyl-5-(tert-butyldimethylsilyloxymethyl)-3-pyrrolidene-2-one (4) (14)
EtOAc was used as the solvent instead of CH2Cl2. Yield 89%; 1H NMR δ 7.21 (dd, 1H, J = 6 and 2 Hz), 6.07 (dd, 1H, J = 6 and 2 Hz), 4.56 (m, 1H), 4.11 (dd, 1H, J = 10 and 4 Hz), 3.67 (q, 1H, J = 10 and 7 Hz), 1.52 (s, 9H), 0.83 (s, 9H), 0.01 (s, 3H), 0.00 (s, 3H).
3-Bromo-[(tert-butyl)dimethylsilyloxymethyl]benzene (5)
Prepared according to the literature procedure in ref (27).
(4R,5S)-N-(tert-Butoxycarbonyl)-5-(((tert-butyl)dimethylsilyloxy)methyl)-4-(3-(((tert-butyl)-dimethylsilyloxy)methyl)phenyl)-pyrrolidin-2-one (6)
To a solution of 5 (1.86 g, 6.50 mmol) in dry THF (22 mL) at −78 °C was added tert-BuLi (7.65 mL, 1.7 M in pentane, 13.0 mmol). After stirring for 15 min, a suspension of CuCN (295 mg, 3.25 mmol) in dry THF (2 mL) was added, and the reaction mixture allowed to warm up to −50 °C by removing dry ice and adding acetone (clear solution). After 5 min, the flask was recooled to −78 °C, and 4 (847 mg, 2.6 mmol) dissolved in dry THF (2.0 mL) was added followed by TMSCl (0.85 mL, 6.50 mmol). The reaction mixture was allowed to warm up to −50 °C and stirred at this temperature for 1 h. The reaction was quenched with sat. NH4Cl and extracted with EtOAc. The organic layer was washed with brine, dried (MgSO4), and concentrated. Purification of the crude product by flash chromatography (heptane, 1.5% EtOAc, Rf = 0.24) gave 7 as a white solid (1,24 g, 90%): mp = 68−69 °C; [α]24589 = −42.85 (c = 0.49, CH2Cl2); 1H NMR δ 7.26 (m, 2H), 7.11 (br s, 1H), 7.05 (br d, 1H, J = 7 Hz), 4.71 (s, 2H), 4.06 (p, 1H, J = 4 and 2 Hz), 3.99 (dd, 1H, J = 10 and 4 Hz), 3.78 (dd, 1H, J = 10 and 2 Hz), 3.44 (dt, 1H, J = 10, 3, and 2 Hz), 3.13 (q, 1H, J = 18 and 10 Hz), 2.53 (dd, 1H, J = 18 and 3 Hz), 1.52 (s, 9H), 0.94 (s, 9H), 0.91 (s, 9H), 0.10 (s, 6H), 0.08 (s, 3H), 0.07 (s, 3H); 13C NMR δ 174.28, 150.00, 144.34, 142.44, 129.14, 125.02, 124.24, 83.16, 66.90, 64.99, 63.82, 40.16, 38.90, 28.25, 26.13, 26.03, −5.05.
(2S,3R)-N-(tert-Butoxycarbonyl)-2-(((tert-butyl)dimethylsilyloxy)methyl)-3-(3—(((tert-butyl)dimethylsilyloxy)methyl)-phenyl)-pyrrolidine (7)
To a solution of 6 (500 mg, 0.938 mmol) in dry THF (4 mL) was added a solution of borane (5.63 mL, 5.63 mmol, 1 M in THF), and the solution was stirred under reflux for 20 h. The flask was then cooled to 0 °C, and 11.5 mL of THF was added. H2O (0.5 mL) was then added carefully, followed by NaOH (7.60 mL, 2 N) and H2O2 (2.35 mL, 35 w/w%). The reaction mixture was then stirred at RT for 1 h and quenched with sat. NaHCO3. The aqueous phase was extracted with EtOAc, and the collective organic layers were washed with brine, dried (MgSO4), and concentrated. Purification of the crude product by flash chromatography (heptane, 5% EtOAc, Rf = 0.23) gave 7 as a colorless oil (345 mg, 71%). [α]24589 = +2.34 (c = 0.76, CH2Cl2); 1H NMR δ 7.22 (m, 1H), 7.13 (br d, 2H, J = 7 Hz), 7.03 (br t, 1H, J = 7 Hz), 4.67 (s, 2H), 4.00 − 3.45 (m, 5H), 3.29 (m, 1H), 2.19 (m, 1 Hz), 1.86 (m, 1H), 1.44 (s, 9H), 1.44 (s, 9H), 0.90 (s, 9H), 0.85 (s, 9H), 0.06 (s, 6H), 0.00 (s, 6H); 13C NMR δ 154.41, 144.01, 143.54, 141.83, 128.63, 126.04, 125.13, 124.41, 79.55, 79.18, 65.74, 65.60, 65.15, 47.22, 46.64, 32.89, 32.05, 28.75, 26.15, −5.04, −5.20.
(2S,3R)-N-(tert-Butoxycarbonyl)-2-(hydroxymethyl)-3-(3-(hydroxymethyl)phenyl)-pyrrolidine (8)
To a solution of 7 (795 mg, 1.53 mmol) in dry THF (13 mL) was added tetrabutylammonium fluoride (4.60 mL, 4.60 mmol, 1 M in THF). The reaction mixture was stirred for 1 h, then quenched with 1/2 sat. NaHCO3. The aqueous phase was extracted with EtOAc and the collective organic layers washed with brine, dried (MgSO4), and concentrated. Purification of the crude product by flash chromatography (heptane/EtOAc 2:3, Rf = 0.23) gave 8 as a colorless oil (417 mg, 94%). [α]24589 = −11.52 (c = 0.59, CH2Cl2); 1H NMR (d-DMSO) δ 7.25 (br t, 1H, J = 7 Hz), 7.14 (br d, 2H, J = 7 Hz), 7.04 (br d, 1H, J = 7 Hz), 5.16 (t, 1H, J = 5 Hz), 4.86 (br t, 1H, J = 5 Hz), 4.48 (br d, 2H, J = 6 Hz), 3.65 (br d, 1H, J = 21 Hz), 3.48 (m, 3H), 3.25 (m, 1H); 2.21 (m, 1H), 1.83 (m, 1H), 1.41 (s, 9H); 13C NMR (CDCl3) δ 156.89, 141.67, 141.04, 129.03, 128.32, 127.04, 126.26, 125.82, 80.70, 67.15, 65.99, 65.16, 47.26, 33.06, 28.60.
(2S,3R)-N-(tert-Butoxycarbonyl)-3-(3-(carboxyphenyl)-pyrrolidine-2-carboxylic acid (9)
To 8 (532 mg, 1.83 mmol) dissolved in MeCN (13.0 mL) and EtOAc (13.0 mL) was added a solution of RuCl3·H2O (7.7 mg, 0.034 mmol) and NaIO4 (3.22 g, 15.05 mmol) in H2O (23.0 mL). The reaction mixture was stirred for 1 h, then filtered on filter paper, and the filter cake washed with EtOAc. The aqueous phase was extracted with EtOAc and the collective organic layers washed with brine, dried (MgSO4), and concentrated. Purification of the crude product by flash chromatography (heptane/EtOAc 2:3, 2% AcOH, Rf = 0.32) gave 9 as a white solid (444 mg, 75%), mp = 106−109 °C [α]24589 = +53.03 (c = 0.69, MeOH); 1H NMR (CD3OD) δ 7.93 (m, 2H), 7.52 (m, 2H), 7.44 (m, 1H), 7.15 (m, 1H), 4.24 (dd, 1H, J = 22 and 6 Hz), 3.69 (m, 1H) 3.55 (m, 2H), 2.36 (m, 1H), 2.09 (m, 1H), 1.53 (s, 9H); 13C NMR (CD3OD, two conformers) δ 175.91, 175.67, 169.60, 156.07, 155.67, 143.11, 142.51, 132.94, 132.80, 132.44, 130.03, 129.69, 129.60, 129.46, 129.32, 81.99, 81.63, 67.36, 66.87, 51.13, 50.07, 47.42, 47.17, 33.96, 33.44, 28.79, 28.60.
(2S,3R)-3-(3-(Carboxyphenyl)-pyrrolidine-2-carboxylic acid (1)
To 9 (416 mg, 1.29 mmol) in 1,4-dioxane (2 mL) at 0 °C was added HCl(g)/1,4-dioxane (3.88 mL, 15.5 mmol, 4.0 M). The reaction mixture was stirred at RT for 1 h then concentrated. The solid was triturated with freezing cold diethyl ether to give the HCl salt of 1 as a white solid. Recrystallization from glacial acetic acid gave the HCl salt of 1 as white crystals (146 mg, 44%): TLC (MeOH/AcOH/EtOAc 1:1:3) Rf = 0.24; mp = 190−193 °C; [α]24589 = +63.71 (c = 0.62, MeOH); 1H NMR (CD3OD, TMS standard ref.) δ 8.05 (br t, 1H, J = 3 and 2 Hz), 7.96 (dt, 1H, J = 8 and 1 Hz), 7.63 (dt, 1H, J = 8 and 1 Hz), 7.49 (t, 1H, J = 15 and 8 Hz), 4.41 (d, 1H, J = 9 Hz), 3.67 (m, 2H), 3.47 (m, 1H), 2.57 (m, 1H), 2.25 (m, 1H); 13C NMR (CD3OD) δ 170.41, 169.19, 140.55, 133.20, 132.55, 130.10, 130.03, 129.77, 65.98, 49.59 (approximated from D2O spectrum.), 47.00, 34.78. Elem. anal. calcd. for C12H14ClNO4, C, 53.05; H, 5.19; N, 5.16; found, C, 52.68; H, 5.03; N, 4.97.
Molecular Modeling
The modeling study was performed using the software package MOE (Molecular Operating Environment, v2009.10, Chemical Computing Group, 2009) using the built-in mmff94x forcefield and the GB/SA continuum solvent model. The compound was submitted to a stochastic conformational search, and with respect to its global minimum (ΔG in kcal/mol), returned conformations above +7 kcal/mol were discarded. The γ-carboxylate group was protonated prior to execution of the conformational search, as this gave a larger and thus more reliable number of output conformations. The superimposition of ligands was carried out using the built-in function in MOE, by fitting the ammonium group and the two carboxylate groups.
Pharmacology
Native Receptor Binding Assays
Affinities for native AMPA, KA, and NMDA receptors in rat cortical synaptosomes were determined using 5 nM (RS)-[3H]AMPA (55.5 Ci/mmol) (28), 5 nM [3H]KA (58.0 Ci/mmol) (29), and 2 nM [3H]CGP 39653 (Kd = 6 nM, 50.0 Ci/mmol) (30), respectively, with minor modifications as previously described (31,32).
Recombinant Receptor Binding Assays
Sf9 cells were cultured and infected with recombinant baculovirus of cloned rat GluA2(R)o or GluK1−3 and membranes prepared and used for binding as previously detailed (33,34). The binding affinity of 1 was determined from competition experiments with 2−5 nM (RS)-[3H]AMPA (42.1 Ci/mmol; PerkinElmer, Waltham, MA) at GluA2(R)o or 1−5 nM [3H]SYM2081 (40 Ci/mmol; ARC, St. Louis, MO) at GluK1(Q)1b, GluK2(V,C,R)a, and GluK3a. The italic letters in parentheses indicate the RNA-edited isoforms of the subunits used. The IC50, Hill coefficient, and Ki values for 1 were evaluated as previously described (35).
TEVC Electrophysiology
X. laevis oocytes were collected, prepared, injected, and maintained as previously described (36). TEVC electrophysiology was also carried out as previously described (36) on nondesensitizing mutants of GluK1−3 (23). Antagonist concentration−response data were fit to the logistic equation: I = Imax/(1 + (10(log[B]−logIC50))nH), where I is the agonist-evoked current, [B] is the antagonist concentration, Imax is the control response in the absence of antagonist, IC50 is the concentration of the antagonist giving 50% inhibition, and nH is the Hill coefficient. (S)-Glutamate was used as the agonist (100 μM at GluK1 and 2; 5 mM at GluK3; EC50 = 83.5 μM, 108 μM, and 9.03 mM, respectively). Kb values were calculated from the IC50 value using the modified Cheng−Prusoff equation (37).
Abbreviations
CNS, central nervous system; D1, domain 1 of the LBD; D2, domain 2 of the LBD; KA, kainic acid; Glu, glutamate; LBD, ligand binding domain; SAR, structure−activity relationship; TBAF, tetrabutylammonium fluoride; TEVC, two electrode voltage clamp.
We thank Carlsberg Foundation, Lundbeck Foundation, and GluTarget for financial support.
References
- Kaczor A. A.; Matosiuk D. (2010) Molecular structure of ionotropic glutamate receptors. Curr. Med. Chem. 17, 2608–2635. [DOI] [PubMed] [Google Scholar]
- Chen P. E.; Wyllie D. J. A. (2006) Pharmacological insights obtained from structure-function studies of ionotropic glutamate receptors. Br. J. Pharmacol. 147, 839–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraguti F.; Shigemoto R. (2006) Metabotropic glutamate receptors. Cell Tissue Res. 326, 483–504. [DOI] [PubMed] [Google Scholar]
- Bartolotto; Clarke Z. A. (1999) Kainate receptors are involved in synaptic plasticity. Nature 402, 297. [DOI] [PubMed] [Google Scholar]
- Mellor J. R. (2006) Synaptic plasticity of kainate receptors. Biochem. Soc. Trans. 34, 949–951. [DOI] [PubMed] [Google Scholar]
- Bloss E. B.; Hunter R. G. (2010) Hippocampal kainate receptors. Vitam. Horm. 82, 167–184. [DOI] [PubMed] [Google Scholar]
- Nistico R.; Dargan S.; Fitzjohn S. M.; Lodge D.; Jane D. E.; Collingridge G. L.; Bortolotto Z. A. (2009) GLUK1 receptor antagonists and hippocampal mossy fiber function. Int. Rev. Neurobiol. 85, 13–27. [DOI] [PubMed] [Google Scholar]
- Lau A.; Tymianski M. (2010) Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 460, 525–542. [DOI] [PubMed] [Google Scholar]
- Jane D. E.; Lodge D.; Collingridge G. L. (2009) Kainate receptors: pharmacology, function and therapeutic potential. Neuropharmacology 56, 90–113. [DOI] [PubMed] [Google Scholar]
- Bunch L.; Krogsgaard-Larsen P. (2009) Subtype selective kainic acid receptor agonists: discovery and approaches to rational design. Med. Res. Rev. 29, 3–28. [DOI] [PubMed] [Google Scholar]
- Gill A.; Birdsey-Benson A.; Jones B. L.; Henderson L. P.; Madden D. R. (2008) Correlating AMPA receptor activation and cleft closure across subunits: crystal structures of the GluR4 ligand-binding domain in complex with full and partial agonists. Biochemistry 47, 13831–13841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong N.; Gouaux E. (2000) Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: Crystal structures of the GluR2 ligand binding core. Neuron 28, 165–181. [DOI] [PubMed] [Google Scholar]
- Mayer M. L.; Ghosal A.; Dolman N. P.; Jane D. E. (2006) Crystal structures of the kainate receptor GluR5 ligand binding core dimer with novel GluR5-selective antagonists. J. Neurosci. 26, 2852–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acevedo C. M.; Kogut E. F.; Lipton M. A. (2001) Synthesis and analysis of the sterically constrained l-glutamine analogues (3S,4R)-3,4-dimethyl-l-glutamine and (3S,4R)-3,4-dimethyl-l-pyroglutamic acid. Tetrahedron 57, 6353–6359.18159221 [Google Scholar]
- Bunch L.; Krogsgaard-Larsen P.; Madsen U. (2002) Mixed cyano-gilman cuprates: advances in conjugate addition to alpha,beta-unsaturated pyroglutaminol. Synthesis 31–33. [Google Scholar]
- Bunch L.; Nielsen B.; Jensen A. A.; Brauner-Osborne H. (2006) Rational design and enantioselective synthesis of (1R,4S,5R,6S)-3-azabicyclo[3.3.0]octane-4,6-dicarboxylic acid - A novel inhibitor at human glutamate transporter subtypes 1, 2, and 3. J. Med. Chem. 49, 172–178. [DOI] [PubMed] [Google Scholar]
- Bunch L.; Norrby P. O.; Frydenvang K.; Krogsgaard-Larsen P.; Madsen U. (2001) Unprecedented migration of N-alkoxycarbonyl groups in protected pyroglutaminol. Org. Lett. 3, 433–435. [DOI] [PubMed] [Google Scholar]
- Carlsen P. H. J.; Katsuki T.; Martin V. S.; Sharpless K. B. (1981) A greatly improved procedure for ruthenium tetraoxide catalyzed oxidations of organic-compounds. J. Org. Chem. 46, 3936–3938. [Google Scholar]
- Dolman N. P.; More J. C. A.; Alt A.; Knauss J. L.; Pentikäinen O. T.; Glasser C. R.; Bleakman D.; Mayer M. L.; Collingridge G. L.; Jane D. E. (2007) Synthesis and pharmacological characterization of N3-substituted willardiine derivatives: role of the substituent at the 5-position of the uracil ring in the development of highly potent and selective GLUK5 kainate receptor antagonists. J. Med. Chem. 50, 1558–1570. [DOI] [PubMed] [Google Scholar]
- Perrais D.; Pinheiro P. S.; Jane D. E.; Mulle C. (2009) Antagonism of recombinant and native GluK3-containing kainate receptors. Neuropharmacology 56, 131–140. [DOI] [PubMed] [Google Scholar]
- Moller E. H.; Egebjerg J.; Brehm L.; Stensbol T. B.; Johansen T. N.; Madsen U.; Krogsgaard-Larsen P. (1999) Resolution, absolute stereochemistry, and enantiopharmacology of the GluR1−4 and GluR5 antagonist 2-amino-3-[5-tert-butyl-3-(phosphonomethoxy)-4-isoxazolyl]propionic acid. Chirality 11, 752–759. [DOI] [PubMed] [Google Scholar]
- Weiss B.; Alt A.; Ogden A. M.; Gates M.; Dieckman D. K.; Clemens-Smith A.; Ho K. H.; Jarvie K.; Rizkalla G.; Wright R. A.; Calligaro D. O.; Schoepp D.; Mattiuz E. L.; Stratford R. E.; Johnson B.; Salhoff C.; Katofiasc M.; Phebus L. A.; Schenck K.; Cohen M.; Filla S. A.; Ornstein P. L.; Johnson K. W.; Bleakman D. (2006) Pharmacological characterization of the competitive GLU(K5) receptor antagonist decahydroisoquinoline LY466195 in vitro and in vivo. J. Pharmacol. Exp. Ther. 318, 772–781. [DOI] [PubMed] [Google Scholar]
- Weston M. C.; Schuck P.; Ghosal A.; Rosenmund C.; Mayer M. L. (2006) Conformational restriction blocks glutamate receptor desensitization. Nat. Struct. Mol. Biol 13, 1120–1127. [DOI] [PubMed] [Google Scholar]
- Perrais D.; Coussen F.; Mulle C. (2009) Atypical functional properties of GluK3-containing kainate receptors. J. Neurosci. 29, 15499–15510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl P.; Anker C.; Traynelis S. F.; Egebjerg J.; Rasmussen J. S.; Krogsgaard-Larsen P.; Madsen U. (1998) Antagonist properties of a phosphono isoxazole amino acid at glutamate R1−4 (R,S)-2-amino-3-(3-hydroxy-5-methyl-4-isoxazolyl)propionic acid receptor subtypes. Mol. Pharmacol. 53, 590–596. [DOI] [PubMed] [Google Scholar]
- Micheli F.; Di Fabio R.; Marchioro C. (1999) Asymmetric synthesis of some substituted-3-phenyl prolines. Farmaco 54, 461–464. [DOI] [PubMed] [Google Scholar]
- Doucet-Personeni C.; Bentley P. D.; Fletcher R. J.; Kinkaid A.; Kryger G.; Pirard B.; Taylor A.; Taylor R.; Taylor J.; Viner R.; Silman I.; Sussman J. L.; Greenblatt H. M.; Lewis T. (2001) A structure-based design approach to the development of novel, reversible AChE inhibitors. J. Med. Chem. 44, 3203–3215. [DOI] [PubMed] [Google Scholar]
- Honore T.; Nielsen M. (1985) Complex structure of quisqualate-sensitive glutamate receptors in rat cortex. Neurosci, Lett. 54, 27–32. [DOI] [PubMed] [Google Scholar]
- Braitman D. J.; Coyle J. T. (1987) Inhibition of [H-3] kainic acid receptor-binding by divalent-cations correlates with ion affinity for the calcium-channel. Neuropharmacology 26, 1247–1251. [DOI] [PubMed] [Google Scholar]
- Sills M. A.; Fagg G.; Pozza M.; Angst C.; Brundish D. E.; Hurt S. D.; Wilusz E. J.; Williams M. (1991) [H-3] Cgp-39653 - A new N-methyl-d-aspartate antagonist radioligand with low nanomolar affinity in rat-brain. Eur. J. Pharmacol. 192, 19–24. [DOI] [PubMed] [Google Scholar]
- Hermit M. B.; Greenwood J. R.; Nielsen B.; Bunch L.; Jorgensen C. G.; Vestergaard H. T.; Stensbol T. B.; Sanchez C.; Krogsgaard-Larsen P.; Madsen U.; Bräuner-Osborne H. (2004) Ibotenic acid and thioibotenic acid: a remarkable difference in activity at group III metabotropic glutamate receptors. Eur. J. Pharmacol. 486, 241–250. [DOI] [PubMed] [Google Scholar]
- Clausen R. P.; Hansen K. B.; Cali P.; Nielsen B.; Greenwood J. R.; Begtrup M.; Egebjerg J.; Brauner-Osborne H. (2004) The respective N-hydroxypyrazole analogues of the classical glutamate receptor ligands ibotenic acid and (RS)-2-amino-2-(3-hydroxy-5-methyl-4-isoxazolyl)acetic acid. Eur. J. Pharmacol. 499, 35–44. [DOI] [PubMed] [Google Scholar]
- Vogensen S. B.; Clausen R. P.; Greenwood J. R.; Johansen T. N.; Pickering D. S.; Nielsen B.; Ebert B.; Krogsgaard-Larsen P. (2005) Convergent synthesis and pharmacology of substituted tetrazolyl-2-amino-3-(3-hydroxy-5-methyl-4-isoxazolyl)propionic acid analogues. J. Med. Chem. 48, 3438–3442. [DOI] [PubMed] [Google Scholar]
- Sagot E.; Pickering D. S.; Pu X.; Umberti M.; Stensbøl T. B.; Nielsen B.; Chapelet M.; Bolte J.; Gefflaut T.; Bunch L. (2008) Chemo-enzymatic synthesis of a series of 2,4-syn-functionalized (S)-glutamate analogues: new insight into the structure-activity-relation of ionotropic glutamate receptor subtypes 5, 6 and 7. J. Med. Chem. 51, 4093–4103. [DOI] [PubMed] [Google Scholar]
- Nielsen B. S.; Banke T. G.; Schousboe A.; Pickering D. S. (1998) Pharmacological properties of homomeric and heteromeric GluR1(o) and GluR3(o) receptors. Eur. J. Pharmacol. 360, 227–238. [DOI] [PubMed] [Google Scholar]
- Greenwood J. R.; Mewett K. N.; Allan R. D.; Martin B. O.; Pickering D. S. (2006) 3-hydroxypyridazine 1-oxides as carboxylate bioisosteres: a new series of subtype-selective AMPA receptor agonists. Neuropharmacology 51, 52–59. [DOI] [PubMed] [Google Scholar]
- Leff P.; Dougall I. G. (1993) Further concerns over Cheng-Prusoff analysis. Trends Pharmacol. Sci. 14, 110–112. [DOI] [PubMed] [Google Scholar]