

Abstract
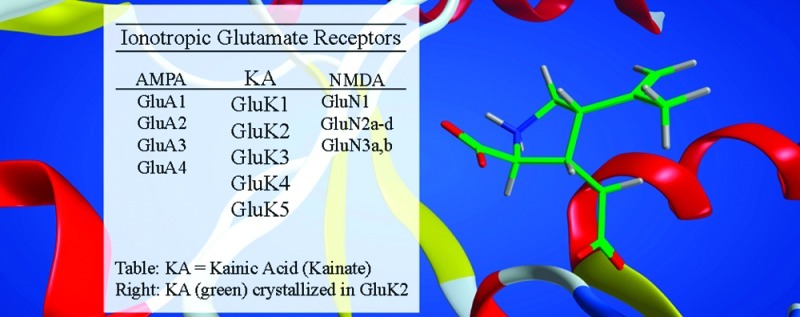
Kainic acid (KA) receptors belong to the group of ionotropic glutamate receptors and are expressed throughout in the central nervous system (CNS). The KA receptors have been shown to be involved in neurophysiological functions such as mossy fiber long-term potentiation (LTP) and synaptic plasticity and are thus potential therapeutic targets in CNS diseases such as schizophrenia, major depression, neuropathic pain and epilepsy. Extensive effort has been made to develop subtype-selective KA receptor antagonists in order to elucidate the physiological function of each of the five subunits known (GluK1−5). However, to date only selective antagonists for the GluK1 subunit have been discovered, which underlines the strong need for continued research in this area. The present review describes the structure−activity relationship and pharmacological profile for 10 chemically distinct classes of KA receptor antagonists comprising, in all, 45 compounds. To the medicinal chemist this information will serve as reference guidance as well as an inspiration for future effort in this field.
Keywords: Glutamate receptors, kainic acid receptors, competitive antagonists, medicinal chemistry, structure−activity relationship studies
The common α-amino acid (S)-glutamate (Glu) functions as the major endogenous excitatory neurotransmitter in the central nervous system (CNS). Upon its release into the synaptic cleft, it activates the Glu receptors. These receptors have been divided into two major classes: the ligand-gated ion channels named the ionotropic receptors (iGluRs) and the slower acting G-protein coupled receptors named the metabotropic receptors (mGluRs). On the basis of ligand affinity studies, the class of iGluRs has been divided into three subgroups: the N-methyl-d-aspartate (NMDA), (S)-2-amino-3-(3-hydroxy-5-methyl-4-isoxazolyl)propionic acid (AMPA) and kainate (KA) receptors (1−3). With focus on the KA receptors, it is believed that this subgroup of receptors are potential therapeutic targets in diseases such as schizophrenia, major depression, pain, and epilepsy. In order to elucidate the role of the KA receptors, medicinal chemists have attempted to develop subtype-selective KA receptor ligands for the last two to three decades. Although extensive effort has been made, only a limited number of subtype-selective KA receptor ligands have been reported. For a comprehensive review on KA receptor agonists, see our recent review (4). The aim of this review is to provide the medicinal chemists (and others) who have an interest in the structure−activity relationship (SAR) of competitive KAR antagonists with an overview of the field. This information may aid the design of new subunit-selective KA receptor antagonists.
KA Receptor Structure, Expression, and Function
The iGluRs are transmembrane-bound proteins comprising four regions: an intracellular carboxylate terminal domain (CTD), a transmembrane domain (TMD), a ligand-binding domain (LBD), and the amino-terminal domain (ATD) (Figure 1A) (5). To this date, five distinct KA subunits have been identified which are named GluK1, GluK2, GluK3 (formerly iGluR5−7), and GluK4 and GluK5 (formerly KA1 and KA2), in accordance with the new IUPHAR nomenclature (6−8). Functional KA receptors comprise four KA subunits which are assembled from two dimers of two homo- or two heteromeric subunits. In control of the assembly process, the ATD is believed to play a pivotal role (9). It has been shown that GluK1−3 may form functional homomeric10 or heteromeric11 receptors, whereas GluK4,5 only form functional receptors in combination with subunits GluK1, GluK2, or GluK3 (Figure 1B) (12). However, homomeric GluK413 and homomeric GluK5 receptors have been expressed in mammalian cells for use in binding studies. Although a higher degree of symmetry (2-fold) is achieved by opposite assembly (heterotetramer B), it remains to be elucidated if uniform assembly (heterotetramer A) is also feasible (Figure 1).
Figure 1.
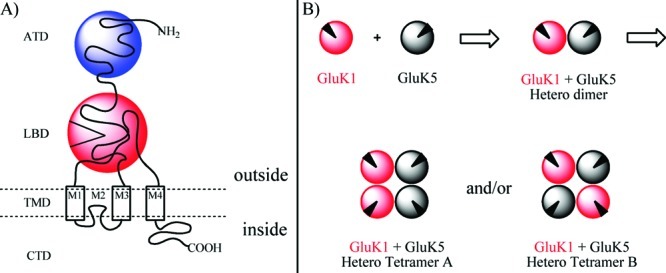
(A) Cartoon illustration of an iGluR showing the four regions: the carboxylate terminal domain (CTD), the transmembrane domain (TMD), the ligand binding domain (LBD), and the amino terminal domain (ATD). (B) Cartoon illustration of uniform assembly (hetero tetramer A) or opposite assembly (hetero tetramer B) of two heterodimers of GluK1 and GluK5. Black triangle illustrates the LBD.
KA receptors are highly expressed in the central nervous system (CNS), particularly in the hippocampal formation (mossy fiber (MF)) (15), and are implicated in several neurological functions (7,16−20), such as long-term potentiation (LTP) (21) and synaptic plasticity5.
Although most studies have focused on AMPA receptors (5), the biostructural mechanism underlying activation of the KA receptors has also been compared to the closure of a clam shell (Figure 2) (22): The ligand-binding domain (LBD) is in its apo state at which the KA agonist4 may approach. This progression facilitates closing of the D1- and D2-domains, rendering the agonist-LBD in an occluded state (agonist state). During this process, the ion channel at the center of the tetrameric receptor complex undergoes a conformational change which allows for calcium ions to flow. Rapidly thereafter the agonist-bound receptor enters a desensitized closed state in which the ion channel is again closed. It has been shown that some agonists promote desensitization at a speed so fast that an ion channel opening cannot be measured (current). This phenomenon has been named functional antagonism and is outside the scope of this review. For KA receptors, desensitization may be inhibited by addition of concanavalin A (ConA) (23) which is a lectin isolated from the jack-bean. Release of the agonist renders the receptor in the desensitized open state which is reorganized into the free apo state. It may be speculated whether the latter process exists in equilibrium (dotted arrow). In that case, it would be possible for a full functional antagonist to facilitate the desensitized closed state. In contrast, the mechanism underlying the functional properties of an antagonist is more straightforward. When in its apo form, the receptor recognizes the antagonist, and upon binding the closure of the clam shell (D1, D2) is blocked (antagonist state) and the ion channel is not opened.
Figure 2.

Schematic representation of the ligand-binding domain (LBD) of GluK1 as a clam shell: In its open free form (apo state), an agonist may bind, which facilitates closure of the shell and opening of the ion channel (agonist state). Depending on the nature of the agonist, the receptor may enter a desensitized closed state in which the ion channel is blocked. Subsequent release of the agonist renders the receptor available in the free apo state via an intermediate desensitized open state. An antagonist captures and stabilizes the apo state of the receptor (antagonist state), thus keeping the ion channel closed.
Therapeutic Potential
Despite evidence that AMPA/KA receptors play a pivotal role in respiratory (24) and cardiovascular (9,25) regulation, AMPA/KA receptor antagonists have been shown to be well-tolerated after systemic administration in animals (26), and in human studies (25,27−29). Although AMPA/KA receptor antagonists have been shown to cause cognitive deficits in rats, such side effects are believed to be temporary and thus acceptable in the case of prevention of permanent neurological damage after, for example, stroke (30).
GluK2-containing KA receptors have been suggested to facilitate propagation of seizures (31,32). In support of this hypothesis, it has been demonstrated that mice lacking the GluK2 gene (GRIK2 knock out) are much less sensitive to KA-induced seizures than wild type mice (33). GluK1 and/or GluK3 may also be involved in seizure activity, since antagonism (by compound 3.1, see Table 3) of receptors containing these subunits provides an anticonvulsant effect in the kindling model of epilepsy. Extracellular glutamate has been shown to increase after physical brain trauma (34,35), and an increased synaptic release of glutamate has been reported during periods of severe global and focal ischemia (36,37). Accordingly, KA receptor antagonists may prove neuroprotective by inhibiting Glu release. On the other hand, KA agonists may be beneficial in accordance with the hypoglutamatergic theory of schizophrenia which builds on the finding that the expression of GluK2 and GluK5 mRNAs is reduced in the hippocampus of schizophrenic patients (38). Finally, activation of AMPA, KA, and NMDA receptors appears to be involved in the development of chronic pain (39−42). As an example, GluK1-selective antagonists have been shown effective in the treatment of migraine (43).
Table 3. Chemical Structures and Binding Affinities (Ki) of Quinoxaline-2,3-dione Analogues 3.1−3.3 at Homomeric iGluR Subtypes (HEK293 Cells)a.

| cmpd | GluA2 | GluK1 | GluK2 | GluK3 |
|---|---|---|---|---|
| 3.1 (LU 136541)b | 0.06 | 1.1 | 1.2 | 0.26 |
| 3.2 (LU 115455)b | 0.018 | 1.3 | 0.37 | 0.21 |
| 3.3 (LU 97175)b | 1.3 | 0.088 | 0.31 | 0.022 |
All values in [μM]. Radioligands used: GluA2, [3H]AMPA; GluK1-3, [3H]KA.
From ref (59).
Quinoxaline-2,3-dione Derivatives
The quinoxaline-2,3-dione parental skeleton is a highly rigid heterocycle with an aromatic benzene ring fused with a nonaromatic heterocyclic moiety. Within the class, analogues 1.1 (DNQX) and 1.2 (CNQX) (Table 1) were the first competitive AMPA receptor antagonists to be discovered. The two compounds show micromolar affinity for AMPA/KA receptors but low receptor group and -subtype selectivity (Table 1). Furthermore, they show affinity for the glycine binding site at NMDA receptors, which may produce unwanted side effects (44,45). From X-ray structure studies in GluA2, it has been shown that it is the dione moiety of compound 1.1 (DNQX) that interacts with the receptor amino acid residues otherwise engaged in α-amino acid recognition and binding (46). The compounds primarily interact with domain D1, preventing agonists from forming initial contacts with the open cleft of the binding pocket (47).
Table 1. Chemical Structures and Pharmacological Profile of Quinoxaline-2,3-dione Analogues 1.1−1.3a.

| AMPA | KA | NMDA | GluK1 |
GluK2 |
GluK3 | GluK2/5 |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| cmpd | func.b | func.b | func.b | bind.c | func.d | bind.e | func.d | bind.c | bind.c | func.d |
| 1.1 (DNQX) | 0.25f | 0.53f | 4.1f | −h | −h | 0.35 | −h | −h | −h | −h |
| 1.2 (CNQX) | 0.40f | 0.27f | 13f | −h | 8.0 | 0.53 | 18 | −h | −h | 72 |
| 1.3 (NBQX) | 0.063g | 0.078g | >300g | 12 | 25 | 0.87 | 21 | 24 | 0.6 | 87 |
Data for KA receptor subunits were obtained in the presence of ConA. All values in [μM].
Ki values for inhibition of currents activated by kainate (100 μM), AMPA (30 μM), or NMDA (30 μM/glycine (3 μM)) in Xenopus oocytes injected with mRNA from rat cortex.
Ki values for displacement of [3H]KA radioligand at human KA receptors expressed in HEK293 cells; from ref (56).
IC50 values for inhibition of 100 μM glutamate-induced Ca2+ influx in HEK293 cells expressing human KA receptors; from ref (50).
Ki values for displacement of [3H]KA radioligand at homomeric receptors in BHK cells; from ref (51).
From ref (57).
From ref (58).
Not tested.
Expanding the quinoxaline-2,3-dione scaffold by a fused benzylsulfonamide ring gave rise to 1.3 (NBQX) which was disclosed as a potent, selective antagonist at AMPA/KA receptors (Table 1) (48). This compound showed a modest selectivity (3-fold) for AMPA receptors versus GluK1-containing KA receptors expressed in dorsal root ganglia (KB values: 0.3 and 0.9 μM, respectively) (49). However, when tested in HEK293 cells expressing GluK1 receptors, 1.3 displayed an IC50 value of 25 μM (50). In a binding study, 1.3 showed micromolar affinity for GluK2 (51), with only modest antagonistic activity at this subtype (IC50 = 21 μM) (50). In a functional study, little antagonistic effect was observed when 1.3 was applied at a concentration of up to 0.30 μM at rat GluK2 or GluK3 receptors (52). However, care should be taken when interpreting this latter finding, as the pharmacological profile of native human GluK3 is very different from that of the rat homologue used in this study (53). Although 1.3 has been shown to exhibit neuroprotective and analgesic effects in rats (45,54), the compound was discontinued as a drug candidate due to nephrotoxicity arising from low water-solubility (55).
By introduction of a substituent at N1 and introduction of a pyrrolyl group in the 7-position, a third generation of quinoxalinedione analogues 2.1−2.7 (Table 2) and 3.1−3.3 (Table 3) was discovered. The class showed selective affinity for AMPA/KA receptors (59). In 2.1−5, a phenylureamethyl group was added in the 3-position of the pyrrolyl moiety, affording high affinity for AMPA receptors but only moderate affinity for KA receptors. By furthermore introducing an ethylester group in the 3- or 4-position of the phenyl ring (2.2 and 2.3, respectively), both AMPA and KA receptor affinity was increased moderately (3−5-fold). Replacing this ester group with a halogen-containing group (2.4 and 2.5) further improved KA receptor affinity (5-fold) while maintaining the level of AMPA receptor affinity. A final attempt in this series was to omit substituents on the pyrrolyl moiety (2.6), which led to the loss of AMPA affinity, decrease in KA affinity, and increase in NMDA receptor affinity. Replacement of the trifluoromethyl group in the 6-positon of 2.6 with a nitro group (compound 2.7) amounted in loss of NMDA affinity and moderate increase of both AMPA and KA receptor affinity.
Table 2. Chemical Structures and Binding Affinities of Quinoxaline-2,3-dione Analogues 2.1−2.7 at Native iGluRs (Rat Synaptosomes)a.

Inspired by 2.1−2.5, compound 3.1 (LU 136541) was designed with a pyrrolyl group carrying a phenylureamethyl substituent in the 3-position. A hydroxyl group was furthermore introduced at N1 and a hexane ring was fused with the aromatic parental skeleton. The compound exhibited moderate to good affinity for subtypes GluA2, GluK1, GluK2, and GluK3 (Table 3). Affinity for GluK2 and GluK3 was improved by replacement of the N1-hydroxyl with a methylacetyl group, and by replacement of the distal acid with a nitro group to yield 3.2 (LU 115455). A noteworthy compound in this series, compound 3.3 (LU 97175), displayed 60-fold higher affinity for recombinant rat GluK3 receptors versus native rat AMPA receptors in radioligand binding assays and within the KA receptor subtypes a 4- and 14-fold higher affinity for GluK3 over GluK1 and GluK2, respectively (59). In in vivo rat models, 3.3 exhibited improved anticonvulsant activity compared to selective AMPA antagonists (63) without inducing motor impairment (59).
The pyrrolylquinoxaline-2,3-dione analogues 4.1−4.6 were characterized in binding assays at homomeric GluK1−3 and homomeric GluK5 receptors (14) (Table 4) (64). The N-benzyl-N-methylamine 4.1 (BSF-84077) was found to bind selectively to GluK1 (Table 4) with low affinity for AMPA receptors (>23 μM). In in vivo models, this analogue protected animals from NMDA-induced convulsions with a median effective dose (ED50) of 14 mg/kg. In 4.2, the N-benzyl-N-methylamine was substituted for a piperidine ring. This retained the same binding profile at KA receptors as compared to 4.1, but induced an increased affinity at the AMPA receptors (>10-fold). In analogue 4.3, a phenyl group was included at the 4-position of the piperidine ring which improved the GluK3 affinity by 10-fold. This phenyl substituent was linked by an ethylene in 4.4 and the piperidine ring replaced by a piperazine, improving GluK1 affinity 19-fold compared to 4.3. However, the optimal linker for high GluK1-affinity appeared to be a methylene group as in analogue 4.5 (BSF-91594), which showed an impressive >170-fold selectivity for human GluK1 (Ki value 3.8 nM) over GluK2, GluK3, and GluK5. Furthermore, 4.5 showed only moderate binding affinity at native AMPA receptors (Ki value 0.25 μM), and no binding to the glycine coagonist site of the NMDA receptors was observed. In an in vivo model, 4.5 also protected animals from NMDA-induced convulsions with a median effective dose (ED50) of 3.3 mg/kg (64). The basic nature of the distal amine was depleted by its inclusion in an aromatic ureyl moiety, compound 4.6. This analogue displayed decreased affinity for GluK1, but higher affinity for GluK2, GluK3 (64), and native AMPA (60) receptors.
Table 4. Chemical Structures and Binding Affinities (Ki) of Quinoxaline-2,3-dione Derivatives 4.1−4.6 at Native AMPA Receptorsa.

Isatinoxime Derivatives
Oxime 5.1 (NS102) has been reported to be a selective GluK2 antagonist with little effect on GluA2/GluA4 receptors (65). However, 5.1 has also been shown to block GluK1-containing KA receptors on DRG neurons with weaker effects at native AMPA receptors expressed in the cortex (Table 5) (49). In mice, 5.1 (10 mg/kg i.p.) (66) antagonized behavioral effects including seizures induced by systemic domoate administration and blocked domoate-induced neurodegeneration. However, 5.1 only offered partial protection of the negative behavioral responses to KA and could not block KA-induced neurodegeneration. The lack of discrimination between KA receptor subunits and low water solubility limits the usefulness of 5.1 (NS-102) as a pharmacological tool.
Table 5. Chemical Structures and Pharmacological Profile of Isatin Oxime Analogues 5.1−5.3a.

| GluA1 |
GluA2 | GluK1 |
GluK2 |
||||
|---|---|---|---|---|---|---|---|
| cmpd | bind. | func. | bind. | bind. | func. | bind. | func. |
| 5.1 (NS 102) | −b | −b | −b | −b | 6c | 1.4d | −b |
| 5.2 (NS 257)e | −b | 0.97 | −b | −b | −b | −b | 2.3 |
| 5.3 ((R/S)-NS 1209) | 0.12g | −b | 0.030g | 0.74g | 0.63f | 6.2g | 65f |
All values in [μM].
Not tested.
KB value for inhibition of Ca2+ influx in rat dorsal root ganglion (DRG) neurons; from ref (49),.
Displacement of [3H]KA radioligand; from ref (65).
IC50 values for inhibition of KA-induced depolarization in the appropriate mRNA-injected Xenopus oocytes; from ref (67).
IC50 values for inhibition of domoate-induced currents in HEK293 cells; from ref (68).
Displacement of [3H]AMPA radioligand; from ref (69).
Compound 5.2 (NS257) exhibited an IC50 value of 1.05 μM estimated from inhibition of KA-induced currents recorded from oocytes injected with total mouse brain mRNA. The compound was shown to have a slight preference (2-fold) for GluA2 antagonism over GluK2 antagonism (Table 5) (67).
Analogue 5.3 ((R/S)-NS1209) showed medium range nanomolar affinity for homomeric GluA1, GluA2, and GluK1 and low micromolar affinity for GluK2 (Table 5). In a functional study, 5.3 was 100-fold more potent at GluK1 versus GluK2 receptors (Table 5) (68,69). The (S)-stereoisomer stabilizes the GluA2 ligand-binding domain in a hyper-extended conformation compared to the apo state (69), but whether this flexibility is also true for KA receptors is unknown. Racemate 5.3 has shown promising results in animal models of status epilepticus (SE) (70), neuropathic pain, and allodynia in humans (71) and as a neuroprotectant against status-induced hippocampal neurodegeneration in a rodent (rat) animal model (70).
Decahydroisoquinoline Derivatives
Decahydroisoquinolines 6.1−6.6 constitute a class of AMPA/KA receptor antagonists which comprise an α-amino acid moiety as part of a rigid skeleton The analogues possess a flexible linker with a distal tetrazolyl group as a carboxylic acid bioisoster which forms hydrogen bonding interactions with the D2 domain of the KA receptor. On the chemical variation of this distal functionality, several interesting pharmacological results were observed. Analogue 6.1 (LY293558 or LY326325 for the HCL salt), which contains a tetrazolyl group as a bioisosteric substitution for a carboxylic acid, is a nonselective AMPA/KA receptor antagonist (72−74). The compound displayed only a 10-fold selectivity for GluK1 receptors versus NMDA receptors (17). Nonetheless 6.1 has been used in the identification and characterization of GluK1 receptors in neurons (75). Activity at neuronal AMPA receptors in acutely isolated cerebellar Purkinje neurons has been previously reported (75) for 6.1 giving an estimated IC50 of 0.5 μM (vs 100 μM KA). The compound was shown to be neuroprotective in a cat focal ischemia model and demonstrated anticonvulsant activity in mice (73) and analgesic activity in rats (54). Furthermore, it has recently been proven effective in preclinical models for acute migraine, which is believed to be due to its GluK1 antagonistic acitivty (76). In rat models, the monohydrate has proven to be antipsychotic with no extra-pyramidal side effects (77) and anxiolytic with no negative locomotor side effects (78). Finally, 6.1 reduced hyperalgesia in a human study on skin (27).
Reverting to a carboxylic acid functionality, compound 6.2 (LY382884) showed good affinity and antagonistic activity at human GluK1 receptors (Table 6) with no significant activity at AMPA or other KA receptor subunits at a concentration of 100 μM (79). However, the quantity of compound required to fully block KA receptors (10 μM) in vivo also weakly antagonized AMPA receptor-mediated synaptic transmission at CA3−CA1 synapses (80). Compound 6.2 displayed only weak activity at NMDA receptors (80), and a number of observations suggest that the compound is also effective as an antagonist of heteromeric compositions of GluK1 and GluK2 (17,50,79,80). 6.2 prevented induction and maintenance of seizures in multiple models of epilepsy (81), and it has been shown to have analgesic effects when tested in vivo in rats (54). This suggests that the GluK1 subunit has a central role in epilepsy and pain sensation. Furthermore, 6.2 produced anxiolytic type behavior in the Vogel conflict test (82).
Table 6. Chemical Structures and Pharmacological Profile of Decahydroisoquinolines 6.1−6.6a.

| GluA1 | GluA2 |
GluA3 | GluA4 | GluK1 |
GluK3 | |||
|---|---|---|---|---|---|---|---|---|
| cmpd | bind. | bind. | funct. | bind. | bind. | bind. | funct. | bind. |
| 6.1 (LY293558)c | 9.2 | 3.2 | 0.40b | 32 | 51 | 4.2 | 0.5b | >100 |
| 6.2 (LY382884)c | >100 | >100 | >100b | >100 | >100 | 4.0 | 1.1b | >100 |
| 6.3 (LY377770)d | >100 | 35 | −f | 48 | 32 | 3.1 | 0.09b | −f |
| 6.4e | 130 | 120 | −f | 250 | 260 | 0.16 | 1.2 | 50 |
| 6.5 (LY466195)c | 75 | 270 | >100b | 310 | 430 | 0.05 | 0.024 | 8.9 |
| 6.6 | −f | −f | >100b | −f | −f | −f | 0.003b | −f |
Binding affinities (Ki) were determined using [3H]AMPA radioligand at AMPA subtypes and [3H]KA radioligand at KA subtypes. No affinity or activity was observed below 100 μM at homomeric GluK2 and GluK5 receptors. All values in [μM].
KB values for inhibition of 100 μM glutamate-induced Ca2+ influx in HEK293 cells expressing human KA receptors. Data for KA receptor subunits were obtained in the presence of ConA; from ref (86), additional experimental data may be found in ref (50).
From ref (87): Binding data in Ki; functional data as KB inhibition of 100 μM Glu induced Ca2+ influx in HEK293 cells.
From ref (43): Binding data as Ki at homomeric receptors in HEK293 cells; functional data as IC50 inhibition of 200 μM Glu invoked Ca2+ influx in HEK293 cells.
Not tested.
Compound 6.3 (LY377770), the active stereoisomer of racemate LY294486, contains a one-atom longer ether linker at the distal region, as compared to 6.1. The compound was used as a pharmacological probe for GluK1-containing KA receptors in studies of synaptic transmission in the rat hippocampus and in studies of cerebral ischemia in the gerbil middle cerebral artery occlusion model (83,84). This suggested a preference for GluK1-containing KA receptors with an approximate 10-fold selectivity for GluK1 receptors over AMPA receptors. Consistent with this, 6.3 was found to be a potent and selective antagonist at homomeric GluK1 receptors (Table 6) with no activity at GluK2 homomeric receptors. Furthermore, NMDA receptor mediated currents were not affected (85). The imidazolyl analogue 6.4 inhibited glutamate-evoked calcium influx in the human GluK1-(Q) receptor transfected in HEK293 cell membranes, with an estimated KB value of 0.26 μM (Table 6) (43). In addition, 6.4 was evaluated at GluK1 receptors isolated from rat neonatal DRG neurons, giving an IC50 value of 0.85 μM (30 μM KA) (72).
Compound 6.5 (LY466195) exhibited a KB value of 0.024 μM for antagonism of human recombinant GluK1 and an estimated KB value of 13 nM for antagonism of GluK1-containing KA receptors expressed in dorsal root ganglion cells (86,87). The compound displayed only weak competitive antagonist activity at NMDA receptors (IC50 = 2.5 μM) and AMPA receptors (IC50 = 54 μM) expressed in hippocampal neurons. It is currently being evaluated as an antimigraine drug further highlighting the involvement of GluK1 in the pathophysiology of pain (88).
Compound 6.6 was reported as the most potent GluK1 antagonist of the decahydroisoquinoline series with a KB value of 3 nM for antagonism of human recombinant GluK1. It is clear that introduction of the bulky hydrophobic tricyclic moiety contributes significantly to this by reducing the water solubility properties of the ligand. Analogue 6.6 exhibited no activity at GluA2 or GluK2 receptors up to a concentration of 100 μM (86).
Based on 6.1 and 6.2 as lead structures, a small series of 6-tetrazolyl decahydroisoquinoline analogues were prepared (Table 7). These compounds were designed with an aromatic linker minimizing the number of possible conformations - i.e. potential loss of entropy upon binding. The aryl ether derivative 7.1 antagonized KA-evoked currents in rat DRG neurons with an IC50 value of 0.15 μM and AMPA- and NMDA-evoked currents in hippocampal neurons with IC50 values of 2.1 μM and 90 μM, respectively (86). In the subtype-specific HEK 293 cell assay, 7.1 was shown to be a potent GluK1 antagonist and a moderate GluA2 antagonist (Table 7). In compound 7.2 the aryl ether functionality was replaced by an aniline moiety. This analogue blocked KA-, AMPA-, and NMDA-evoked currents with IC50 values of 0.18, 1.4, and 1.9 μM, respectively (86). In HEK 293 cells, 7.2 was shown to be a slightly more potent (13-fold) GluK1 over GluA2 antagonist. In analogue 7.3, a chlorine substituent was added in the ortho position. This compound acted as a very potent GluK1 antagonist with a 400-fold selectivity over GluA2 (86).
Table 7. Chemical Structures and Pharmacological Profile of Decahydroisoquionoline Derivatives 7.1−7.3 at Recombinant AMPA/KA Receptorsa.
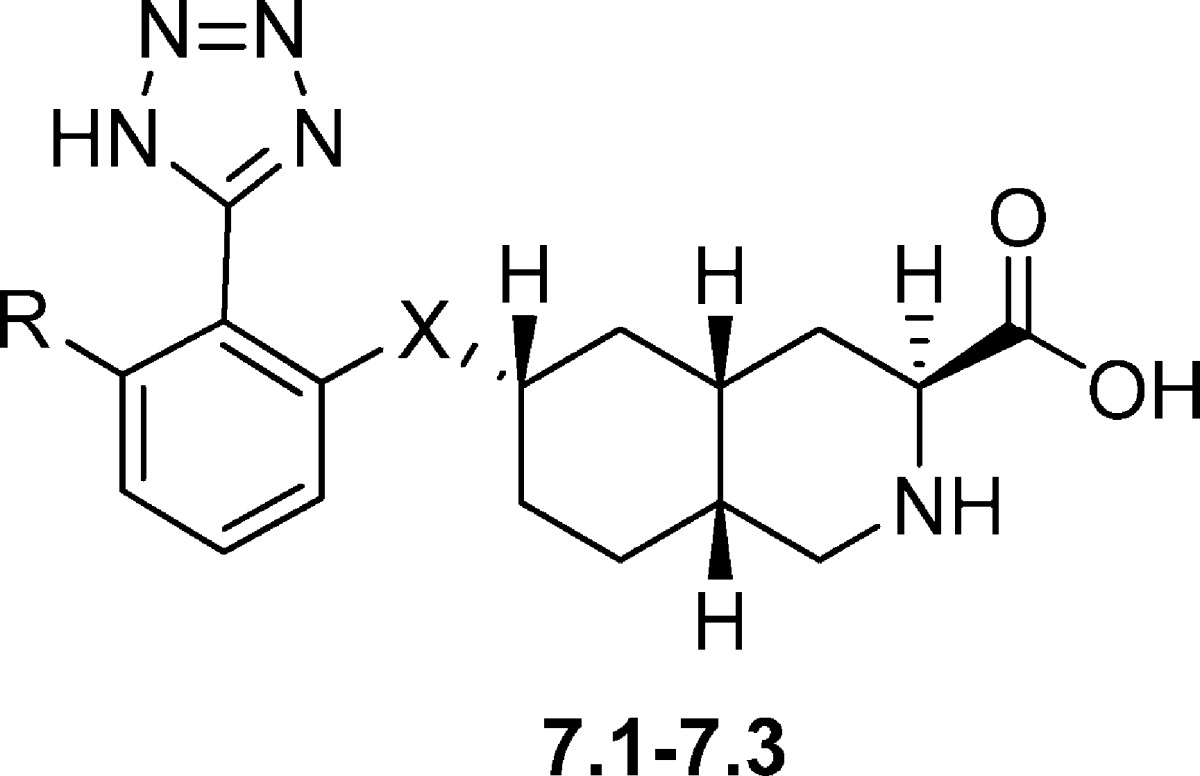
| GluA2 |
GluK1 |
GluK1/2 | GluK1/5 | |||||
|---|---|---|---|---|---|---|---|---|
| cmpd | X | R | bind.b | funct.c | bind.b | funct.c | bind.b | bind.b |
| 7.1 (LY458545) | O | H | 8.3 | 1.1 | 1.7 | 0.04 | 1.0 | 7.9 |
| 7.2 (LY457691) | NH | H | 5.5 | 0.4 | 1.6 | 0.03 | 1.4 | 9.9 |
| 7.3 | NH | Cl | −d | 2.0 | −d | 0.005 | −d | −d |
Functional data represent KB values for inhibition of 100 μM glutamate-induced Ca2+ influx in HEK 293 cells expressing human homomeric AMPA or KA receptor subtypes. Binding data represent Ki values. [3H]AMPA was used as radioligand for GluA2; others, [3H]KA. The compounds showed no affinity for GluK2 or GluK2/5. All values in [μM].
From ref (90).
From ref (86).
Not tested.
A major focus of recent research has been to improve the oral bioavailability of this class of GluK1 antagonists by formation of ester prodrugs (43,86,87,90). A number of successful prodrugs have been prepared: The diethyl ester prodrug of 6.4 was orally active in two animal models of migraine pain (43). The 2-isobutylester prodrug of compound 7.1 showed only moderate affinity for GluK1 receptors (Ki of 7.4 μM) (90). However, when administered orally, this prodrug produced dose-related antihyperalgesic effects in the Carrageenan test and formalin-induced paw-licking and capsaicin-induced allodynia was reversed (86,90), at doses that did not show performance deficits in the rotarod test (90). The 2-ethylbutylester prodrug of compound 7.2 was largely devoid of affinity for GluK1 receptors (Ki of 32 μM) (90), but upon oral administration this prodrug produced dose-related antihyperalgesic effects in the Carrageenan test, reversed formalin-induced paw-licking and reversed capsaicin-induced allodynia (86,90). These effects occurred at doses that did not show performance deficits in the rotarod test as well (90). The 2-ethylbutylester prodrug of compound 7.3 produced dose-related antihyperalgesic effects in carrageenan-induced thermal hyperalgesia and reversal of capsaicin-induced allodynia, although the prodrug was largely devoid of affinity for GluK1 receptors (86).
Willardiine Derivatives
A series of selective GluK1 antagonists were developed by addition of substituents at the N3 position of the uracil ring of the natural product willardiine (91−95). The lead compound 8.1 (3-CBW or UBP282) contains a 4-carboxybenzyl substituent in the N3 position and was shown to be a competitive antagonist of AMPA receptors expressed on motor neurons and of GluK1-containing KA receptors (Table 8) expressed in dorsal root C-fibers (96). The X-ray structure of 8.1 in complex with the LBD of GluA2 revealed a hyper-extended conformation of the protein compared to its apo state (97). The 2-carboxybenzyl analogue 8.2 (UBP296) was found to be 100-fold selective for GluK1 versus AMPA receptors when tested on cloned human and native rat AMPA receptors (93,95). In addition, 8.2 was >300-fold selective for homomeric GluK1 versus homomeric GluK2 and GluK3 and displayed similar potency on the heteromeric GluK1 receptors, GluK1/GluK2 and GluK1/GluK5, compared to that observed for GluK1 alone (Table 8) (93,95). The antagonist activity was found to reside in the S-enantiomer 8.3 (UBP302) which is in agreement with other amino acid-based KA receptor antagonists (Tables 7−9) (98). Compound 8.3 was the only compound in the UBP series to be tested in a binding assay on human GluK3 (Ki value> 100 μM). The 5-iodo-substituted analogue of 8.3 (structure not shown) was also tested at GluK1 receptors, and it was found to have slightly increased antagonism of KA-induced responses in the dorsal root preparation (KD = 0.21 μM)95, but the compound was not evaluated further. By replacement of the benzene ring in 8.3 with a thiophene ring, to give 8.4 (UBP304), potency and selectivity for the GluK1 subtype was further increased (92,99). Variation of the substituent in the 5-position of the uracil ring revealed that a methyl group, as in compound 8.5 (UBP310), led to low nanomolar potent GluK1 antagonist activity (10-fold increase) while also increasing selectivity for GluK1 versus GluK2 and AMPA receptors (91,94,98). However, this analogue was furthermore a potent antagonist at GluK3 (IC50 = 0.023 μM) (52).
Table 8. Chemical Structures and Functional as Well as Binding Data for Willardiine Analogues 8.1−8.6a.

| cmpd | GluA2 | GluK1 | GluK2 | GluK3 | GluK1/2 | GluK1/5 | GluK2/5 |
|---|---|---|---|---|---|---|---|
| 8.1 (UBP282)b | −h | 9.3 | >1000d | −h | −h | −h | >1000d |
| 8.2 (UBP296)c | >300 | 0.6 | >1000d | >100d | 0.8 | 1.0 | >100d |
| 8.3 (UBP302)e | >300 | 0.6 | >100 | 4.0f | 0.8 | 1.0 | >300 |
| 8.4 (UBP304)e | >100 | 0.12 | >100 | −h | 0.12 | 0.18 | >100 |
| 8.5 (UBP310)e | >100 | 0.010 | >100 | 0.023f | −h | 0.008 | >100 |
| 8.6 (ACET)e | >100 | 0.007g | >100 | 0.092f | −h | 0.005 | >100 |
All values in [μM].
From ref (93): KD values for inhibition of KA-induced currents in neonatal rat dorsal root C fibers.
KB values for inhibition of 10−100 μM glutamate-induced Ca2+ influx in HEK 293 cells expressing human homomeric AMPA or KA receptor subtypes; from ref (95).
From ref (93): Binding study Ki or IC50 from displacement of [3H]KA at human homomeric KA receptors expressed in HEK 293 cells.
From ref (91): Ki or IC50 value for inhibition of glutamate-evoked currents in HEK 293 cells expressing homomeric KA receptors.
From ref (52): IC50 values in an Xenopus oocyte assay.
In another study, this value was measured to be 0.0014l; ref (100).
Not tested.
Table 9. Chemical Structures and Binding Affinities of 9.1−9.5 at Native AMPA, KA, and NMDA Receptors (Rat Synaptosomes) and at Cloned Homomeric Rat GluA2 and GluK1-3 Receptorsa.

X-ray crystal structures were obtained of the LBD dimer of GluK1 complexed with 8.3 and 5-methyl analogue 8.5, respectively (94). The X-ray crystal structure of 8.3 revealed the largest domain opening reported so far for the iGluRs, where the α-amino moiety of the antagonist was not able to form a direct contact with Glu723, as it has been seen in all previously solved AMPA and KA receptor agonist and antagonist complexes (94). The improved selectivity for GluK1 versus AMPA receptors and other KA receptor subunits could be explained by the steric clashes imposed by substitution of Val670 for Leu650 and Ser720 for Met708 in GluA2. The low affinity of N3-substituted willardiines for GluK2 could be explained by the replacement of Thr503 and Ser674 for two alanine groups in this particular subunit, preventing favorable interaction with the α-ammonium and terminal carboxyl groups of the antagonist. Similarly, replacement of Ser674 in GluK1 by an Ala residue in GluK3 may explain the low affinity of 8.2 and 8.3 for GluK3. Furthermore, Ser760 in GluK1 is replaced with an Asp residue in GluK3, possibly hindering the placement of the benzene or the thiophene ring bearing the carboxyl group in the optimal orientation for interaction with a Thr residue in D2. The residues Val670 and Ser726 in GluK1 are replaced by the more bulky Ile and Met side chains in GluK4 and GluK5 explaining the lower affinity for these subunits.
With inspiration from the X-ray crystal structure of the LBD of GluK1 in complex with 8.5, the new antagonist 8.6 (UBP316 or ACET), was designed. A phenyl group was introduced at the 2-position of the thiophene ring of 8.5 to increase the hydrophobic interaction with Val670 and the CH2 group of Asn705. The improved selectivity for native GluK1 over AMPA receptors is likely due to the steric clash of the phenyl group of 8.6 with the Leu residue that replaces Val690 in AMPA receptors (91). As predicted, compound 8.6 exhibited improved potency and selectivity for native and cloned human GluK1 receptors versus native AMPA and cloned human GluA2 receptors (91,98,100). It was shown that 8.6 at a concentration of 100 nM completely blocked GluK1-containing dorsal root C-fiber responses to KA (10 μM), with and in the absence of ConA (101). As for 8.4, 8.5 and 8.6 showed excellent selectivity for human recombinant GluK1 over GluK2 subtypes and no effect on GluK2/GluK5 heteromers up to a concentration of 100 μM (Table 8).
The most potent GluK1 antagonists 8.4 (UBP304), 8.5 (UBP310), and 8.6 (UBP316 or ACET) were tested on native AMPA receptors expressed on spinal motor neurons giving KD values of 71.4, 83.4, and 108 μM, respectively, for blocking (S)-5-fluorowillardiine (5-F-will)-induced depolarization, suggesting that these antagonists were highly selective for the GluK1 subtype over AMPA receptors (91,92,95). Pharmacological characterization of 8.2 (50 μM), 8.4 (50 μM), 8.5 (10 μM), and 8.6 (1 μM) at NMDA and group I metabotropic glutamate receptors expressed in neonatal rat motor neurons demonstrated that these compounds displayed no activity at these receptors (91−93). Thus, these willardiine derivatives, and in particular 8.6, are useful pharmacological tools for studying the roles of GluK1 in CNS function (91,93,99), though care should be taken in interpreting the data for compounds 8.5 and 8.6 given the GLUK3 activity of these compounds.
Other Chemical Classes
Based on structure-based design, the compound (2S,3R)-m-carboxyphenylproline 9.1 was recently synthesized as a novel class of selective iGluR antagonists (102). In binding studies, 9.1 exhibited medium range micromolar affinity for native AMPA, and KA receptors and low micromolar affinity for native NMDA receptors. At cloned homomeric GluK1 and GluK3 receptors, 9.1 exhibited low micromolar affinity (3 and 8.1 μM respectively) whereas it was without affinity for GluK3 (Table 9). At the mGluRs, 9.1 has been shown to be without activity (rat mGluR1, Ki > 10 000 μM) (103). Functional characterization at nondesensitizing mutants of homomeric GluK1−3 receptors revealed that the compound is an antagonist of GluK1,3 (Table 10) (102).
Table 10. Functional Characterization (KB) of Antagonists 9.1 and 9.2 in a TEVC Oocyte Assay at Cloned Non-Desensitizing Homomeric Subtypes GluK1-3a.
A phosphonic acid analogue of AMPA, compound 9.2 ((S)-ATPO), was shown to have antagonistic activity at both recombinant GluA1−4 (Ki values of 2.0, 3.6, 3.6, and 6.7 μM, respectively) and GluK1 subtypes (KB = 24 μM), but no activity at the GluK2 subtype (Tables 9 and 10) (104). X-ray crystal structures of (S)-ATPO in complex with GluA2 (46) and GluK1 (105), respectively, show that (S)-ATPO stabilizes the open form of the LBD by binding to residues in domains D1 and D2 otherwise engaged in agonist binding.
A recent SAR study on a number of 3-substituted phenylalanine compounds focused on exploring compounds based on an alanine-substituted biphenyl core (106). The study concluded that a 3-carboxylic acid functionality was essential for GluK1 affinity as in compounds 9.3−9.5 (Table 9). Furthermore, the two aromatic rings had to be directly attached, that is, not connected by a methyl linker (structures not shown). The GluK1 affinity observed for 9.3 was found to reside in the S-isomer (9.4) which was to be expected from earlier reported α-amino acid-based KA receptor antagonists (Tables 7−9). The GluK1 affinity could be further increased (3-fold) by the addition of two chlorine atoms, as in 9.5. None of the mentioned compounds showed affinity for homomeric GluK2 or GluK3 receptors or even native AMPA, KA, or NMDA receptors. Compounds 9.3 and 9.5 were docked into the LBD of GluK1 complexed with 7.3 (UBP302), and, as expected, the compounds adopted a binding mode similar to that for 7.3. In this in silico study, the chlorine atom in the 5-position of 9.5 filled a partly hydrophobic cavity, which may explain the improved affinity compared to 9.3. On the other hand, the chlorine atom in the 4-position filled a hydrophilic cavity.
Miscellaneous
Early SAR studies suggested that the d-glutamine analogue 10.1 (GAMS) (108) and the benzaldehyde-substituted piperazines 10.2 (p-BB-PzDA) and 10.3 (p-CB-PzDA) (109) (Figure 3) were selective antagonists for AMPA/KA receptors over NMDA receptors. However, in these studies, quisqualate was used as the AMPA/KA receptor agonist and it is now known that this compound also activates group I metabotropic glutamate receptors (22,110,111). Compound 10.1 has been reported to be a weak antagonist of KA receptors on DRG neurons (49) and to preferentially antagonize KA receptors (112). In assays on recombinant human KA receptors, weak antagonist activity was observed at GluK1, GluK1/GluK2, and GluK1/GluK5, but not GluK2 or GluK2/GluK5 (IC50 values in the millimolar range at each subtype) (50). While compounds 10.2 and 10.3 blocked KA-induced responses, they blocked NMDA-induced responses with greater potency on CA1 hippocampal neurons (113). Both of these compounds have been shown to display moderate antagonist activity at GluK1-containing receptors on dorsal root C-fibres (114), but they have not been tested across recombinant AMPA/KA receptor subtypes.
Figure 3.

Chemical structures of compounds 10.1−10.4.
The tryptophan metabolite 10.4 (kynurenic acid or KYNA) (Figure 3) has often been referred to as a broad-spectrum Glu receptor antagonist (115), although at low levels the compound is an antagonist of only the strychnine-insensitive glycine site of the NMDA receptor (IC50 of 8−15 μmol/L) (116). However, the compound also antagonizes the glutamate recognition site of the NMDA receptor. At higher concentrations (IC50 in the millimolar range), 10.4 is a competitive antagonist at AMPA and KA receptors (117) and exhibits IC50 values in the 500 μM range for blocking homomeric and heteromeric GluK1 and homomeric GluK2 receptors, but does not block GluK2/GluK5 receptors at concentrations up to 3 mM (101). The metabolite 10.4 is thought to act as a tonic modulatory agent in the healthy CNS and has been shown to be involved in pathological states such as schizophrenia and cognitive deficits (118).
Conclusion and Perspectives
This review provides the reader with an overview of the field medicinal chemistry of competitive KA receptor antagonists. From this overview, it is evident that despite extensive efforts only selective antagonists for the GluK1 subunit have been developed. The discovery of selective antagonists for subunits GluK2−5 is yet to be accomplished and may call for an alternative strategy such as development of negative allosteric modulators. In perspective, selective KA receptor antagonists which discriminate between heteromeric receptor compositions may be of high value. Such pharmacological tools will allow for the investigation of the function and distribution of heteromeric KA receptors in the CNS.
We are deeply thankful to The Lundbeck Foundation for the financial support.
References
- Watkins J. C.; Evans R. H. (1981) Excitatory amino-acid transmitters. Annu. Rev. Pharmacool. Toxicol. 21, 165–204. [DOI] [PubMed] [Google Scholar]
- Lodge D. (2009) The history of the pharmacology and cloning of ionotropic glutamate receptors and the development of idiosyncratic nomenclature. Neuropharmacology 56, 6–21. [DOI] [PubMed] [Google Scholar]
- Watkins J. C.; Jane D. E. (2006) The glutamate story. Br. J. Pharmacol. 147, S100–S108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch L.; Krogsgaard-Larsen P. (2009) Subtype Selective Kainic Acid Receptor Agonists: Discovery and Approaches to Rational Design. Med. Res. Rev. 29, 3–28. [DOI] [PubMed] [Google Scholar]
- Mellor J. R. (2006) Synaptic plasticity of kainate receptors. Biochem. Soc. Trans. 34, 949–951. [DOI] [PubMed] [Google Scholar]
- Collingridge G. L.; Olsen R. W.; Peters J.; Spedding M. (2009) A nomenclature for ligand-gated ion channels. Neuropharmacology 56, 2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro P.; Mulle C. (2006) Kainate receptors. Cell Tissue Res. 326, 457–482. [DOI] [PubMed] [Google Scholar]
- Hollmann M.; Heinemann S. (1994) Cloned Glutamate Receptors. Annu. Rev. Neurosci. 17, 31–108. [DOI] [PubMed] [Google Scholar]
- van Giersbergen P. L.; Palkovits M.; De Jong W. (1992) Involvement of neurotransmitters in the nucleus tractus solitarii in cardiovascular regulation. Physiol. Rev. 72, 789–824. [DOI] [PubMed] [Google Scholar]
- Sommer B.; Burnashev N.; Verdoorn T. A.; Keinanen K.; Sakmann B.; Seeburg P. H. (1992) A glutamate receptor channel with high-affinity for domoate and kainate. EMBO J. 11, 1651–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C. H.; Mayer M. L. (1999) Heteromeric kainate receptors formed by the coassembly of GluR5, GluR6, and GluR7. J. Neurosci. 19, 8281–8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herb A.; Burnashev N.; Werner P.; Sakmann B.; Wisden W.; Seeburg P. H. (1992) The KA-2 subunit of excitatory amino-acid receptors shows widespread expression in brain and forms ion channels with distantly related subunits. Neuron 8, 775–785. [DOI] [PubMed] [Google Scholar]
- Kamboj R. K.; Schoepp D. D.; Nutt S.; Shekter L.; Korczak B.; True R. A.; Rampersad V.; Zimmerman D. M.; Wosnick M. A. (1994) Molecular cloning, expression, and pharmacological characterization of humEAA1, a human kainate receptor subunit. J. Neurochem. 62, 1–9. [DOI] [PubMed] [Google Scholar]
- Kamboj R. K.; Schoepp D. D.; Nutt S.; Shekter L.; Korczak B.; True R. A.; Zimmerman D. M.; Wosnick M. A. (1992) Molecular structure and pharmacological characterization of humEAA2, a novel human kainate receptor subunit. Mol. Pharmacol. 42, 10–15. [PubMed] [Google Scholar]
- Nistico R.; Dargan S.; Fitzjohn S. M.; Lodge D.; Jane D. E.; Collingridge G. L.; Bortolotto Z. A. (2009) GLUK1 receptor antagonists and hippocampal mossy fiber function. Int. Rev. Neurobiol. 85, 13–27. [DOI] [PubMed] [Google Scholar]
- Chittajallu R.; Braithwaite S. P.; Clarke V. R. J.; Henley J. M. (1999) Kainate receptors: subunits, synaptic localization and function. Trends Pharmacol. Sci. 20, 26–35. [DOI] [PubMed] [Google Scholar]
- Bleakman D.; Gates M. R.; Ogden A. M.; Mackowiak M. (2002) Kainate receptor agonists, antagonists and allosteric modulators. Curr. Pharm. Design 8, 873–885. [DOI] [PubMed] [Google Scholar]
- Bleakman D.; Lodge D. (1998) Neuropharmacology of AMPA and kainate receptors. Neuropharmacology 37, 1187–1204. [DOI] [PubMed] [Google Scholar]
- Lerma J.; Paternain A. V.; Rodriguez-Moreno A.; Lopez-Garcia J. C. (2001) Molecular physiology of kainate receptors. Physiol. Rev. 81, 971–998. [DOI] [PubMed] [Google Scholar]
- Lerma J. (2003) Roles and rules of kainate receptors in synaptic transmission. Nat. Rev. Neurosci. 4, 481. [DOI] [PubMed] [Google Scholar]
- Bortolotto Z. A.; Nistico R.; More J. C.; Jane D. E.; Collingridge G. L. (2005) Kainate receptors and mossy fiber LTP. Neurotoxicology 26, 769–777. [DOI] [PubMed] [Google Scholar]
- Mayer M. L. (2005) Crystal structures of the GluR5 and GluR6 ligand binding cores: Molecular mechanisms underlying kainate receptor selectivity. Neuron 45, 539–552. [DOI] [PubMed] [Google Scholar]
- Huettner J. E. (1990) Glutamate receptor channels in rat DRG neurons: activation by kainate and quisqualate and blockade of desensitization by Con A. Neuron 5, 255–266. [DOI] [PubMed] [Google Scholar]
- Morgado-Valle C.; Feldman J. L. (2007) NMDA receptors in preBotzinger complex neurons can drive respiratory rhythm independent of AMPA receptors. J.Physiol. (Oxford, U.K.) 582, 359–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang C. N.; Ramadan N. M.; Wallihan R. G.; Chappell A. S.; Freitag F. G.; Smith T. R.; Silberstein S. D.; Johnson K. W.; Phebus L. A.; Bleakman D.; Ornstein P. L.; Arnold B.; Tepper S. J.; Vandenhende F. (2004) LY293558, a novel AMPA/GluR5 antagonist, is efficacious and well-tolerated in acute migraine. Cephalalgia 24, 596–602. [DOI] [PubMed] [Google Scholar]
- Lees G. J. (2000) Pharmacology of AMPA/kainate receptor ligands and their therapeutic potential in neurological and psychiatric disorders. Drugs 59, 33–78. [DOI] [PubMed] [Google Scholar]
- Sang C. N.; Hostetter M. P.; Gracely R. H.; Chappell A. S.; Schoepp D. D.; Lee G.; Whitcup S.; Caruso R.; Max M. B. (1998) AMPA/kainate antagonist LY293558 reduces capsaicin-evoked hyperalgesia but not pain in normal skin in humans. Anesthesiology 89, 1060–1067. [DOI] [PubMed] [Google Scholar]
- Umemura K.; Kondo K.; Ikeda Y.; Teraya Y.; Yoshida H.; Homma M.; Uematsu T.; Nakashima M. (1997) Pharmacokinetics and safety of the novel amino-3-hydroxy-5-methylisoxazole-4-propionate receptor antagonist YM90K in healthy men. J. Clin. Pharmacol. 37, 719–727. [DOI] [PubMed] [Google Scholar]
- Kobayashi T.; Caringi D.; Mokler D. J.; Ally A. (1997) Effects of ventrolateral medullary AMPA-receptor antagonism on pressor response during muscle contraction. Am. J. Physiol. Heart Circ. Physiol. 272, H2774–H2781. [DOI] [PubMed] [Google Scholar]
- Danysz W.; Zajaczkowski W.; Parsons C. G. (1995) Modulation of learning-processes by ionotropic glutamate-receptor ligands. Behav. Pharmacol. 6, 455–474. [PubMed] [Google Scholar]
- Melyan Z.; Lancaster B.; Wheal H. V. (2004) Metabotropic regulation of intrinsic excitability by synaptic activation of kainate receptors. J. Neurosci. 24, 4530–4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A.; Sachidhanandam S.; Utvik J. K.; Coussen F.; Mulle C. (2005) Distinct subunits in heteromeric kainate receptors mediate ionotropic and metabotropic function at hippocampal mossy fiber synapses. J. Neurosci. 25, 11710–11718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulle C.; Sailer A.; Perez-Otano I.; Dickinson-Anson H.; Castillo P. E.; Bureau I.; Maron C.; Gage F. H.; Mann J. R.; Bettler B.; Heinemann S. F. (1998) Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature 392, 601–605. [DOI] [PubMed] [Google Scholar]
- Bittigau P.; Ikonomidou C. (1997) Topical Review: Glutamate in Neurologic Diseases. J. Child Neurol. 12, 471–485. [DOI] [PubMed] [Google Scholar]
- Obrenovitch T. P.; Urenjak J. (1997) Is High Extracellular Glutamate the Key to Excitotoxicity in Traumatic Brain Injury?. J. Neurotrauma 14, 677–698. [DOI] [PubMed] [Google Scholar]
- Boris-Moller F.; Wieloch T. (1998) Changes in the extracellular levels of glutamate and aspartate during ischemia and hypoglycemia - Effects of hypothermia. Exp. Brain Res. 121, 277–284. [DOI] [PubMed] [Google Scholar]
- Caragine L. P.; Park H. K.; Diaz F. G.; Phillis J. W. (1998) Real-time measurement of ischemia-evoked glutamate release in the cerebral cortex of four and eleven vessel rat occlusion models. Brain Res. 793, 255–264. [DOI] [PubMed] [Google Scholar]
- Porter R. H. P.; Eastwood S. L.; Harrison P. J. (1997) Distribution of kainate receptor subunit mRNAs in human hippocampus, neocortex and cerebellum, and bilateral reduction of hippocampal GluR6 and KA2 transcripts in schizophrenia. Brain Res. 751, 217–231. [DOI] [PubMed] [Google Scholar]
- Harris J. A.; Corsi M.; Quartaroli M.; Arban R.; Bentivoglio M. (1996) Upregulation of spinal glutamate receptors in chronic pain. Neuroscience 74, 7–12. [DOI] [PubMed] [Google Scholar]
- Li P.; Kerchner G. A.; Sala C.; Wei F.; Huettner J. E.; Sheng M.; Zhuo M. (1999) AMPA receptor-PDZ interactions in facilitation of spinal sensory synapses. Nature Neurosci. 2, 972–977. [DOI] [PubMed] [Google Scholar]
- Li P.; Zhuo M. (1998) Silent glutamatergic synapses and nociception in mammalian spinal cord. Nature 393, 695–698. [DOI] [PubMed] [Google Scholar]
- Sorkin L. S.; Yaksh T. L.; Doom C. M. (1999) Mechanical allodynia in rats is blocked by a Ca2+ permeable AMPA receptor antagonist. NeuroReport 10, 3523–3526. [DOI] [PubMed] [Google Scholar]
- Filla S. A.; Winter M. A.; Johnson K. W.; Bleakman D.; Bell M. G.; Bleisch T. J.; Castano A. M.; Clemens-Smith A.; del Prado M.; Dieckman D. K.; Dominguez E.; Escribano A.; Ho K. H.; Hudziak K. J.; Katofiasc M. A.; Martinez-Perez J. A.; Mateo A.; Mathes B. M.; Mattiuz E. L.; Ogden A. M. L.; Phebus L. A.; Stack D. R.; Stratford R. E.; Ornstein P. L. (2002) Ethyl (3S,4aR,6S,8aR)-6-(4-ethoxycarbonylimidazol-1-ylmethyl)decahydroiso-quin oline-3-carboxylic ester: A prodrug of a GluR5 kainate receptor antagonist active in two animal models of acute migraine. J. Med. Chem. 45, 4383–4386. [DOI] [PubMed] [Google Scholar]
- Kew J. N. C.; Kemp J. A. (2005) Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology 179, 4–29. [DOI] [PubMed] [Google Scholar]
- Carta A.; Piras S.; Loriga G.; Paglietti G. (2006) Chemistry, biological properties and SAR analysis of quinoxalinones. Mini-Rev. Med Chem 6, 1179–1200. [DOI] [PubMed] [Google Scholar]
- Hogner A.; Greenwood J. R.; Liljefors T.; Lunn M. L.; Egebjerg J.; Larsen I. K.; Gouaux E.; Kastrup J. S. (2003) Competitive antagonism of AMPA receptors by ligands of different classes: Crystal structure of ATPO bound to the GluR2 ligand-binding core, in comparison with DNQX. J. Med. Chem. 46, 214–221. [DOI] [PubMed] [Google Scholar]
- Armstrong N.; Gouaux E. (2000) Mechanisms for activation and antagonism of an AMPA-Sensitive glutamate receptor: Crystal structures of the GluR2 ligand binding core. Neuron 28, 165–181. [DOI] [PubMed] [Google Scholar]
- Sheardown M. J.; Nielsen E. O.; Hansen A. J.; Jacobsen P.; Honore T. (1990) 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science 247, 571–574. [DOI] [PubMed] [Google Scholar]
- Wilding T. J.; Huettner J. E. (1996) Antagonist pharmacology of kainate- and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-preferring receptors. Mol. Pharmacol. 49, 540–546. [PubMed] [Google Scholar]
- Alt A.; Weiss B.; Ogden A. M.; Knauss J. L.; Oler J.; Ho K.; Large T. H.; Bleakman D. (2004) Pharmacological characterization of glutamatergic agonists and antagonists at recombinant human homomeric and heteromeric kainate receptors in vitro. Neuropharmacology 46, 793–806. [DOI] [PubMed] [Google Scholar]
- Tygesen C. K.; Rasmussen J. S.; Jones S. V.; Hansen A.; Hansen K.; Andersen P. H. (1994) Stable expression of a functional GluR6 homomeric glutamate receptor channel in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 91, 13018–13022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrais D.; Pinheiro P. S.; Jane D. E.; Mulle C. (2009) Antagonism of recombinant and native GluK3-containing kainate receptors. Neuropharmacology 56, 131–140. [DOI] [PubMed] [Google Scholar]
- Nutt S. L.; Hoo K. H.; Rampersad V.; Deverill R. M.; Elliott C. E.; Fletcher E. J.; Adams S. L.; Korczak B.; Foldes R. L.; Kamboj R. K. (1994) Molecular characterization of the human EAA5 (GluR7) receptor: a high-affinity kainate receptor with novel potential RNA editing sites. Recept. Channels 2, 315–326. [PubMed] [Google Scholar]
- Simmons R. M. A.; Li D. L.; Hoo K. H.; Deverill M.; Ornstein P. L.; Iyengar S. (1998) Kainate GluR5 receptor subtype mediates the nociceptive response to formalin in the rat. Neuropharmacology 37, 25–36. [DOI] [PubMed] [Google Scholar]
- Nordholm L. S. M., Honoré T. (1997) In Excitatory amino acids: Clinical results with antagonists. (Herrling P. L., Ed.), pp 89−97, 129−152, Academic Press, London. [Google Scholar]
- Bleakman D.; Ogden A. M.; Ornstein P. L.; Hoo K. (1999) Pharmacological characterization of a GluR6 kainate receptor in cultured hippocampal neurons. Eur. J. Pharmacol. 378, 331–337. [DOI] [PubMed] [Google Scholar]
- Randle J. C. R.; Guet T.; Bobichon C.; Moreau C.; Curutchet P.; Lambolez B.; Decarvalho L. P.; Cordi A.; Lepagnol J. M. (1992) Quinoxaline derivatives - structure-activity-relationships and physiological implications of inhibition of N-methyl-D-aspartate and non-N-methyl-D-aspartate receptor-mediated currents and synaptic potentials. Mol. Pharmacol. 41, 337–345. [PubMed] [Google Scholar]
- Randle J. C.; Guet T.; Cordi A.; Lepagnol J. M. (1992) Competitive inhibition by NBQX of kainate/AMPA receptor currents and excitatory synaptic potentials: importance of 6-nitro substitution. Eur. J. Pharmacol. 215, 237–244. [DOI] [PubMed] [Google Scholar]
- Löscher W.; Lehmann H.; Behl B.; Seemann D.; Teschendorf H. J.; Hofmann H. P.; Lubisch W.; Höger T.; Lemaire H. G.; Gross G. (1999) A new pyrrolyl-quinoxalinedione series of non-NMDA glutamate receptor antagonists: pharmacological characterization and comparison with NBQX and valproate in the kindling model of epilepsy. Eur. J. Neurosci. 11, 250–262. [DOI] [PubMed] [Google Scholar]
- Lubisch W.; Behl B.; Hofmann H. P. (1997) Pyrrolylquinoxalinediones: The importance of pyrrolic substitution on AMPA receptor binding. Bioorg. Med. Chem. Lett. 7, 1101–1106. [Google Scholar]
- Lubisch W.; Behl B.; Hofmann H. P.; Teschendorf H. J. (1997) Pyrrolylquinoxalinediones: Dicarboxylates as highly potent AMPA receptor antagonists. Bioorg. Med. Chem. Lett. 7, 2441–2446. [Google Scholar]
- Lubisch W.; Behl B.; Hofmann H. P. (1996) Pyrrolylquinoxalinediones: A new class of AMPA receptor antagonists. Bioorg. Med. Chem. Lett. 6, 2887–2892. [Google Scholar]
- Madsen U.; Stensbol T. B.; Krogsgaard-Larsen P. (2001) Inhibitors of AMPA and kainate receptors. Curr. Med. Chem. 8, 1291–1301. [DOI] [PubMed] [Google Scholar]
- Lubisch W.; Behl B.; Henn C.; Hofmann H. P.; Reeb J.; Regner F.; Vierling M. (2002) Pyrrolylquinoxalinediones carrying a piperazine residue represent highly potent and selective ligands to the homomeric kainate receptor GluR5. Bioorg. Med. Chem. Lett. 12, 2113–2116. [DOI] [PubMed] [Google Scholar]
- Verdoorn T. A.; Johansen T. H.; Drejer J.; Nielsen E. O. (1994) Selective block of recombinant glur6 receptors by NS-102, a novel non-NMDA receptor antagonist. Eur. J. Pharmacol. 269, 43–49. [DOI] [PubMed] [Google Scholar]
- Nielsen E. O.; Johansen T. H.; Tasker R. A. R.; Strain S. M.; Jensen L. H.; Watjen F.; Drejer J. (1992) Selective displacement of low affinity tritiated kainate binding correlates with antagonism of domoic acid toxicity. Soc. Neurosci. Abstr. 18, 86. [Google Scholar]
- Nijholt I.; Blank T.; Grafelmann B.; Cepok S.; Kugler H.; Spiess J. (1999) NS-257, a novel competitive AMPA receptor antagonist, interacts with kainate and NMDA receptors. Brain Res. 821, 374–382. [DOI] [PubMed] [Google Scholar]
- Christensen J. K.; Varming T.; Ahring P. K.; Jorgensen T. D.; Nielsen E. O. (2004) In Vitro Characterization of 5-Carboxyl-2,4-di-benzamidobenzoic Acid (NS3763), a Noncompetitive Antagonist of GLUK5 Receptors. J. Pharmacol. Exp. Ther. 309, 1003–1010. [DOI] [PubMed] [Google Scholar]
- Kasper C.; Pickering D. S.; Mirza O.; Olsen L.; Kristensen A. S.; Greenwood J. R.; Liljefors T.; Schousboe A.; Wätjen F.; Gajhede M.; Sigurskjold B. W.; Kastrup J. S. (2006) The Structure of a Mixed GluR2 Ligand-binding Core Dimer in Complex with (S)-Glutamate and the Antagonist (S)-NS1209. J. Mol. Biol. 357, 1184–1201. [DOI] [PubMed] [Google Scholar]
- Pitkänen A.; Mathiesen C.; Rønn L. C. B.; Møller A.; Nissinen J. (2007) Effect of novel AMPA antagonist, NS1209, on status epilepticus: An experimental study in rat. Epilepsy Res. 74, 45–54. [DOI] [PubMed] [Google Scholar]
- Gormsen L.; Finnerup N. B.; Almqvist P. M.; Jensen T. S. (2009) The Efficacy of the AMPA Receptor Antagonist NS1209 and Lidocaine in Nerve Injury Pain: A Randomized, Double-Blind, Placebo-Controlled, Three-Way Crossover Study. Anesth. Analg. 108, 1311–1319. [DOI] [PubMed] [Google Scholar]
- Bleakman R.; Schoepp D. D.; Ballyk B.; Bufton H.; Sharpe E. F.; Thomas K.; Ornstein P. L.; Kamboj R. K. (1996) Pharmacological discrimination of GluR5 and GluR6 kainate receptor subtypes by (3S,4aR,6R,8aR)-6-[2-(1(2)H-tetrazole-5-yl)ethyl]decahyd roisdoquinoline-3 carboxylic-acid. Mol. Pharmacol. 49, 581–585. [PubMed] [Google Scholar]
- Ornstein P. L.; Arnold M. B.; Augenstein N. K.; Lodge D.; Leander J. D.; Schoepp D. D. (1993) 3SR,4aRS,6RS,8aRS)-6-[2-(1H-Tetrazol-5-yl)ethyl]decahydroisoquinoline-3-carboxylic acid: a structurally novel, systemically active, competitive AMPA receptor antagonist. J. Med. Chem. 36, 2046–2048. [DOI] [PubMed] [Google Scholar]
- Schoepp D. D.; Lodge D.; Bleakman D.; Leander J. D.; Tizzano J. P.; Wright R. A.; Palmer A. J.; Salhoff C. R.; Ornstein P. L. (1995) In-vitro and in vivo antagonism of AMPA receptor activation by (3S,4AR,6R,8AR)-6-[2-(1(2)H-tetrazole-5-yl)ethyl]decahydroisoquinoline-3 -carboxylic acid. Neuropharmacology 34, 1159–1168. [DOI] [PubMed] [Google Scholar]
- Bleakman D.; Ballyk B. A.; Schoepp D. D.; Palmer A. J.; Bath C. P.; Sharpe E. F.; Woolley M. L.; Bufton H. R.; Kamboj R. K.; Tarnawa I.; Lodge D. (1996) Activity of 2,3-benzodiazepines at native rat and recombinant human glutamate receptors in vitro: Stereospecificity and selectivity profiles. Neuropharmacology 35, 1689–1702. [DOI] [PubMed] [Google Scholar]
- Doucet-Personeni C.; Bentley P. D.; Fletcher R. J.; Kinkaid A.; Kryger G.; Pirard B.; Taylor A.; Taylor R.; Taylor J.; Viner R.; Silman I.; Sussman J. L.; Greenblatt H. M.; Lewis T. (2001) A structure-based design approach to the development of novel, reversible AChE inhibitors. J. Med. Chem. 44, 3203–3215. [DOI] [PubMed] [Google Scholar]
- Mathe J. M.; Fagerquist M. V.; Svensson T. H. (1999) Antipsychotic-like effect of the AMPA receptor antagonist LY326325 as indicated by suppression of conditioned avoidance response in the rat. J. Neural Transm. 106, 1003–1009. [DOI] [PubMed] [Google Scholar]
- Kotlinska J.; Liljequist S. (1998) The putative AMPA receptor antagonist, LY326325, produces anxiolytic-like effects without altering locomotor activity in rats. Pharmacol., Biochem. Behav. 60, 119–124. [DOI] [PubMed] [Google Scholar]
- Bartolotto; Clarke Z. A. (1999) Kainate receptors are involved in synaptic plasticity. Nature 402, 297. [DOI] [PubMed] [Google Scholar]
- Bortolotto Z. A.; Clarke V. R. J.; Delany C. M.; Parry M. C.; Smolders I.; Vignes M.; Ho K. H.; Miu P.; Brinton B. T.; Fantaske R.; Ogden A.; Gates M.; Ornstein P. L.; Lodge D.; Bleakman D.; Collingridge G. L. (1999) Kainate receptors are involved in synaptic plasticity. Nature 402, 297–301. [DOI] [PubMed] [Google Scholar]
- Smolders I.; Bortolotto Z. A.; Clarke V. R. J.; Warre R.; Khan G. M.; O’Neill M. J.; Ornstein P. L.; Bleakman D.; Ogden A.; Weiss B.; Stables J. P.; Ho K. H.; Ebinger G.; Collingridge G. L.; Lodge D.; Michotte Y. (2002) Antagonists of GLU(K5)-containing kainate receptors prevent pilocarpine-induced limbic seizures. Nat. Neurosci. 5, 796–804. [DOI] [PubMed] [Google Scholar]
- Alt A.; Weiss B.; Ornstein P. L.; Gleason S. D.; Bleakman D.; Stratford R. E.; Witkin J. M. (2007) Anxiolytic-like effects through a GLUKS kainate receptor mechanism. Neuropharmacology 52, 1482–1487. [DOI] [PubMed] [Google Scholar]
- Clarke V. R. J.; Ballyk B. A. (1997) A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature 389, 599. [DOI] [PubMed] [Google Scholar]
- O’Neill M. J.; Bond A.; Ornstein P. L.; Ward M. A.; Hicks C. A.; Hoo K.; Bleakman D.; Lodge D. (1998) Decahydroisoquinolines: novel competitive AMPA/kainate antagonists with neuroprotective effects in global cerebral ischaemia. Neuropharmacology 37, 1211–1222. [DOI] [PubMed] [Google Scholar]
- Vignes M.; Clarke V. R. J.; Parry M. J.; Bleakman D.; Lodge D.; Ornstein P. L.; Collingridge G. L. (1998) The GluR5 subtype of kainate receptor regulates excitatory synaptic transmission in areas CA1 and CA3 of the rat hippocampus. Neuropharmacology 37, 1269–1277. [DOI] [PubMed] [Google Scholar]
- Jones C. K.; Alt A.; Ogden A. M.; Bleakman D.; Simmons R. M. A.; Iyengar S.; Dominguez E.; Ornstein P. L.; Shannon H. E. (2006) Antiallodynic and Antihyperalgesic Effects of Selective Competitive GLUK5 (GluR5) Ionotropic Glutamate Receptor Antagonists in the Capsaicin and Carrageenan Models in Rats. J. Pharmacol. Exp. Ther. 319, 396–404. [DOI] [PubMed] [Google Scholar]
- Weiss B.; Alt A.; Ogden A. M.; Gates M.; Dieckman D. K.; Clemens-Smith A.; Ho K. H.; Jarvie K.; Rizkalla G.; Wright R. A.; Calligaro D. O.; Schoepp D.; Mattiuz E. L.; Stratford R. E.; Johnson B.; Salhoff C.; Katofiasc M.; Phebus L. A.; Schenck K.; Cohen M.; Filla S. A.; Ornstein P. L.; Johnson K. W.; Bleakman D. (2006) Pharmacological characterization of the competitive GLU(K5) receptor antagonist decahydroisoquinoline LY466195 in vitro and in vivo. J. Pharmacol. Exp. Ther. 318, 772–781. [DOI] [PubMed] [Google Scholar]
- Andreou A. P.; Goadsby P. J. (2009) LY466195, a clinically active compound in the acute treatment of migraine, inhibits activation in the trigeminocervical complex and the ventroposteromedial thalamus after nociceptive trigeminovascular activation. Cephalalgia 29, 132–132. [Google Scholar]
- Partin K. M.; Patneau D. K.; Winters C. A.; Mayer M. L.; Buonanno A. (1993) Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin A. Neuron 11, 1069–1082. [DOI] [PubMed] [Google Scholar]
- Dominguez E.; Iyengar S.; Shannon H. E.; Bleakman D.; Alt A.; Arnold B. M.; Bell M. G.; Bleisch T. J.; Buckmaster J. L.; Castano A. M.; Del Prado M.; Escribano A.; Filla S. A.; Ho K. H.; Hudziak K. J.; Jones C. K.; Martinez-Perez J. A.; Mateo A.; Mathes B. M.; Mattiuz E. L.; Ogden A. M. L.; Simmons R. M. A.; Stack D. R.; Stratford R. E.; Winter M. A.; Wu Z.; Ornstein P. L. (2005) Two Prodrugs of Potent and Selective GluR5 Kainate Receptor Antagonists Actives in Three Animal Models of Pain. J. Med. Chem. 48, 4200–4203. [DOI] [PubMed] [Google Scholar]
- Dolman N. P.; More J. C. A.; Alt A.; Knauss J. L.; Pentikäinen O. T.; Glasser C. R.; Bleakman D.; Mayer M. L.; Collingridge G. L.; Jane D. E. (2007) Synthesis and Pharmacological Characterization of N3-Substituted Willardiine Derivatives: Role of the Substituent at the 5-Position of the Uracil Ring in the Development of Highly Potent and Selective GLUK5 Kainate Receptor Antagonists. J. Med. Chem. 50, 1558–1570. [DOI] [PubMed] [Google Scholar]
- Dolman N. P.; More J. C. A.; Alt A.; Knauss J. L.; Troop H. M.; Bleakman D.; Collingridge G. L.; Jane D. E. (2006) Structure-Activity Relationship Studies on N3-Substituted Willardiine Derivatives Acting as AMPA or Kainate Receptor Antagonists. J. Med. Chem. 49, 2579–2592. [DOI] [PubMed] [Google Scholar]
- More J. C. A.; Nistico R.; Dolman N. P.; Clarke V. R. J.; Alt A. J.; Ogden A. M.; Buelens F. P.; Troop H. M.; Kelland E. E.; Pilato F.; Bleakman D.; Bortolotto Z. A.; Collingridge G. L.; Jane D. E. (2004) Characterisation of UBP296: a novel, potent and selective kainate receptor antagonist. Neuropharmacology 47, 46–64. [DOI] [PubMed] [Google Scholar]
- Mayer M. L. (2006) Glutamate receptors at atomic resolution. Nature 440, 456–462. [DOI] [PubMed] [Google Scholar]
- Dolman N. P.; Troop H. M.; More J. C.; Alt A.; Knauss J. L.; Nistico R.; Jack S.; Morley R. M.; Bortolotto Z. A.; Roberts P. J.; Bleakman D.; Collingridge G. L.; Jane D. E. (2005) Synthesis and pharmacology of willardiine derivatives acting as antagonists of kainate receptors. J. Med. Chem. 48, 7867–7881. [DOI] [PubMed] [Google Scholar]
- More J. C. A.; Troop H. M.; Jane D. E. (2002) The novel antagonist 3-CBW discriminates between kainate receptors expressed on neonatal rat motoneurones and those on dorsal root C-fibres. Br. J. Pharmacol. 137, 1125–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed A. H.; Thompson M. D.; Fenwick M. K.; Romero B.; Loh A. P.; Jane D. E.; Sondermann H.; Oswald R. E. (2009) Mechanisms of Antagonism of the GluR2 AMPA Receptor: Structure and Dynamics of the Complex of Two Willardiine Antagonists with the Glutamate Binding Domain. Biochemistry 48, 3894–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- More J. C. A.; Troop H. M.; Dolman N. P.; Jane D. E. (2003) Structural requirements for novel willardiine derivatives acting as AMPA and kainate receptor antagonists. Br. J. Pharmacol. 138, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxter J. R.; Zinyuk L. E.; Roloff E. V. L.; Clarke V. R. J.; Dolman N. P.; More J. C. A.; Jane D. E.; Collingridge G. L.; Muller R. U. (2007) Inhibition of kainate receptors reduces the frequency of hippocampal theta oscillations. J. Neurosci. 27, 2212–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dargan S. L.; Clarke V. R. J.; Alushin G. M.; Sherwood J. L.; Nistico R.; Bortolotto Z. A.; Ogden A. M.; Bleakman D.; Doherty A. J.; Lodge D.; Mayer M. L.; Fitzjohn S. M.; Jane D. E.; Collingridge G. L. (2009) ACET is a highly potent and specific kainate receptor antagonist: Characterisation and effects on hippocampal mossy fibre function. Neuropharmacology 56, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jane D. E.; Lodge D.; Collingridge G. L. (2009) Kainate receptors: Pharmacology, function and therapeutic potential. Neuropharmacology 56, 90–113. [DOI] [PubMed] [Google Scholar]
- Larsen A. M., Venskutonyté R., Nielsen B., Pickering D. S., and Bunch L. (2010) Discovery of a New Class of Ionotropic Glutamate Receptor Antagonists by the Rational Design of (2S,3R)-3-(3-Carboxyphenyl)-pyrrolidine-2-carboxylic Acid, ACS Chem. Neurosci. DOI: 10.1021/cn100093f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheli F.; Di Fabio R.; Marchioro C. (1999) Asymmetric synthesis of some substituted-3-phenyl prolines. Farmaco 54, 461–464. [DOI] [PubMed] [Google Scholar]
- Moller E. H.; Egebjerg J.; Brehm L.; Stensbol T. B.; Johansen T. N.; Madsen U.; Krogsgaard-Larsen P. (1999) Resolution, absolute stereochemistry, and enantiopharmacology of the GluR1−4 and GluR5 antagonist 2-amino-3-[5-tert-butyl-3-(phosphonomethoxy)-4-isoxazolyl]propionic acid. Chirality 11, 752–759. [DOI] [PubMed] [Google Scholar]
- Hald H.; Naur P.; Pickering D. S.; Sprogoe D.; Madsen U.; Timmermann D. B.; Ahring P. K.; Liljefors T.; Schousboe A.; Egebjerg J.; Gajhede M.; Kastrup J. S. (2007) Partial agonism and antagonism of the ionotropic glutamate receptor iGLuR5 - Structures of the ligand-binding core in complex with domoic acid and 2-amino-3-[5-tert-butyl-3-(phosphonomethoxy)-4-isoxazolyl]propionic acid. J. Biol. Chem. 282, 25726–25736. [DOI] [PubMed] [Google Scholar]
- Szymanska E.; Pickering D. S.; Nielsen B.; Johansen T. N. (2009) 3-Substituted phenylalanines as selective AMPA- and kainate receptor ligands. Bioorg. Med. Chem. 17, 6390–6401. [DOI] [PubMed] [Google Scholar]
- Wahl P.; Anker C.; Traynelis S. F.; Egebjerg J.; Rasmussen J. S.; Krogsgaard-Larsen P.; Madsen U. (1998) Antagonist properties of a phosphono isoxazole amino acid at glutamate R1−4 (R,S)-2-amino-3-(3-hydroxy-5-methyl-4-isoxazolyl)propionic acid receptor subtypes. Mol. Pharmacol. 53, 590–596. [DOI] [PubMed] [Google Scholar]
- Davies J.; Watkins J. C. (1985) Depressant Actions of Gamma-D-Glutamylaminomethyl Sulfonate (Gams) on Amino Acid-Induced and Synaptic Excitation in the Cat Spinal-Cord. Brain Res. 327, 113–120. [DOI] [PubMed] [Google Scholar]
- Davies J.; Jones A. W.; Sheardown M. J.; Smith D. A. S.; Watkins J. C. (1984) Phosphono dipeptides and piperazine derivatives as antagonists of amino acid-included and synaptic excitation in mammalian and amphibian spinal-cord. Neurosci. Lett. 52, 79–84. [DOI] [PubMed] [Google Scholar]
- Abe T.; Sugihara H.; Nawa H.; Shigemoto R.; Mizuno N.; Nakanishi S. (1992) Molecular characterization of a novel metabotropic glutamate receptor mGluR5 coupled to inositol phosphate/Ca2+ signal transduction. J. Biol. Chem. 267, 13361–13368. [PubMed] [Google Scholar]
- Aramori I.; Nakanishi S. (1992) Signal transduction and pharmacological characteristics of a metabotropic glutamate receptor, mGluR1, in transfected CHO cells. Neuron 8, 757–765. [DOI] [PubMed] [Google Scholar]
- Zhou N.; Hammerland L. G.; Parks T. N. (1993) Gamma-D-glutamylaminomethyl sulfonic-acid (GAMS) distinguishes kainic acid-induced from AMPA-induced responses in xenopus-oocytes expressing chick brain glutamate receptors. Neuropharmacology 32, 767–775. [DOI] [PubMed] [Google Scholar]
- Ganong A. H.; Jones A. W.; Watkins J. C.; Cotman C. W. (1986) Parallel antagonism of synaptic transmission and kainate/quisqualate responses in the hippocampus by piperazine-2,3-dicarboxylic acid analogs. J. Neurosci. 6, 930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R. H.; Evans S. J.; Pook P. C.; Sunter D. C. (1987) A comparison of excitatory amino-acid antagonists acting at primary afferent c-fibers and motoneurons of the isolated spinal-cord of the rat. Br. J. Pharmacol. 91, 531–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone T. W. (1993) Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 45, 309–379. [PubMed] [Google Scholar]
- Parsons C. G.; Danysz W.; Quack G. n.; Hartmann S.; Lorenz B.; Wollenburg C.; Baran L.; Przegalinski E.; Kostowski W.; Krzascik P.; Chizh B.; Max. Headley P. (1997) Novel Systemically Active Antagonists of the Glycine Site of the N-Methyl-D-aspartate Receptor: Electrophysiological, Biochemical and Behavioral Characterization. J. Pharmacol. Exp. Ther. 283, 1264–1275. [PubMed] [Google Scholar]
- Kessler M.; Terramani T.; Lynch G. (1989) A glycine site associated with N-methyl-D-aspartic acid receptors: characterization and identification of a new class of antagonists. J. Neurochem. 52, 1319–1328. [DOI] [PubMed] [Google Scholar]
- Erhardt S.; Olsson S. K.; Engberg G. r. (2009) Pharmacological Manipulation of Kynurenic Acid: Potential in the Treatment of Psychiatric Disorders. CNS Drugs 23, 91–101. [DOI] [PubMed] [Google Scholar]