

Abstract
Multiple sclerosis (MS) is a neuroinflammatory and neurodegenerative disorder involving demyelination, axonal transection, and neuronal loss in the brain. Recent studies have indicated that active MS lesions express elevated levels of butyrylcholinesterase (BuChE). BuChE can hydrolyze a wide variety of esters, including fatty acid esters of protein. Proteolipid protein (PLP), an important transmembrane protein component of myelin, has six cysteine residues acylated, via thioester linkages, with fatty acids, usually palmitic, that contribute to the stability of myelin. Experimental chemical deacylation of PLP has been shown to lead to decompaction of myelin. Because of elevated levels of BuChE in active MS lesions and its propensity to catalyze the hydrolysis of acylated protein, we hypothesized that this enzyme may contribute to deacylation of PLP in MS, leading to decompaction of myelin and contributing to demyelination. To test this hypothesis, a series of increasing chain length (C2−C16) acyl thioester derivatives of N-acetyl-l-cysteine methyl ester were synthesized and examined for hydrolysis by human cholinesterases. All N-acetyl-l-cysteine fatty acyl thioester derivatives were hydrolyzed by BuChE but not by the related enzyme acetylcholinesterase. In addition, it was observed that the affinity of BuChE for the compound increased the longer the fatty acid chain, with the highest affinity for cysteine bound to palmitic acid. This suggests that the elevated levels of BuChE observed in active MS lesions could be related to the decompaction of myelin characteristic of the disorder.
Keywords: Cholinesterase, multiple sclerosis, donepezil, acylproteins, palmitic acid
Multiple sclerosis (MS) is a chronic neuroinflammatory and neurodegenerative disorder of the central nervous system, exhibiting demyelination, axonal transection, and neuronal loss that can give rise to sensory, motor, autonomic, and cognitive dysfunction (1). The cause of MS remains unknown, but neuroinflammation and demyelination are well documented aspects of the disease (1,2).
Myelin is mainly composed of lipids and a number of proteins (3). One of the most abundant proteins associated with myelin is proteolipid protein (PLP) (3,4). PLP is a 30 kDa, transmembrane protein containing 276 amino acids and is bonded to fatty acids (mainly palmitic) through thioester linkages at six cysteine residues, C5, C6, C9, C108, C138, and C148. These fatty acyl residues are embedded in the myelin membrane, imbuing stability (4).
It has been observed that abnormalities in PLP lead to abnormal myelin and impairment of the function of the central nervous system. For example, in Pelizaeus−Merzbacher disease, PLP genetic abnormalities lead to formation of abnormal myelin that is manifested in motor and cognitive dysfunction (5). Furthermore, it has been observed in an experimental rat optic nerve model that chemical deacylation of PLP fatty acid moieties leads to decompaction of myelin and abnormal functioning of this nerve while reacylation of PLP with palmitic acid restores its integrity and function (6).
Earlier observations (7) indicated the presence of an enzyme with esterase activity associated with white matter lesions in MS brain tissue. Recently, a particular esterase, butyrylcholinesterase (BuChE), has been observed to be associated with active MS lesions (8). BuChE is an enzyme that has the ability to hydrolyze a wide variety of esters, including long chain choline thioesters (9,10) and the long chain fatty acid ester of the polypeptide growth hormone secretagogue, ghrelin (11). BuChE and the related enzyme, acetylcholinesterase (AChE), are widely distributed throughout the brain and are coregulators of the neurotransmitter acetylcholine (12−14). However, only BuChE is able to efficiently catalyze the hydrolysis of esters larger than acetylcholine (9). Because of elevated levels of BuChE in active MS lesions and its propensity to catalyze the hydrolysis of longer fatty acyl esters, including acylated proteins, we hypothesized that this enzyme may contribute to deacylation of PLP, leading to the decompaction of myelin and the demyelination characteristic of MS. To test this hypothesis, a series of increasing chain length (C2−C16) fatty acyl thioester derivatives of N-acetyl-l-cysteine methyl ester were synthesized and examined for hydrolysis by human BuChE and AChE.
Results and Discussion
Since PLP is acylated as a thioester at cysteine residues, derivatives of this amino acid were synthesized as model analogues of PLP and tested to determine whether they were susceptible to catalytic hydrolysis by BuChE. The amino group of cysteine was protected as an acetyl amide, while the carboxylic acid functionality was converted to a methyl ester. The thiol was subsequently acylated with acid chlorides of different alkyl chain lengths. The synthesized thioesters were examined for susceptibility to hydrolysis by human BuChE. The ability of the related enzyme AChE was also tested for comparison. A total of seven l-cysteine fatty acyl thioester derivatives were synthesized and tested.
Synthetic Chemistry
Commercially available N-acetyl-l-cysteine (1) was converted to N-acetyl-l-cysteine methyl ester (2) using thionyl chloride in methanol (15), as depicted in Scheme 1. Thioester derivatives of 2 were prepared by mixing this compound with the corresponding acid chloride in dichloromethane as depicted in Scheme 1, in the presence of triethylamine to remove the HCl byproduct. Briefly, compound 2 was dissolved in dichloromethane, and to this solution triethylamine was added dropwise followed by the acid chloride, all at 0 °C. The course of the reaction was monitored by thin layer chromatography (TLC) until starting material (2) was consumed. After workup, the desired product was purified by recrystallization from hexanes/ethyl acetate (3) or silica gel flash chromatography (4−9).
Scheme 1. Scheme Depicting the Two Step General Synthesis of Cysteine Thioester Analogues (3−9) from N-Acetyl-l-cysteine (1) by Forming the Methyl Ester First Then Acylating the Thiol with Acid Chlorides of Different Alkyl Chain Lengths.

All purified compounds were homogeneous on TLC, and HPLC analysis revealed all to be more than 98% pure (see the Supporting Information). Analytical data, including IR, 1H and 13C NMR spectroscopy, as well as high resolution (accurate mass) mass spectrometry, were consistent with the chemical structure of each synthesized compound (2−9).
Enzyme Kinetic Studies
Hydrolysis of the thioester linkages in compounds 3−9 releases a thiol functionality; therefore, a modification (16) of the Ellman (17) spectrophotometric method could be employed to detect this cleavage. In this method, a released thiol group reacts with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) to produce a yellow colored nitro thiophenolate (Scheme 2). Since the formation of the yellow product is not a rate-limiting step in the process, the hydrolysis of the thioesters was monitored and kinetic parameters determined spectrophotometrically.
Scheme 2. Scheme Demonstrating a Modification to the Ellman Spectrophotometric Method Showing the Formation of the Yellow Colored Nitro Thiophenolate That Allowed for Detection of Hydrolysis of Thioesters to Be Monitored and Kinetic Parameters to Be Determined.

All seven thioester derivatives (3−9) were susceptible to BuChE-catalyzed hydrolysis. AChE, on the other hand, was not able to hydrolyze of any of these derivatives. As has been observed for other molecules susceptible to hydrolysis (18,19), the decanoyl derivative (6) was found to undergo some buffer-catalyzed hydrolysis in the presence of 0.1 M phosphate, pH 8.0, in the absence of the enzyme. To circumvent this problem, kinetic assays for this derivative were performed in 0.06 M Tris buffer at pH 8.0. Also, to maintain substrate solubility in the aqueous environment, assays for the palmityl thioester (9) were performed at 40 °C, while all others were carried out at 23 °C.
Affinity Constants
Lineweaver−Burk double reciprocal plots (see Figure 1 for examples) were generated to determine the affinity constant (Km value) for each thioester substrate in the presence of human BuChE. The Km value is inversely proportional to the affinity of the enzyme for the substrate so that a lower Km value represents a higher affinity of the enzyme for the substrate. As indicated in Table 1, the affinity of BuChE for the thioester derivatives (3−9) was found to increase as the carbon chain length of the acyl group lengthened from 2C (3) to 16C (9). The ability of BuChE to hydrolyze long chain fatty acyl derivatives of cysteine that are analogous to the thioester linkages in myelin PLP, together with the association of this enzyme with active MS lesions (8), suggests its potential to disrupt myelin, under pathological conditions.
Figure 1.

Lineweaver−Burk double reciprocal plots of cysteine thioester derivatives 4, 5, and 9 generated to determine the affinity constants for the thioester substrates in the presence of human BuChE.
Table 1. Affinity Constants (Km) of Butyrylcholinesterase for Synthesized N-Acetyl-l-cysteine Fatty Acyl Thioester Derivativesb.
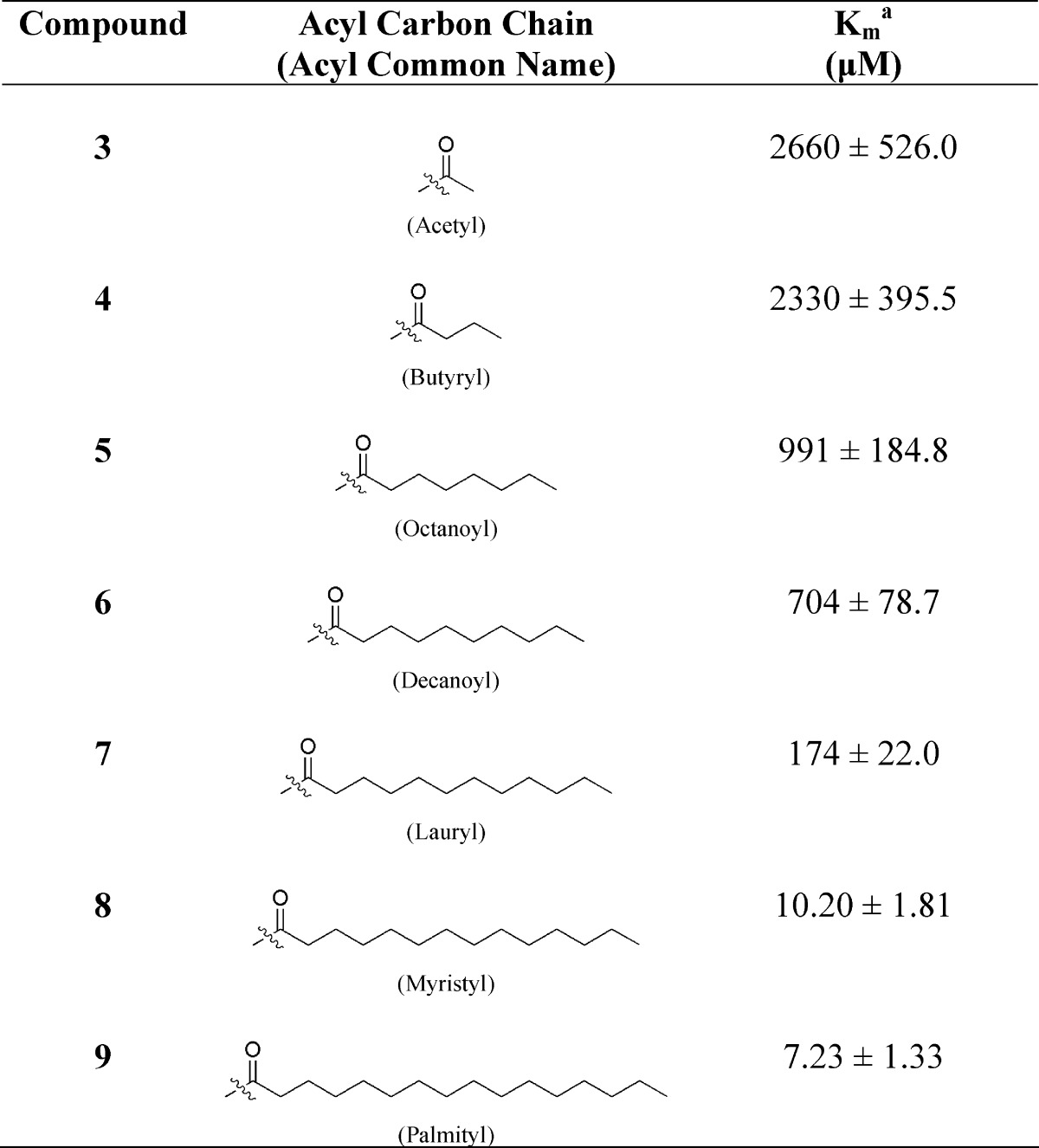 |
Km values were determined at 23 °C for compounds 3−8 and 40 °C for compound 9.
There was no hydrolysis of any of these compounds by acetylcholinesterase.
Structure−Activity Considerations
Examination of the Km values for the compounds 3−9 (Table 1) indicated a marked increase in the affinity between compound 6 and compound 7. This change in affinity became more evident by plotting the negative log of the affinity constant (pKm) against the carbon chain length of the acyl group of the thioester (Figure 2). This figure indicates an exponential increase in affinity beyond the decanoyl derivative (6) that must be related to the lengthening of the acyl chain, since this is the only altered moiety of the substrate.
Figure 2.

Relationship between negative log affinity constant (pKm) and number of carbons in the acyl chain of the cysteine thioester derivatives (3−9). As the number of carbons increases, substrate affinity of BuChE for the substrate increases, with the highest affinity for the 16C palmityl derivative (9).
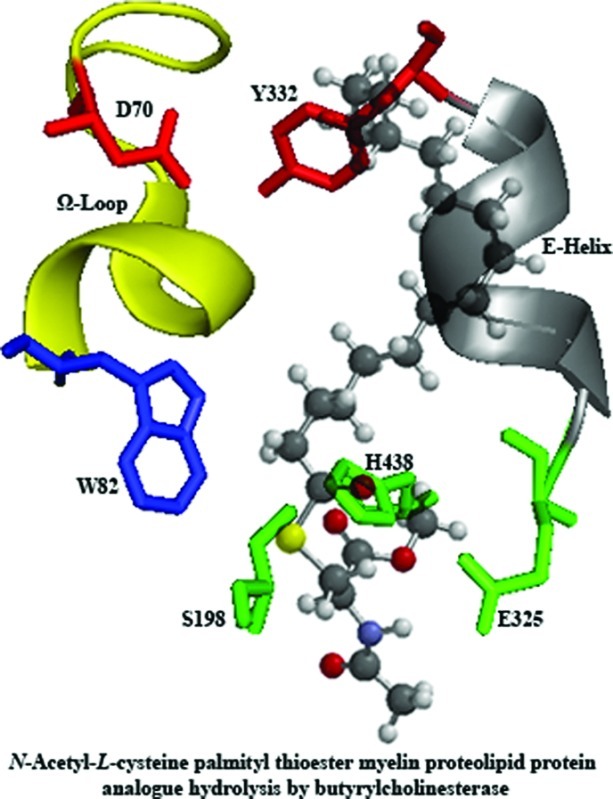
To help explain this behavior, the preferred geometry of each derivative was determined, at the molecular mechanics level, using Spartan '06 (20). The single difference between the compounds is the length of the hydrocarbon chain of the acyl group. As can be seen (Figure 3), the hydrocarbon chain remains roughly linear for compounds 3−5. However, a significant change in the general conformation begins at C10 with thioester 6, and this change becomes more pronounced as the chain lengthens through compounds 7−9 (arrows, Figure 3). It is significant that the relationship between affinity and chain length changes most beyond derivative 6 where this hairpin loop in the conformation of the hydrocarbon chain takes place. This substrate conformational change at derivative 6 that results in the sharp increase in BuChE affinity must be related to a more favorable interaction of thioester with the enzyme active site. The BuChE active site gorge, the volume of which is estimated to be 500 Å3, almost twice that of AChE (21), contains many amino acid residues that could be affected by changes in the structures of cysteine thioesters. In addition to the catalytic triad of serine (S198), histidine (H438), and glutamate (E325), that would bind to the thioester carbonyl, the crystal structure of BuChE (22) indicates that it has a peripheral anionic site composed of aspartate (D70) and tyrosine (Y332) residues. The peripheral anionic site functions to guide substrates down into the active site gorge and is linked by the Ω-loop to the π-cationic site composed of tryptophan (W82). The gorge also has an E-helix containing tyrosine, phenylalanine, alanine, and glutamate residues (Y332, F329, A328, and E325). The acyl pocket is composed of lysine and valine residues (L286 and V288) known to accommodate the acyl portion of choline esters. The lengthening of the hydrocarbon chain from 2C (3) to 16C (9) should increase the number of secondary, such as hydrophobic, attractions between substrate and amino acid side chains in the active site gorge. This supports the observed increased substrate affinity (smaller Km values) for BuChE as the fatty acyl hydrocarbon chain lengthens (Table 1). The lengthening of the acyl chain provides additional interactions between enzyme and substrate, produced by the hairpin loop after C10 has been reached (6) and surpassed (7−9). This hairpin loop remains at C10 as the hydrocarbon chain is lengthened in compounds 7−9. It may be reasonable to assume that this altered conformation favors the formation of additional hydrophobic interactions between substrate and enzyme that results in the observed increase in affinity. Thus, the high affinity of BuChE for the cysteine thioester of palmitic acid supports the notion that this enzyme could be involved in the deacylation of PLP contributing to the pathogenesis of MS.
Figure 3.

Preferred geometries (Spartan ’06) of cysteine thioester derivatives (3−9). Arrows point toward C10 of the fatty acid side chain. The hydrocarbon chain remains roughly linear for compounds 3−5 with a hairpin loop of the hydrocarbon chain at C10 for compounds 6−9. Thioester carbonyls are depicted by an asterisk (∗).
Inhibition Studies
Hydrolysis of fatty acid residues from PLP, in addition to degrading myelin structure, may cause the exposure of antigenic peptide sequences (4), and this epitope spreading phenomenon could accentuate the neuroinflammatory response characteristic of MS. It has been shown previously (23) that inhibition of cholinesterase activity dampens neuroinflammatory responses in the experimental autoimmune encephalomyelitis (EAE) model of MS. Donepezil is known to inhibit both BuChE and AChE (16), and this compound has been shown to improve cognitive symptoms of MS (24). Therefore, we examined the ability of donepezil to interfere with hydrolysis of fatty acyl thioesters in a manner comparable to its effect on butyrylthiocholine hydrolysis. Donepezil was able to inhibit the hydrolysis of the octanoyl thioester (5) with an IC50 value (8.84 ± 0.73 × 10−6 M) that was comparable to that obtained for the same inhibitor on BuChE hydrolysis of butyrylthiocholine under the same conditions (IC50 = 6.77 ± l.39 × 10−6 M). Thus, the effects of donepezil in MS may be, in part, mediated through BuChE inhibition.
Conclusions
We have previously observed that active MS lesions show intense staining for BuChE activity. In the present study, we have shown that BuChE is able to catalyze the hydrolysis of fatty acyl thioester derivatives of l-cysteine that represent simple analogues of PLP, a protein component contributing to the structural integrity of myelin. This hydrolytic ability suggests a possible role for BuChE in the decompaction of myelin and in the formation of, and propagation of, demyelinated white matter lesions that are characteristic of MS. In addition to BuChE hydrolysis destabilizing degrading myelin structure, hydrolysis of fatty acid residues from PLP may cause the exposure of antigenic peptide sequences and this epitope spreading phenomenon could accentuate the neuroinflammatory response contributing to the pathogenesis of MS.
Although these simple cysteine thioester derivatives provide preliminary evidence as to the ability of BuChE to hydrolyze the type of linkage found in myelin PLP, it does not answer the question as to whether the same catalysis is possible with the fatty acyl group attached to a relatively large transmembrane protein. This remains to be determined with fatty acyl derivatives of larger peptide sequences. Nonetheless, this study suggests that inhibitors of BuChE activity may prove valuable for slowing the progression of the MS disease process.
Methods
Materials
Thionyl chloride, acetyl chloride, butyryl chloride, octanoyl chloride, decanoyl chloride, lauroyl chloride, myristyl chloride, palmityl chloride, N-acetyl-l-cysteine, phosphomolybdic acid, 5,5′-dithio-bis(2-nitrobenzoic acid), and human recombinant AChE were purchased from Sigma-Aldrich (St. Louis, MO). BuChE purified from human plasma was a gift from Dr. Oksana Lockridge (Eppley Institute, University of Nebraska Medical Center). Donepezil was synthesized as previously described (16).
Organic Synthesis and Purification
The synthesis of the starting material (2) is shown in Scheme 1, and the syntheses of the thioester compounds 3−9 depicted in Scheme 1 were carried out as follows.
Methyl 2-(acetylamino)-3-sulfanylpropanoate (2)
This compound was synthesized by a method described previously (15). Briefly, thionyl chloride (13.5 mmol) was added dropwise to a solution of N-acetyl-l-cysteine (12.3 mmol) in methanol (50 mL). The reaction was stirred for 90 min at room temperature under a drying tube. The reaction was monitored using TLC on Alugram Sil G/UV254 sheets. When a distinct product spot was present on the TLC plate, showing no presence of starting material, the dichloromethane was removed under reduced pressure and elevated temperature. The residue obtained was a yellow oil and was extracted with ethyl acetate (3 × 40 mL) and washed with distilled water (20 mL). The combined organic layers were dried over anhydrous magnesium sulfate for 20 min, and the solution was gravity filtered and concentrated to dryness under reduced pressure and elevated temperature. The product was used without further purification.
General Synthesis and Purification of Thioester Analogues (3−9)
For the synthesis of the cysteine thioester compounds, the starting material (2) was dissolved in dichloromethane (20 mL). Triethylamine was added dropwise followed by the appropriate acid chloride. The starting material (2), triethylamine, and acid chloride were present in equimolar quantities. The actual amounts are given in the Analytical Data section. The reaction solution was stirred at 0 °C under a drying tube. All reactions were monitored using TLC and were stopped when there was no starting material (2) remaining. The developing solvents were ethyl acetate/dichloromethane mixtures, and TLC plates were visualized using phosphomolybdic acid. Reaction periods were typically 2−4 h. The reaction mixture was extracted with 0.1 M HCl (20 mL), saturated sodium carbonate (20 mL), distilled water (20 mL) and was washed with brine (20 mL). The organic layer was dried over anhydrous magnesium sulfate, gravity filtered, and concentrated to dryness under reduced pressure and elevated temperature.
Compound 3 was recrystallized using a mixed solvent technique with ethyl acetate and hexanes. The crude material was dissolved in the minimum amount of ethyl acetate and precipitated by the addition of hexanes.
Compounds 4−9 were purified through flash chromatography on silica gel using ethyl acetate or ethyl acetate/dichloromethane mixtures as the eluting solvent. Fractions obtained from flash chromatography were analyzed by mp, IR, 1H and 13C NMR, and HRMS.
Characterization of Synthesized Compounds
Melting points were measured using a Mel-Temp II apparatus from Laboratory Services Inc. and are uncorrected. Infrared spectra were recorded as Nujol mulls between sodium chloride plates on a Nicolet Avatar 330 FT-IR spectrometer. Peak positions were reproducible within 1−2 cm−1. NMR spectra were recorded at the Atlantic Region Magnetic Resonance Centre, Dalhousie University on a Bruker AVANCE 500 instrument operating at 500.1 MHz for proton and 125.8 MHz for carbon-13, or utilizing the additional distorsionless enhancement by polarization transfer-quaternary (DEPT-Q) experiment. Proton chemical shifts are reported in ppm relative to tetramethylsilane (TMS) in CDCl3 or dimethyl sulfoxide (DMSO) solution. Carbon chemical shifts are reported in ppm relative to CDCl3 or DMSO solvent. Reported multiplicities are apparent. Accurate mass positive-ion electrospray ionization (ESI) measurements were recorded at the Mass Spectrometry Laboratory at Dalhousie University using a Bruker Daltonics microTOF instrument with a flow rate of 2 uL/min and spray voltage of 4500 V, and the tray temperature was 180 °C. Mass measurements were routinely within 3 ppm of the calculated value. The purity of the synthesized compounds was also determined using an Agilent Technologies 1200 series HPLC system with a C18 reverse phase column with methanol as the mobile phase.
Analytical Data
Methyl 2-(Acetylamino)-3-sulfanylpropanoate (2)
Yield 74%; colorless crystals; mp 77−79 °C. IR (Nujol): 3301, 2360, 1737, 1643, 1223 cm−1. 1H NMR (CHCl3-d3) δ ppm 1.40 (t, J = 8.9 Hz, 1H), 2.10 (s, 3H), 3.01−3.05 (m, 2H), 3.81 (s, 3H), 4.89−4.92 (m, 1H), 6.53 (d, J = 4.7 Hz, 1H). 13C NMR (CHCl3-d3) δ ppm 170.6, 169.9, 53.5, 52.8, 26.8, 23.1. HRMS (ESI) M+ found, 200.0343; calcd, 200.0352 (for C6H11NNaO3S).
Methyl 2-Acetamido-3-(acetylthio)propanoate (3)
Yield 45% (based on 1.97 mmol reaction); colorless crystals; mp 83−85 °C. IR (Nujol): 3252, 1731, 1703, 1634, 1227 cm−1. 1H NMR (CHCl3-d3) δ ppm 2.05 (s, 3H), 2.39 (s, 3H), 3.35−3.43 (m, 2H), 3.78 (s, 3H), 4.81 (ddd, J = 7.5, 6.0, 4.7 Hz, 1H), 6.32 (d, J = 6.6 Hz, 1H). 13C NMR (CHCl3-d3) δ ppm 195.3, 170.7, 169.9, 52.8, 52.0, 30.9, 30.4, 23.0. HRMS (ESI) M+ found, 242.0447; calcd, 242.0457 (for C8H13NNaO4S).
Methyl 2-Acetamido-3-(butyrylthio)propanoate (4)
Yield 46% (based on 2.00 mmol reaction); colorless crystals; mp 60−61 °C. IR (Nujol): 3280, 1734, 1700, 1687, 1228 cm−1. 1H NMR (DMSO-d6) δ ppm 0.89 (t, J = 7.3 Hz, 3H), 1.59 (sxt, J = 7.3 Hz, 2H), 1.84 (s, 3H), 2.55 (t, J = 7.2 Hz, 2H), 3.05 (dd, J = 13.7, 8.2 Hz, 1H), 3.31 (dd, J = 13.4, 5.4 Hz, 1H), 3.62 (s, 3H), 4.38 (td, J = 8.2, 5.3 Hz, 1H), 8.34 (d, J = 7.8 Hz, 1H). 13C NMR (DMSO-d6) δ ppm 198.2, 171.0, 169.7, 52.5, 51.8, 45.4, 29.8, 22.5, 18.9, 13.5. HRMS (ESI) M+ found, 270.0788; calcd, 270.0770 (for C10H17NNaO4S).
Methyl 2-Acetamido-3-(octanoylthio)propanoate (5)
Yield 70% (based on 1.50 mmol reaction); colorless crystals; mp 33−35 °C. IR (Nujol): 3320, 1740, 1701, 1651 cm−1. 1H NMR (CHCl3-d3) δ ppm 0.88 (t, J = 6.9 Hz, 3H), 1.27−1.31 (m, 8H), 1.65 (quin, J = 7.4 Hz, 2H), 2.01 (s, 3H), 2.57 (t, J = 5.0 Hz, 2H), 3.37 (d, J = 5.4 Hz, 2H), 3.76 (s, 3H), 4.80 (dt, J = 7.6, 5.4 Hz, 1H), 6.26 (d, J = 7.1 Hz, 1H). 13C NMR (CHCl3-d3) δ ppm 199.2, 170.7, 169.9, 52.8, 52.2, 44.0, 31.6, 30.5, 28.84, 28.81, 25.6, 23.0, 22.5, 14.0. HRMS (ESI) M+ found, 326.1383; calcd, 326.1397 (for C14H25NNaO4S).
Methyl 2-Acetamido-3-(decanoylthio)propanoate (6)
Yield 43% (based on 1.00 mmol reaction); colorless crystals; mp 39−41 °C. IR (Nujol): 3317, 1740, 1702, 1650 1249 cm−1. 1H NMR (CHCl3-d3) δ ppm 0.92 (t, J = 6.9 Hz, 3H), 1.28−1.34 (m, 12H), 1.67 (quin, J = 7.2 Hz, 2H), 2.04 (s, 3H), 2.59 (t, J = 7.4 Hz, 2H), 3.38 (d, J = 5.3 Hz, 2H), 3.77 (s, 3H), 4.81 (dt, J = 7.5, 5.4 Hz, 1H), 6.32 (d, J = 7.2 Hz, 2H). 13C NMR (CHCl3-d3) δ ppm 199.1, 170.7, 169.9, 52.7, 52.1, 43.9, 31.8, 30.5, 29.3, 29.16, 29.14, 28.8, 25.6, 23.0, 22.6, 14.0. HRMS (ESI) M+ found, 354.1704; calcd, 354.1710 (for C16H29NNaO4S).
Methyl 2-Acetamido-3-(dodecanoylthio)propanoate (7)
Yield 81% (based on 1.50 mmol reaction); colorless crystals; mp 56−59 °C. IR (Nujol): 3314, 1740, 1717, 1690, 1258 cm−1. 1H NMR (CHCl3-d3) δ ppm 0.93 (t, J = 6.9 Hz, 3H), 1.30−1.33 (m, 16H), 1.68 (quin, J = 5.0 Hz, 2H), 2.05 (s, 3H), 2.60 (t, J = 7.5 Hz, 2H), 3.39 (d, J = 5.5 Hz, 2H), 3.78 (s, 3H), 4.82 (dt, J = 7.6, 5.4 Hz, 1H), 6.29 (d, J = 6.6 Hz, 1H). 13C NMR (CHCl3-d3) δ ppm 199.2, 170.7, 169.9, 52.7, 52.2, 44.0, 31.9, 30.5, 29.5 (2), 29.4, 29.3, 29.2, 28.9, 25.6, 23.0, 22.6, 14.1. HRMS (ESI) M+ found, 382.2018; calcd, 382.2023 (for C18H33NNaO4S).
Methyl 2-Acetamido-3-(tetradecanoylthio)propanoate (8)
Yield 43% (based on 1.07 mmol reaction); colorless crystals; mp 66−68 °C. IR (Nujol): 3311, 1740, 1702, 1650 cm−1. 1H NMR (DMSO-d6) δ ppm 0.87 (t, J = 6.7 Hz, 3H), 1.24−1.29 (m, 20H), 1.55 (quin, J = 7.3 Hz, 2H), 1.84 (s, 3H), 2.56 (t, J = 7.2 Hz, 2H), 3.05 (dd, J = 13.6, 8.2 Hz, 1H), 3.30 (dd, J = 13.7, 5.3 Hz, 1H), 3.62 (s, 3H), 4.37 (td, J = 8.0, 5.3 Hz, 1H), 8.34 (d, J = 7.8 Hz, 1H). 13C NMR (DMSO-d6) δ ppm 197.8, 170.5, 169.1, 51.9, 51.3, 43.1, 31.1, 29.3, 28.82, 28.79 (2), 28.74, 28.6, 28.5, 28.4, 27.9, 24.8, 22.0, 21.9, 13.7. HRMS (ESI) M+ found, 410.2335; calcd, 410.2336 (for C20H37NNaO4S).
Methyl 2-Acetamido-3-(palmitoylthio)propanoate (9)
Yield 61% (based on 1.02 mmol reaction); colorless crystals; mp 52−54 °C. IR (Nujol): 3287, 1739, 1718, 1690, 1227 cm−1. 1H NMR (CHCl3-d3) δ ppm 0.93 (t, J = 6.9 Hz, 3H), 1.30−1.35 (m, 24H), 1.68 (quin, J = 7.2 Hz, 2H), 2.05 (s, 3H), 2.59 (t, J = 7.5 Hz, 2H), 3.39 (d, J = 5.3 Hz, 2H), 3.78 (s, 3H), 4.82 (dt, J = 7.5, 5.4 Hz, 1H), 6.30 (d, J = 7.3 Hz, 1H). 13C NMR (CHCl3-d3) δ ppm 198.9, 170.4, 169.7, 52.5, 51.9, 43.7, 31.6, 30.3, 29.39 (2), 29.37 (2), 29.34, 29.28, 29.11, 29.06, 28.9, 28.6, 25.3, 22.7, 22.4, 13.8. HRMS (ESI) M+ found, 438.2617; calcd, 438.2649 (for C22H41NNaO4S).
Biochemical Analyses
The ability of AChE and BuChE to catalyze the hydrolysis at the thioester linkage of each synthesized compound was studied using a modification (16) of the method described by Ellman et al. (17). Briefly, in a 3 mL cuvette of 1 cm light path was placed 1.41 mL of buffered DTNB solution (pH 8.0). Stock DTNB solution consisted of 10 mM DTNB with 18 mM sodium bicarbonate in 0.1 M phosphate buffer, pH 7.0 or in 0.06 M Tris buffer, pH 8.0. The working DTNB solution was prepared by mixing 3.6 mL of 10 mM stock DTNB solution with 96.4 mL of 0.1 M phosphate buffer at pH 8.0 or 0.06 M Tris buffer, pH 8.0. Tris buffer was only used in assays for compound 6, as this compound was somewhat susceptible to hydrolysis by phosphate buffer in the absence of enzyme. After adding 0.04 mL of BuChE (15 units), except compounds 5 and 6 which required only 0.04 mL of a 1:10 dilution of BuChE in 0.1% aqueous gelatin (1.5 units), or AChE (6.64 units), the stoppered cuvette was mixed by inversion, the absorbance at 412 nm calibrated to zero, and 0.05 mL of substrate in acetonitrile was added and mixed to initiate the reaction. Decreasing working substrate concentrations were then prepared by diluting stock substrates with acetonitrile. The final volume in the cuvette was 1.5 mL. For compounds 3−8, the assays were performed at 23 °C. The absorbance was measured at 412 nm every 15 s for 10 min, after an initial 10 s delay. For compound 9, the reaction was performed at 40 °C to maintain substrate solubility and the absorbance was measured at 412 nm after 10 min. The rate of change of absorbance (ΔA/min) was determined from these assays, that were performed in triplicate for each of five working substrate concentrations (4.65 × 10−4−1.28 × 10−5 M), and the Km values obtained from Lineweaver−Burk plots were averaged.
Determination of IC50 for Cholinesterase Inhibition
Experiments were performed to compare the effect of a known cholinesterase inhibitor, donepezil, on the hydrolysis of cysteine thioester 5 with that for the usual BuChE substrate, butyrylthiocholine. To make the comparison, the inhibitor concentration that gave half-maximum inhibition (IC50) was determined in each case.
The highest concentration of donepezil, due to solubility limits, was 5 mM in 50% aqueous acetonitrile. Serial (1:10) dilutions in the same solvent were done to obtain an inhibition−concentration profile. Briefly, the assays were carried out by mixing 1.36 mL of buffered DTNB working solution (pH 8.0), 0.05 mL of butyrylthiocholine (4.8 × 10−3 M) or compound 5 (5.77 × 10−3 M), 0.05 mL of 50% aqueous acetonitrile or donepezil (5.0 × 10−3−5.0 × 10−6 M) in the same solvent, and 0.04 mL of BuChE (1.5 units) in a quartz cuvette (1 cm path length). The final volume was 1.5 mL, and the reaction was performed at 23 °C. The absorbance at 412 nm was measured every 3 s for 5 min, after an initial 3 s delay. The rate of change of absorbance (ΔA/min) was obtained using these assays, performed in triplicate.
The rate of change of absorbance related to the concentration of inhibitor was plotted, and IC50 values were determined. Briefly, the average change in absorbance with inhibitor divided by the average change in absorbance of the uninhibited enzyme activity generates the relative activity. To determine experimental relative inhibition, the relative activity is subtracted from 1. The plot employed a relative inhibition enzyme interaction assumption to model the decrease in enzyme activity. This assumption was minimized to the experimental data using the Solver add-in of Microsoft Excel (25,26) to yield the IC50 values.
Computational Chemistry
The ground states, and thus most stable geometries, of representative thioester molecules (3−9) were calculated using the Merck molecular force field (MMFF) in Spartan ’06 (20). The graphical abstract was constructed using the crystal structure of BuChE which was obtained from the protein data bank (PDB ID: 1POI) (22,27). Only amino acids associated with the active site gorge were depicted using PyMOL Executable Build 2006 (28). The preferred geometry of compound 9 was overlaid within the active site gorge using Adobe Photoshop CS3.
Acknowledgments
We would like to thank Drs. J. Fisk and V. Bhan for critically reviewing this manuscript.
Abbreviations
AChE, acetylcholinesterase; BuChE, butyrylcholinesterase; °C, degrees centigrade; DTNB, 5,5′-dithiobis-(2-nitrobenzoic acid); HPLC, high performance liquid chromatography; IC50, 50% inhibition concentration; mL, milliliter; μM, micromolar; mM, millimolar; min, minute; MS, multiple sclerosis; PLP, proteolipid protein; %, percentage; rt, room temperature; TLC, thin layer chromatography; ΔA/min, rate of change of absorbance per minute.
Supporting Information Available
Yields and purities of synthesized analogues along with characterization spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
I.R.P.: Conduct experiments, data analysis, first draft manuscript, and revisions. E.A.H.: Conduct experiments, data analysis, and manuscript revisions. R.A.B.: Conduct experiments and data analysis. I.R.M.: Conduct experiments, data analysis, first draft manuscript, and revisions. E.M.: Conceptualization, conduct experiments, data analysis, first draft manuscript, and revisions. S.D.: Conceptualization, conduct experiments, data analysis, first draft manuscript, and revisions.
Canadian Institutes of Health Research, Canada Foundation for Innovation, Vascular Health and Dementia Initiative (DOV-78344) (through partnership of Canadian Institutes of Health Research, Heart & Stroke Foundation of Canada, the Alzheimer Society of Canada and Pfizer Canada Inc.), Natural Sciences and Engineering Research Council of Canada, Multiple Sclerosis Society of Canada, endMS Research and Training Network, Capital District Health Authority Research Fund, Nova Scotia Health Research Foundation, Brain Tumor Foundation of Canada, the Committee on Research and Publications of Mount Saint Vincent University.
Supplementary Material
References
- Noseworthy J. H.; Lucchinetti C.; Rodriguez M.; Weinshenker B. G. (2000) Medical progress: Multiple sclerosis. N. Engl. J. Med. 343, 938–952. [DOI] [PubMed] [Google Scholar]
- Frohman E. M.; Racke M. K.; Raine C. S. (2006) Multiple sclerosis—the plaque and its pathogenesis. N. Engl. J. Med. 354, 942–955. [DOI] [PubMed] [Google Scholar]
- Tzakos A. G.; Kursula P.; Troganis A.; Theodorou V.; Tselios T.; Svarnas C.; Matsoukas J.; Apostolopoulos V.; Gerothanassis I. P. (2005) Structure and function of the myelin proteins: Current status and perspectives in relation to multiple sclerosis. Curr. Med. Chem. 12, 1569–1587. [DOI] [PubMed] [Google Scholar]
- Greer J. M.; Lees M. B. (2002) Myelin proteolipid protein - the first 50 years. Int. J. Biochem. Cell Biol. 34, 211–215. [DOI] [PubMed] [Google Scholar]
- Gow A.; Lazzarini R. A. (1996) A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat. Genet. 13, 422–428. [DOI] [PubMed] [Google Scholar]
- Bizzozero O. A.; Bixler H. A.; Davis J. D.; Espinosa A.; Messier A. M. (2001) Chemical deacylation reduces the adhesive properties of proteolipid protein and leads to decompaction of the myelin sheath. J. Neurochem. 76, 1129–1141. [DOI] [PubMed] [Google Scholar]
- Lumsden C. E. (1951) Fundamental problems in the pathology of multiple sclerosis and allied demyelinating diseases. Br. Med. J. 1, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvesh S.; Leblanc A. M.; Macdonald I. R.; Reid G. A.; Bhan V.; Macaulay R. J.; Fisk J. D. (2010) Butyrylcholinesterase activity in multiple sclerosis neuropathology. Chem. Biol. Interact. 187, 425–431. [DOI] [PubMed] [Google Scholar]
- Silver A. (1974) The Biology of Cholinesterases, 1st ed., Elsevier, Amsterdam. [Google Scholar]
- Darvesh S.; McDonald R. S.; Darvesh K. V.; Mataija D.; Mothana S.; Cook H.; Carneiro K. M.; Richard N.; Walsh R.; Martin E. (2006) On the active site for hydrolysis of aryl amides and choline esters by human cholinesterases. Bioorg. Med. Chem. 14, 4586–4599. [DOI] [PubMed] [Google Scholar]
- De Vriese C.; Gregoire F.; Lema-Kisoka R.; Waelbroeck M.; Robberecht P.; Delporte C. (2004) Ghrelin degradation by serum and tissue homogenates: Identification of the cleavage sites. Endocrinology 145, 4997–5005. [DOI] [PubMed] [Google Scholar]
- Mesulam M.-M.; Guillozet A.; Shaw P.; Levey A.; Duysen E. G.; Lockridge O. (2002) Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience 110, 627–639. [DOI] [PubMed] [Google Scholar]
- Giacobini E. (2003) Butyrylcholinesterase: Its Function and Inhibitors, 1st ed., Martin Dunitz, London and New York. [Google Scholar]
- Darvesh S.; Hopkins D. A.; Geula C. (2003) Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 4, 131–138. [DOI] [PubMed] [Google Scholar]
- Conchillo A.; Camps F.; Messeguer A. (1990) 3,4-Epoxyprecocenes as Models of Cytotoxic Epoxides - Synthesis of the Cis Adducts Occurring in the Glutathione Metabolic Pathway. J. Org. Chem. 55, 1728–1735. [Google Scholar]
- Darvesh S.; Walsh R.; Kumar R.; Caines A.; Roberts S.; Magee D.; Rockwood K.; Martin E. (2003) Inhibition of human cholinesterases by drugs used to treat Alzheimer disease. Alzheimer Dis. Assoc. Disord. 17, 117–126. [DOI] [PubMed] [Google Scholar]
- Ellman G. L.; Courtney K. D.; Andres V. J.; Feather-Stone R. M. (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95. [DOI] [PubMed] [Google Scholar]
- Holland J. M.; Miller J. G. (1961) Quantitative Studies on Lipolytic Enzyme Activity in Degenerating and Regenerating Nerve. J. Phys. Chem. 65, 463–466. [DOI] [PubMed] [Google Scholar]
- Darvesh S.; Kumar R.; Roberts S.; Walsh R.; Martin E. (2001) Butyrylcholinesterase-mediated enhancement of the enzymatic activity of trypsin. Cell. Mol. Neurobiol. 21, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wavefunction Inc. (2006) Spartan ’06 for Windows and Linux: Tutorial and User’s Guide, Wavefunction Inc. Irvine CA, USA and Japan Branch Office Chiyoda-Ku, Tokyo, Japan.
- Saxena A.; Redman A. M.; Jiang X.; Lockridge O.; Doctor B. P. (1997) Differences in active site gorge dimensions of cholinesterases revealed by binding of inhibitors to human butyrylcholinesterase. Neuroscience 36, 14642–14651. [DOI] [PubMed] [Google Scholar]
- Nicolet Y.; Lockridge O.; Masson P.; Fontecilla-Camps J. C.; Nachon F. (2003) Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 278, 41141–41147. [DOI] [PubMed] [Google Scholar]
- Nizri E.; Irony-Tur-Sinai M.; Faranesh N.; Lavon I.; Lavi E.; Weinstock M.; Brenner T. (2008) Suppression of neuroinflammation and immunomodulation by the acetylcholinesterase inhibitor rivastigmine. J. Neuroimmunol. 203, 12–22. [DOI] [PubMed] [Google Scholar]
- Krupp L. B.; Christodoulou C.; Melville P.; Scherl W. F.; MacAllister W. S.; Elkins L. E. (2004) Donepezil improved memory in multiple sclerosis in a randomized clinical trial. Neurology 63, 1579–1585. [DOI] [PubMed] [Google Scholar]
- Fylstra D.; Lasdon L.; Watson J.; Waren A. (1998) Design and use of the microsoft excel solver. Interfaces 28, 29–55. [Google Scholar]
- Nenov I. P.; Fyltstra D. H. (2003) Interval methods for accelerated global search in the microsoft excel solver. Reliab. Comput. 9, 143–159. [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L. (2006) The PyMOL Molecular Graphics System, version 0.99rc6. DeLano Scientific, San Carlos, CA, http://www.pymol.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.