

Abstract
In vivo imaging using two-photon microscopy 1 in mice that have been genetically engineered to express fluorescent proteins in specific cell types 2-3 has significantly broadened our knowledge of physiological and pathological processes in numerous tissues in vivo4-7. In studies of the central nervous system (CNS), there has been a broad application of in vivo imaging in the brain, which has produced a plethora of novel and often unexpected findings about the behavior of cells such as neurons, astrocytes, microglia, under physiological or pathological conditions 8-17. However, mostly technical complications have limited the implementation of in vivo imaging in studies of the living mouse spinal cord. In particular, the anatomical proximity of the spinal cord to the lungs and heart generates significant movement artifact that makes imaging the living spinal cord a challenging task.
We developed a novel method that overcomes the inherent limitations of spinal cord imaging by stabilizing the spinal column, reducing respiratory-induced movements and thereby facilitating the use of two-photon microscopy to image the mouse spinal cord in vivo. This is achieved by combining a customized spinal stabilization device with a method of deep anesthesia, resulting in a significant reduction of respiratory-induced movements. This video protocol shows how to expose a small area of the living spinal cord that can be maintained under stable physiological conditions over extended periods of time by keeping tissue injury and bleeding to a minimum. Representative raw images acquired in vivo detail in high resolution the close relationship between microglia and the vasculature. A timelapse sequence shows the dynamic behavior of microglial processes in the living mouse spinal cord. Moreover, a continuous scan of the same z-frame demonstrates the outstanding stability that this method can achieve to generate stacks of images and/or timelapse movies that do not require image alignment post-acquisition. Finally, we show how this method can be used to revisit and reimage the same area of the spinal cord at later timepoints, allowing for longitudinal studies of ongoing physiological or pathological processes in vivo.
Keywords: Neuroscience, Issue 59, Spinal cord imaging, in vivo two photon microscopy, axons, microglia, blood vessels
Protocol
1. Building the spinal stabilization device
Order the Narishige STS-A Compact Spinal Cord Clamps and the Narishige MA-6N head holding adaptor.
Custom design and make a stainless steel base plate to hold the two Narishige parts in alignment so that the animal's head is supported while its spinal column and tail are clamped. Keep in mind that the entire device should fit under the microscope lens usually on a lowered microscope stage.
2. Animal surgery
Pre-heat the microscope chamber with an air heating device and maintain it at 37 °C.
Weigh the animal and make the anesthetic mix using 100 mg ketamine, 15 mg xylazine and 2.5 mg acepromazine per kg of body weight, diluted in 0.9% NaCl solution under sterile conditions.
Anesthetize the animal by injecting the anesthetic mix (KXA) intraperitoneally. Regular toe pinching should be performed to ensure deep anesthesia throughout the experiment. Supplement with half the original dose hourly or as needed.
Use a small animal heating pad to provide thermal support while performing the surgical procedure.
Apply artificial tears ointment over the eyes to prevent corneal dehydration and damage.
Shave the back of the animal first with a trimming device and then with a sharp blade to completely remove hair from the area around the incision.
Remove shaved hairs and disinfect the shaved skin surface with an alcohol pad.
Perform a small (~1.5 cm) longitudinal incision of the skin over the desired spinal level (thoracic vertebrae shown here).
Expose the back musculature by carefully retracting the subcutaneous connective tissue.
Carefully separate the paravertebral muscles from the spinal column at the desired level. Perform the fewest possible incisions to detach the muscles from both sides of each spinous process and then scrape the detached muscles away from the laminar surface.
Choose the lamina that will be removed and prepare the sites of entry for the spinal clamps on each side of the spinal column, immediately rostral and caudal to the laminectomy. Carefully displace muscular tissue to make four pockets where the clamps will be inserted.
Lift the spinal column with the straight toothed-tip forceps to allow safe insertion of the curved blunt-tip scissors under the lamina without touching the spinal cord. Perform the laminectomy by slowly cutting the bone on one side and then on the other side.
Use a cotton swab or insert precut small gelfoam absorbable gelatin sponge pieces next to the incisions to limit minor bleeding. Use a small vessel cauterizer to control further bleeding if necessary. Use preheated sterile artificial cerebrospinal fluid (ACSF) to wash tissue.
3. Stabilization of the spinal column and preparation for in vivo imaging
Place the animal on an elevation pad on the base plate of the spinal stabilization device and secure the head in the head holding adaptor.
Place the spinal clamps of the STS-A device along the anterior-posterior axis of the animal in the pre-prepared pockets flanking the laminectomy (Figure 1). The two clamps should be placed at an angle of ~45° relative to the animal's rostro-caudal axis to allow enough space for lowering a water immersion lens over the exposed spinal cord (Figure 1).
Place the third clamp of the STS-A device at the base of the tail so the animal's body can be suspended in the air after removal of the elevation pad for the duration of the imaging experiments (Figure 1).
Build a small well of Gelseal (Amersham Biosciences Corp.) around the exposed spinal cord to facilitate the maintenance of the spinal cord in ACSF and the immersion of the microscope lens in this solution for in vivo imaging.
4. In vivo imaging of the mouse spinal cord with two-photon microscopy
Transfer the animal on the spinal stabilization device inside the preheated chamber covering the microscope and secure it on a lowered microscope stage placing the laminectomy straight under the lens (Figure 1).
Lower a water-immersion lens carefully into the ACSF solution making sure that it does not touch the spinal clamps or the Gelseal.
Use epifluorescence to identify the area of interest and focus on it. Switch to laser scanning mode and perform in vivo imaging using the appropriate two-photon laser excitation wavelength, dichroics and bandpass filters for the fluorophores present in the imaged tissue.
5. Repetitive imaging and post-operative care
At the end of imaging experiments, remove the mouse from the spinal stabilization device and carefully wipe the Gelseal from the area around the laminectomy. Clean the area well.
Restore and suture the back muscles over the laminectomy.
Restore and suture the skin over the laminectomy, swab it with betadine.
Provide 1 ml Lactated Ringers solution (Baxter Healthcare) as a nutrient and hydration supplement, as well as analgesic treatment subcutaneously (0.1 mg/kg buprenorphine).
Administer antiseptic treatment intraperitoneally (0.03 ml per mouse, 2.27% enrofloxacin injectable antibacterial solution).
Place animal on a heating pad until full recovery from anesthesia and subsequently house it individually.
Repeat antiseptic administration daily for the first 3-5 days after surgery and analgesic treatment every 8-12 hours for 2-3 days post-operatively. Monitor animal daily to ensure normal behavior and full recovery.
For re-imaging through the same laminectomy, reopen the sutured skin and muscles and repeat the steps described in sections 2 - 4.
Re-locate the previously imaged area by using the blood vasculature as a map as previously described 10.
6. Representative results
All animal procedures were performed under the guidelines set by institutional Animal Care and Use Committees at the University of California, San Francisco and are in accordance with Federal regulations. A picture of the spinal stabilization device and a schematic showing the positioning of a mouse on the device under a microscope lens is shown in Figure 1. Allowing adequate room for breathing movements underneath the animal's body ensures stable in vivo imaging in the spinal cord. Figure 2 shows the close relationship between microglia and the vasculature as it was imaged in vivo in the spinal cord of Cx3cr1GFP/+ transgenic mice18, in which microglia are endogenously labeled with GFP. Figure 3 shows examples of repetitive in vivo imaging as it was performed in the same spinal cord areas in mice expressing a fluorescent protein in spinal axons (YFP-H line 3) and microglia (in the Cx3cr1GFP/+ mice).
 Figure 1. Stabilization of the mouse spinal column for in vivo imaging using two-photon microscopy. (A) A custom-made steel base plate is used to support and align the STS-A Narishige compact spinal cord clamps and the MA-6N Narishige head holding adaptor as shown here. (B) Proper positioning of adult transgenic mice anesthetized with a KXA anesthetic on the spinal stabilization device. The insert shows the placement of the spinal clamps immediately rostrally and caudally to the laminectomy and the exposed spinal cord tissue.
Figure 1. Stabilization of the mouse spinal column for in vivo imaging using two-photon microscopy. (A) A custom-made steel base plate is used to support and align the STS-A Narishige compact spinal cord clamps and the MA-6N Narishige head holding adaptor as shown here. (B) Proper positioning of adult transgenic mice anesthetized with a KXA anesthetic on the spinal stabilization device. The insert shows the placement of the spinal clamps immediately rostrally and caudally to the laminectomy and the exposed spinal cord tissue.
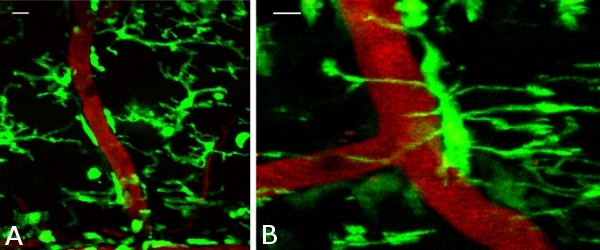 Figure 2. In vivo imaging of high density microglial cells and blood vessels in the spinal cord of anesthetized mice. Projected but non-aligned z-stacks of (A) highly dense GFP-positive microglia (green) in the spinal cord of a CX3CR1GFP/+ mouse in close proximity with blood vessels (red, labeled with i.v. injection of rhodamine dextran). (B) High magnification image labeled as in (A) of a single microglial cell attached to the wall of a blood vessel with processes extended around the vessel and towards the spinal cord parenchyma. Scale bars, 10μm.
Figure 2. In vivo imaging of high density microglial cells and blood vessels in the spinal cord of anesthetized mice. Projected but non-aligned z-stacks of (A) highly dense GFP-positive microglia (green) in the spinal cord of a CX3CR1GFP/+ mouse in close proximity with blood vessels (red, labeled with i.v. injection of rhodamine dextran). (B) High magnification image labeled as in (A) of a single microglial cell attached to the wall of a blood vessel with processes extended around the vessel and towards the spinal cord parenchyma. Scale bars, 10μm.
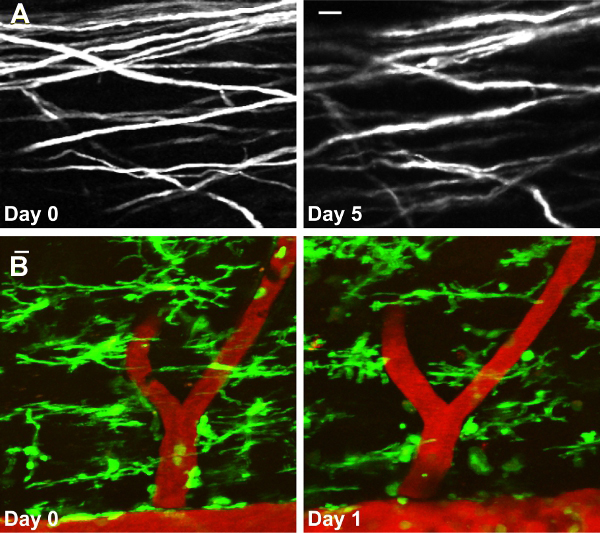 Figure 3. Repetitive in vivo imaging of the same axonal segments and microglia as they were relocated and reimaged in the spinal cord of anesthetized mice on different days. (A) YFP-labeled axons on days 0 and 5. (B). The same vascular structures and microglial cells around them imaged in vivo in the spinal cord of Cx3cr1GFP/+ mice on days 0 and 1. Scale bars, 10μm.
Figure 3. Repetitive in vivo imaging of the same axonal segments and microglia as they were relocated and reimaged in the spinal cord of anesthetized mice on different days. (A) YFP-labeled axons on days 0 and 5. (B). The same vascular structures and microglial cells around them imaged in vivo in the spinal cord of Cx3cr1GFP/+ mice on days 0 and 1. Scale bars, 10μm.
Movie 1. Representative timelapse sequence acquired in vivo from the mouse spinal cord. This sequence shows in detail fine microglial process dynamics (green) and their interactions with the vasculature (red, labeled with i.v. injection of rhodamine dextran) over time within a tissue densely populated with fluorescent structures. The raw images were only corrected for background noise, brightness and contrast and the timelapse movie was constructed by z-projecting sequentially acquired image stacks, without image alignment, averaging or z-selection of individual planes. Blood vessels go through a similar range of z planes as the images of microglia. Z plane depth: 38 μm. Click here to watch the movie.
Movie 2. Timelapse sequence demonstrating the raw stability of the imaging method at the level of a single z-plane. Fast acquisition of the same single z-plane in the spinal cord of CX3CR1GFP/+ mice placed on the spinal stabilization device under KXA anesthesia, demonstrates minimal image displacement between consecutive frames at a scanning rate of 1 frame/s that is faster than the breathing rate of the mouse. The minor residual displacement is probably due to heartbeat. Click here to watch the movie.
Discussion
The method described here allows for stable and repetitive in vivo imaging of densely populated fluorescent cellular structures in the spinal cord of anesthetized mice using two-photon microscopy. The achieved stability is a result of a custom-made spinal stabilization device and an anesthetic regimen that reduces respiratory-induced movement artifact. The spinal stabilization device allows breathing space underneath the mouse body and can be built using commercially available spinal clamps and head mounting piece (Fig. 1). The method is highly reproducible, minimally invasive and consistently generates raw data that can be used for experimental analyses without extensive image post-processing (Fig.2 and Video 1). This technique can therefore be used for studies of cell-cell interactions in the abundance of commercially available fluorescent mouse lines (Figs. 1-3 and Video 1). This method can resolve not only densely populated cellular structures but also rapidly occurring cellular functions or responses (Videos 1, 2). For example, it can be used to quantitatively measure microglial process dynamics over time as well as distance travelled during chemotactic behavior. Quantification can be performed as we previously described for imaging microglial responses in the living brain8. Moreover, it can be used in combination with other experimental approaches such as the use of fluorescently labeled microspheres to measure microglial phagocytosis in the spinal cord in vivo.
The ability to perform repetitive in vivo imaging of the exact same area in the spinal cord of the same animals on different days allows studying the progression of biological phenomena and the dissection of the sequence of events that could for example be involved with the development of disease. Successful application of this method for longitudinal studies relies on the availability of tissue landmarks for reliably relocating the areas that were previously imaged. This needs to be taken into account in particular for spinal cord injury studies. For example in some of the commonly used spinal cord injury models such as spinal cord contusion or dorsal hemisection the massive necrotic lesion that is generated at the epicenter of the injury causes significant axonal and vascular rearrangement that might hinder relocation of the previously imaged area. This can be overcome by either performing repetitive in vivo imaging at the lesion edge or by selecting an injury model that causes a targeted, smaller lesion, such as the thin needle transection model19.
The method described here exposes a small segment of the spinal cord, by performing a single laminectomy over the target imaging area. The underlying dura matter has been left intact where possible. This ensures minimal pertubation of the imaged tissue underneath the meninges and minimizes the injury incurred to the animal's spine. The overall stabilization scheme can easily be adapted to image either through the interlaminar space without performing a laminectomy 20 or through multiple serial laminectomies if a smaller or a larger imaging window is preferred for a given study respectively.
As is common with most in vivo techniques, the results obtained by using this in vivo imaging method may greatly depend on the use of proper anesthesia. The KXA anesthetic mix that is recommended here has also been used in imaging studies of different tissues before 21-24 and was selected exclusively based on its ability to significantly reduce breathing movements. Other anesthetic approaches may produce comparable results to this KXA mix.
The spinal cord imaging technique described in this protocol provides a powerful tool for spinal cord research, since the ability to record cell-cell interactions in real time can greatly facilitate in vivo studies of the spinal cord in physiology and pathology.
Disclosures
We have nothing to disclose.
Acknowledgments
This work was supported by the National Multiple Sclerosis Society grant RG4595A1/T to DD and the NIH/NINDS grants NS051470, NS052189 and NS066361 to K.A. Figures and movies adapted and/or reprinted from Davalos et al., J Neurosci Methods. 2008 Mar 30;169(1):1-7 Copyright 2008, with permission from Elsevier.
References
- Denk W, Strickler JH, Webb WW. Two-photon laser scanning fluorescence microscopy. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- Tsien RY. The green fluorescent protein. Annu. Rev. Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- Feng G. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Denk W. Deep tissue two-photon microscopy. Nat. Methods. 2005;2:932–940. doi: 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- Germain RN, Miller MJ, Dustin ML, Nussenzweig MC. Dynamic imaging of the immune system: progress, pitfalls and promise. Nat. Rev. Immunol. 2006;6:497–507. doi: 10.1038/nri1884. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kerschensteiner M. In vivo imaging of the diseased nervous system. Nat. Rev. Neurosci. 2006;7:449–463. doi: 10.1038/nrn1905. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Yasuda R. Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron. 2006;50:823–839. doi: 10.1016/j.neuron.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Davalos D. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Grutzendler J, Kasthuri N, Gan WB. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420:812–816. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Denk W, Kleinfeld D, Tank DW. In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature. 1997;385:161–165. doi: 10.1038/385161a0. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- Wang X. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat. Neurosci. 2006;9:816–823. doi: 10.1038/nn1703. [DOI] [PubMed] [Google Scholar]
- Christie RH. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed by in vivo multiphoton microscopy. J. Neurosci. 2001;21:858–864. doi: 10.1523/JNEUROSCI.21-03-00858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat. Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Grutzendler J, Gan WB. Two-photon imaging of synaptic plasticity and pathology in the living mouse brain. NeuroRx. 2006;3:489–496. doi: 10.1016/j.nurx.2006.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Han X, Deane R, Zlokovic B, Nedergaard M. Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer's disease. Ann. N. Y. Acad. Sci. 2007;1097:40–50. doi: 10.1196/annals.1379.004. [DOI] [PubMed] [Google Scholar]
- Jung S. Analysis of Fractalkine Receptor CX3CR1 Function by Targeted Deletion and Green Fluorescent Protein Reporter Gene Insertion. Mol. Cell. Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat. Med. 2005;11:572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- Kim JV. Two-photon laser scanning microscopy imaging of intact spinal cord and cerebral cortex reveals requirement for CXCR6 and neuroinflammation in immune cell infiltration of cortical injury sites. J. Immunol. Methods. 2010;352:89–100. doi: 10.1016/j.jim.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakhar G. Stable T cell-dendritic cell interactions precede the development of both tolerance and immunity in vivo. Nat. Immunol. 2005;6:707–714. doi: 10.1038/ni1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadokoro CE. Regulatory T cells inhibit stable contacts between CD4+ T cells and dendritic cells in vivo. J Exp Med. 2006;203:505–511. doi: 10.1084/jem.20050783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist RL. Visualizing dendritic cell networks in vivo. Nat. Immunol. 2004;5:1243–1250. doi: 10.1038/ni1139. [DOI] [PubMed] [Google Scholar]
- Schwickert TA. vivo imaging of germinal centres reveals a dynamic open structure. Nature. 2007;446:83–87. doi: 10.1038/nature05573. [DOI] [PubMed] [Google Scholar]