

Abstract
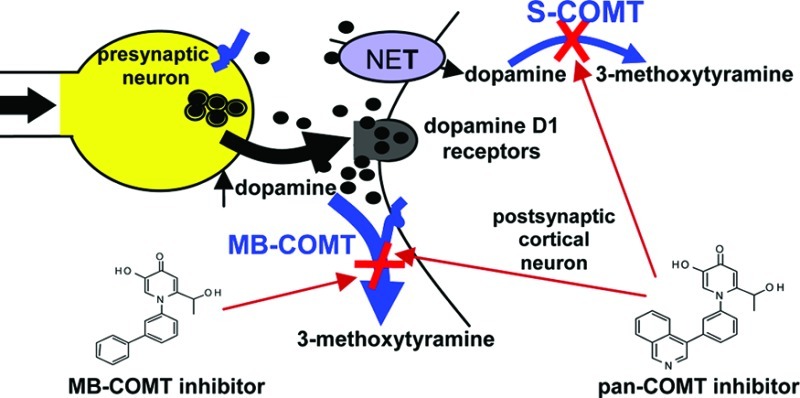
Reduced dopamine neurotransmission in the prefrontal cortex has been implicated as causal for the negative symptoms and cognitive deficit associated with schizophrenia; thus, a compound which selectively enhances dopamine neurotransmission in the prefrontal cortex may have therapeutic potential. Inhibition of catechol-O-methyltransferase (COMT, EC 2.1.1.6) offers a unique advantage, since this enzyme is the primary mechanism for the elimination of dopamine in cortical areas. Since membrane bound COMT (MB-COMT) is the predominant isoform in human brain, a high throughput screen (HTS) to identify novel MB-COMT specific inhibitors was completed. Subsequent optimization led to the identification of novel, non-nitrocatechol COMT inhibitors, some of which interact specifically with MB-COMT. Compounds were characterized for in vitro efficacy versus human and rat MB and soluble (S)-COMT. Select compounds were administered to male Wistar rats, and ex vivo COMT activity, compound levels in plasma and cerebrospinal fluid (CSF), and CSF dopamine metabolite levels were determined as measures of preclinical efficacy. Finally, novel non-nitrocatechol COMT inhibitors displayed less potent uncoupling of the mitochondrial membrane potential (MMP) compared to tolcapone as well as nonhepatotoxic entacapone, thus mitigating the risk of hepatotoxicity.
Keywords: Catechol-O-methyltransferase, high throughput screen, dihydroxyphenylacetic acid, homovanillic acid, fluorescence polarization and hepatotoxicity
Schizophrenia is a chronic, debilitating disease estimated to occur in 1% of the population.1 Effects of the disease are manifested in patients as any one or combination of three symptom domains; positive symptoms, negative symptoms and cognitive deficit.2
The negative symptoms and cognitive impairment present in individuals with schizophrenia represent an unmet medical need.3,4 Reduced dopamine neurotransmission in the prefrontal cortex (PFC) has been implicated as causal for these symptoms.5−7 Catechol-O-methyltransferase (COMT) is the preferential mechanism for catabolism of dopamine in cortical areas.8−11 The enzyme exists as two major isoforms where the membrane (MB) form is more predominant in human brain and the soluble (S) form is more predominant in peripheral tissues.12−15 Preferential expression of MB versus S-COMT in human brain is an evolutionary change and in contrast to rat; where S-COMT predominates.16 The only difference between human MB and S-COMT is the inclusion of an extra 50 hydrophobic amino acids (43 in rat) in MB-COMT. As a critical player in the manipulation of cortical dopamine levels, inhibition of COMT may serve as a promising adjunct therapy for schizophrenia.9,17
There are numerous functional single nucleotide polymorphisms (SNPs) in COMT. The majority of human studies have focused on a SNP common in the COMT gene which leads to the replacement of valine (Val) with methionine (Met) at codon 108 of S-COMT and codon 158 of MB-COMT.18 Val-COMT isolated from human PFC is ∼40% more catalytically active in vitro than enzyme containing the Met allele.19 Various investigations indicate that healthy controls with the Met allele perform better on certain working memory tasks,20−22 but not always.23 Improvements on working memory tasks have been reported in Val 158 homozygote healthy subjects treated with the COMT inhibitor tolcapone.24,25 However, based on a number of haplotypes that influence COMT expression and activity, it seems likely that associated phenotypes result from a multifaceted set of genetic variations.26
The nitrocatechol COMT inhibitors tolcapone and entacapone are successfully utilized as an adjunct therapy for Parkinson’s disease, where inhibition of COMT in the periphery improves the bioavailability of L-dopa in brain. Despite successful clinical use, idiosyncratic toxicity is associated with tolcapone27 and although entacapone is not associated with hepatotoxicity; like tolcapone, its use is sometimes limited by severe diarrhea due to peripheral inhibition.28 Inhibition of S-COMT in the periphery has been proposed to contribute to tolcapone linked hepatotoxicity,17 thus highlighting an advantage of identifying and characterizing MB-COMT specific inhibitors.
Due to the idiosyncratic hepatotoxicity associated with tolcapone and adverse effects associated with peripheral inhibition of COMT, we sought to identify a non-nitrocatechol, brain penetrant MB-COMT inhibitor for the treatment of schizophrenia.29 Additionally, to alleviate side-effects associated with peripheral inhibition, the human brain predominant isoform MB-COMT was utilized to conduct an HTS and complete lead optimization efforts to discover novel, non-nitrocatechol COMT inhibitors. Reported here is the identification and characterization of a series of 4-pyridinone compounds, some of which specifically or preferentially inhibit MB-COMT.
Results and Discussion
COMT purification analysis
Protein expression and purification methods were conducted to individually purify human MB-COMT, human S-COMT, rat MB-COMT, and rat S-COMT. Although S-COMT is soluble in the absence of detergent, all COMT preparations were prepared with detergent to avoid any false positive hits due to the presence of detergent. Additionally, preparation of S-COMT in the absence of detergent led to similar IC50 values when a subset of compounds were screened (data not shown). The recombinant COMT samples were visually compared following gel electrophoresis using staining or Western blotting. The protein bands observed on the stained gel indicated that isoform specific COMT was purified (Figure 1A). S-COMT was not detected in the human or rat MB-COMT and similarly MB-COMT was not observed in the human or rat S-COMT purifications. To affirm the homogeneity of the purified COMT samples, the recombinant proteins were also analyzed by Western blotting using an antibody with a similar affinity for MB and S-COMT (Figure 1B). These results substantiated that each of the recombinant pools were composed of only one of the COMT forms. Additionally, light scattering as well as gel filtration revealed COMT as unaggregated (data not shown) and in vitro enzyme assays demonstrated that all of the COMT forms exhibited catalytic activity.
Figure 1.
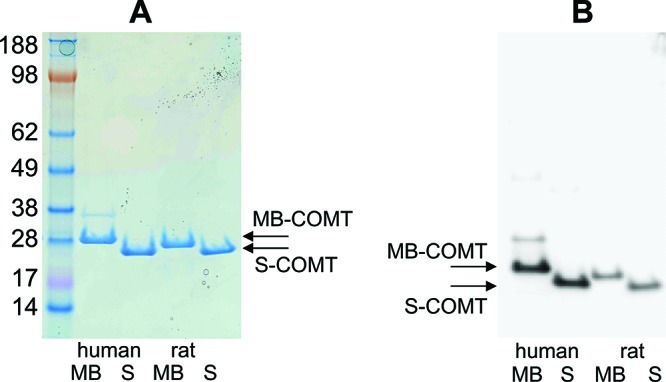
(A) 3.5 μg recombinant proteins were resolved on a 4–12% Bis-Tris gel and stained with Instant Blue. From left to right: SeeBlue Plus2 Pre-Stained standard (molecular weight in kDa, Invitrogen), human MB-COMT (M51A, V158) with 3′ EK and 6-HIS, recombinant human S-COMT (V158) with 3′ EK and 6-HIS, recombinant rat MB-COMT (M44A) with 3′ 10-HIS and recombinant rat S-COMT with 3′ 10-HIS. (B) ∼150 ng recombinant proteins were resolved on 4–12% Bis-Tris gels and subjected to Western blotting with a primary antibody to COMT from Millipore (AB5873).
COMT Inhibitor Characterization
As part of our drug discovery effort to treat cognitive deficit and negative symptoms associated with schizophrenia, an in vitro fluorescence polarization (FP) assay30 was established to define the ability of novel compounds to inhibit the recombinant COMT forms. There are a number of advantages to the FP assay versus previously described HTS assays such as the fluorescence based esculetin assay,31 including use of a long-wavelength rhodamine fluorophore, TAMRA, to avoid interference from fluorescent compounds as well as use of a physiologically relevant substrate. A series of 4-pyridinones possessing robust MB-COMT inhibitory activity were identified, and IC50 values are presented in Table 1 (compounds 1–14). IC50 values versus human MB-COMT ranged from 41 to 291 nM for this group of compounds. These 4-pyridinone compounds were also potent rat MB-COMT inhibitors. Compound IC50 values against the rat enzyme varied from 13 nM to 388 nM (Table 1).
Table 1. Tolcapone and Novel Compound in Vitro IC50 Results (nM) versus Recombinant COMT Forms As Determined Using Fluorescence Polarization Based Assaysa.

Displayed are mean IC50 values ± SEM (n); where applicable.
Relative to MB-COMT, the ability to inhibit S-COMT proved more variable among the novel compounds. IC50 values for compounds 3, 4, 8, 11, and 12 as determined versus human S-COMT were within 3-fold of their human MB-COMT IC50 values. Therefore, these compounds are deemed pan-COMT inhibitors. Compounds 2, 7, and 10 are considered MB-COMT specific since the human and rat S-COMT IC50 values were all >5000 nM in the FP assay. Compounds 1, 9, 13, and 14 are MB-COMT preferring compounds since potencies in FP assays decreased at least 5-fold (compounds 9 and 13) or at least 17-fold (compounds 1 and 14) versus human S-COMT. Inhibition potency versus S-COMT was not defined for compounds 5 and 6 since these compounds selectively served as substrates for rat and human S-COMT (see COMT substrate evaluation below).
We find it intriguing that compounds identified here may either selectively or preferentially inhibit human and rat MB-COMT over human and rat S-COMT. We are unaware of the existence of COMT isoform specific non-nitrocatechol inhibitors. The ability of these novel molecules to differentiate between MB and S-COMT is somewhat surprising considering these two enzymes possess identical catalytic domains and the lone difference is a 50 amino acid hydrophobic region present on MB-COMT. The molecular interactions between MB-COMT and the new compounds remain uncharacterized until an MB-COMT crystal structure becomes available. The fact that we identified MB-COMT specific compounds provides a possibility for reducing the adverse effects associated with pan-COMT inhibition, but clinical data demonstrating efficacy and superior tolerability of MB-COMT specific inhibition versus pan-COMT inhibition is necessary.
Tolcapone and related methylated derivatives were additionally evaluated versus the recombinant COMT enzymes (Table 1). Compared to the aforementioned non-nitrocatechol compounds, tolcapone ranked as the most potent inhibitor of human MB-COMT. Subsequent studies demonstrated that rather than behaving as a true inhibitor, tolcapone served as a substrate for the other COMT forms (described below). 4-methoxy-tolcapone was a pan-COMT inhibitor while 3-methoxy-tolcapone had no effect on human and rat MB or S-COMT activity.
COMT Substrate Evaluation
Dopamine was excluded in a modified version of the FP assay to provide a means of determining whether compounds served as COMT substrates. In Figure 2, each of the recombinant COMT forms was incubated with a 5 μM concentration of novel compound sans dopamine and the conversion of S-adenosyl-l-methionine (SAM) to S-adenosyl-l-homocysteine (SAH) was monitored over time. Routine FP assays which included COMT and 2 μM dopamine, but excluded novel compounds, provided positive controls of product formation. In the absence of dopamine, a decrease in the polarization of the SAH antibody/S-adenosyl-l-cysteine (SAC) TAMRA tracer complex indicated SAH production and therefore compound methylation. No accumulation of SAH was detected when compounds 1 and 2 were tested in these assays. This confirmed that these two compounds were not utilized as substrates by any of the four COMT forms. The aminopyridinone compounds 5 and 6 were unusual because they were potent MB-COMT inhibitors, but also served as S-COMT substrates. SAH increase was not evident when the compounds were tested versus human and rat MB-COMT. However, compounds 5 and 6 (like dopamine) resemble traditional enzyme substrates when exposed to human or rat S-COMT as a gradual accumulation of product formation was observed over time until presumably the substrate and/or cofactor was exhausted. In particular, these two molecules were efficient substrates for rat S-COMT as 5 μM concentrations of the compounds led to a more rapid depletion of SAM than was observed using 2 μM dopamine.
Figure 2.

Modified fluorescence polarization based time course assays were used to identify novel compounds as COMT substrates. A 5 μL mix comprising 40 μM compound stocks, 8 μM SAM, and 40 mM MgCl2 in assay buffer was placed into assay wells (black 96-well round-bottom polystyrene plates from Costar; catalog # 3792). Enzyme reactions were initiated at room temperature by adding 35 μL of one of the COMT forms (1–6 ng of protein) also diluted into assay buffer. The final concentrations of the test compounds and SAM were 5 and 1 μM, respectively. Additional assays replacing the compounds with a final 2 μM concentration of dopamine were also included as a positive COMT substrate reference. The enzyme assays were quenched at various time points with 5 μL of 250 mM EDTA and subsequently 20 μL of a preformed complex containing SAC TAMRA tracer and anti-SAH antibody was added (described in the Methods section). The extent of SAH production was used to evaluate compounds as COMT substrates. Increasing levels of SAH inversely lowered polarization measurements of the SAC TAMRA/SAH antibody complex by displacing the fluorescence label from the antibody. Graphs A through D show time course assay results using 2 μM dopamine or 5 μM novel compounds as substrates, respectively, for human MB-COMT, rat MB-COMT, human S-COMT, and rat S-COMT.
Tolcapone was also analyzed as a substrate for the recombinant COMT preparations. Graphs A through D in Figure 3 compare 1 μM tolcapone and 2 μM dopamine as substrates in FP time course assays. No substantial decrease in polarization was observed by incubating tolcapone with human MB-COMT indicating the drug is not an effective substrate for this COMT form. At the onset of combining tolcapone with human S-COMT or rat MB-COMT a modest increase in SAH levels was observed suggesting a limited capability to be utilized as a substrate. However, the rates of SAH accumulation visualized following the combination of tolcapone with rat MB-COMT or human S-COMT become notably diminished prior to the depletion of substrate or SAM. Evidently, it is not simply the accumulation of methylated tolcapone which explains the time dependent reduction of rat MB-COMT or human S-COMT enzyme activity. A possible explanation comes from studies demonstrating that COMT inhibition potency of tolcapone increases the longer the enzyme and drug are allowed to interact and that tolcapone and COMT combine in a tight binding manner.32 Taken together, this may initially allow for some product formation before enzyme catalytic activity ceases. Conversely, rat S-COMT incubated with tolcapone caused a substantial rapid decrease in assay polarization values. Based on the velocity of SAH accumulation, it appears that 1 μM tolcapone serves more readily as a substrate for rat S-COMT compared to dopamine tested at twice the concentration.
Figure 3.

Modified fluorescence polarization (FP) based time course assays to evaluate tolcapone and methylated derivatives as COMT substrates. (A–D) 1 μM tolcapone or 2 μM dopamine provided as substrates for human MB-COMT, rat MB-COMT, human S-COMT, or rat S-COMT. (E) 1 μM 4-methoxy-tolcapone or 1 μM 3-methoxy-tolcapone provided as substrates for rat S-COMT.
In hopes of utilizing a nonsubstrate tool compound, methylated versions of tolcapone were also evaluated. 4-methoxy- and 3-methoxy-tolcapone variants were allowed to react with rat S-COMT and then were examined for substrate potential (Figure 3E). Polarization readings remained essentially stable for the duration of the time course demonstrating that the modified tolcapone derivatives were no longer utilized as rat S-COMT substrates. Unfortunately, subsequent studies with 4-methoxy-tolcapone demonstrated that it had no effect in biomarker assays due to poor pharmacokinetic properties (data not shown). In fact, the principal tolcapone metabolite in vivo is 3-methoxy-tolcapone rather than 4-methoxy-tolcapone.33 Thus, 4-methoxy-tolcapone does not contribute to the in vivo efficacy of tolcapone due in part to unfavorable methylation by COMT at this site as well as poor pharmacokinetic properties once formed.
Biomarker and ex Vivo Assays
Although free levels of dopamine are very low in CSF, the CSF dopamine metabolites homovanillic acid (HVA) and dihydroxyphenylacetic acid (DOPAC) have been used historically to assess central dopaminergic function.34,35 Following central COMT inhibition, HVA levels are expected to decrease and DOPAC increase, since COMT converts DOPAC to form HVA. Thus, DOPAC and HVA serve as preclinical biomarkers of COMT inhibition, while a COMT ex vivo assay in brain, blood, and liver serves as a direct measure of target engagement centrally and peripherally. However, based on the evolutionary progression moving from mouse to human of increased MB-COMT expression,16 we expected, and observed, a small proportion of MB-COMT relative to S-COMT in fractions utilized for the COMT ex vivo rat brain assay. Total protein was utilized in ex vivo assays where analysis of representative fractions by Western blotting revealed that blood appears to solely express S-COMT, and rat brain and liver fractions contained a clear S-COMT majority (Figure 4).
Figure 4.

Representative Western blot demonstrating the ratio of MB to S-COMT in blood (lanes 1–2), brain (lanes 3–4), or liver (lanes 5–6) fractions utilized in the COMT ex vivo assay from male Wistar rat. To demonstrate similar immunoreactivity for rat MB versus S-COMT, equal amounts of recombinant rat MB (lane 7) or S-COMT (lane 8) were also resolved. Immunoblotting with COMT antibody (Santa Cruz-135872, dilution 1:200) revealed immunoreactivity for MB (indicated by upper arrow) and S-COMT (lower arrow).
The effects of tolcapone or novel compounds 3–14 on total COMT activity and CSF dopamine metabolites were examined in male Wistar rats following 1 or 1.5 h treatment as detailed in Table 2. A separate set of vehicle treated rats were processed similarly for each time point and utilized for data normalization (data not shown). Plasma and CSF compound concentrations were also determined following compound administration. Rats treated with 30 mg/kg tolcapone exhibited over a 3-fold increase in CSF DOPAC levels and an 80% reduction of HVA. Changes in CSF dopamine metabolites were significant as determined by two-way analysis of variance (ANOVA) followed by Dunnett posthoc comparison (p < 0.001 versus vehicle), thus demonstrating central COMT inhibition following tolcapone treatment. To directly confirm COMT inhibition, ex vivo assays demonstrated the presence of minimal, if any, COMT activity in tissue and blood samples; p < 0.001 versus vehicle using an unpaired t test. Tolcapone concentrations averaged 65 μM in plasma and 0.9 μM in CSF, which exceed the concentration of compound needed to completely inhibit MB-COMT in vitro. After confirming biomarker and ex vivo enzyme inhibition effects in our assays with tolcapone, we proceeded to analyze the novel non-nitrocatechol compounds in an attempt to establish a similar relationship.
Table 2. Effect of Single Dose Administration of Tolcapone or Novel Non-Nitrocatechol Compounds 3–14 on COMT Activity in Blood, Brain, or Liver as well as CSF DOPAC and HVA Levels in Male Wistar Rata.

Data were normalized to separate vehicle treated rats for each respective time point and data expressed as 100% of control. CSF biomarker values that significantly differed from vehicle were determined using regular two-way ANOVA followed by post-hoc tests using a Dunnett correction factor for individual group comparisons as indicated; *p <0.05, **p < 0.01, ***p < 0.001. A significant effect on COMT ex vivo activity versus vehicle was determined using an unpaired t test as indicated; *p <0.05, **p < 0.01, ***p < 0.001.
Among the novel compounds, 100 mg/kg administration with S-COMT substrate compounds 5 and 6 or 100 mg/kg treatment using the preferential MB-COMT inhibitor compounds 13 and 14 induced significant biomarker changes that were closest to those observed with tolcapone (Table 2; at least p < 0.05). In the ex vivo assay, each of these compounds resulted in >65% COMT inhibition in the periphery that was significant to at least p < 0.05 by unpaired t test analysis. Despite significant changes in CSF biomarkers for these four compounds, ex vivo COMT activity in brain was only significantly affected following administration of compounds 6 and 14 (COMT activity 50.2 ± 11.7 and 48 ± 12.2%, respectively). Administration of compound 14 resulted in a CSF concentration of 400 ± 100 nM, while 52% inhibition was observed in the rat brain COMT ex vivo assay. These results are consistent with a CSF concentration within 2-fold of the average in vitro S-COMT IC50 values. Thus, despite achieving significant effects on CSF biomarkers reminiscent of effects following 30 mg/kg administration of tolcapone, the effect of compounds 5, 6, 13, and 14 on COMT ex vivo activity were less robust than tolcapone.
Assays validated through the use of tolcapone as a tool compound were utilized to demonstrate preclinical efficacy of novel COMT inhibitors. Unlike tolcapone, significant changes in the levels of CSF dopamine metabolites did not necessarily result in the anticipated amount of brain COMT inhibition following administration of the non-nitrocatechol compounds. Changes in biomarker levels that rivaled those observed with tolcapone treatment were observed with compounds 5, 6, 13, and 14. However, rat brain COMT activity remained at 50% or higher relative to the near complete inhibition observed with tolcapone. For compounds 5 and 6, this was despite the fact that CSF levels were nearly 9-fold above in vitro IC50 values for rat MB-COMT. This may in part be explained by an evolutionary species change where the concentration of MB-COMT increases in brain moving from mouse to human.16 Additionally, compounds 5 and 6 are readily metabolized by S-COMT and are less likely to be efficacious in an S-COMT predominant environment. Compounds 13 and 14, which preferably recognize MB-COMT, also produced a response in ex vivo assays that may be explained by the preponderance of S-COMT activity measured in the assay. For example, 400 ± 100 nM of compound 14 in CSF led to 48 ± 12.2% (p < 0.01) brain activity in the ex vivo assay which corresponded more favorably with the rat S-COMT IC50 potency range of 881 ± 220 nM than with the rat MB-COMT IC50 of 27 ± 3 nM.
Treatment with pan-COMT inhibitors 3, 4, and 8 resulted in significant changes in CSF biomarkers, while administration of pan-COMT inhibitor compounds 11 and 12 did not. Plasma exposure for compounds 11 and 12 exceeded the IC50 values for both rat MB and S-COMT and a significant change in measurements of peripheral COMT activity were observed. However, CSF concentrations for both compounds (∼200 nM) were below in vitro rat S-COMT IC50 values and no effect on brain COMT ex vivo activity was observed for compound 11. Based on lower in vitro S-COMT IC50 values, compound 12 effects on ex vivo COMT brain activity were not determined. In addition to significant effects on CSF dopamine metabolite levels, administration of compounds 3, 4, and 8 significantly reduced ex vivo COMT activity in blood and liver. In line with these data, plasma concentrations for compounds 3, 4, and 8 (18.2 ± 4.5, 15 ± 1 and 8.7 ± 0.5 μM, respectively) exceeded rat S-COMT in vitro IC50 values (483, 565, and 356 nM, respectively). Ex vivo COMT brain activity was significantly decreased following administration of compound 3, but not compound 4. Similarly, CSF concentrations for compound 3 approached the in vitro IC50 value for rat S-COMT (300 ± 90 nM vs 483 nM) while compound 4 CSF concentrations were below the IC50 measurement for rat S-COMT (100 ± 6 nM vs 565 nM).
MB-COMT specific compound 7 significantly increased DOPAC (195 ± 26.5%; p < 0.01) and reduced HVA (41.7 ± 7.5%; p < 0.01), while compound 10 only significantly reduced HVA (70.3 ± 4.1%; p < 0.001). Additionally, compound 10 administration differed from the other non-nitrocatechol test compounds since the dose was 250 mg/kg p.o., whereas the remainder of novel compounds were administered 100 mg/kg i.p. Plasma concentrations (2.9 μM) following compound 10 treatment were the lowest observed and COMT activity was not significantly reduced in blood. Based on low compound levels in CSF, COMT activity in liver and brain were not determined. In contrast, plasma concentration among novel compounds was greatest for compound 7; 321 ± 121 μM while CSF concentration was 3.4 ± 1.1 μM. Compound 7 administration resulted in an ∼40% reduction of COMT activity in blood and brain that was significant (p < 0.05).
In view of the fact S-COMT was the primary isoform in the ex vivo assays, a rational in vitro/in vivo correlation emerged for several of the novel compounds. The pan-COMT inhibitors 3, 4, 8, 11, and 12 achieved plasma concentrations that exceeded their in vitro rat S-COMT IC50 values by at least 20-fold and ex vivo COMT inhibition in blood and/or liver was 85% or better, except for compound 12 (∼50%). CSF concentrations observed for compounds 3 and 4 were near or below their rat S-COMT IC50 potencies so brain COMT inhibition levels observed were reasonable. Compound 7, which was considered MB-COMT specific since in vitro IC50 values versus rat S-COMT were greater than 5 μM, produced weak ex vivo COMT inhibition of ∼40% in brain and blood that was significant. In fact, these data are consistent with plasma and CSF concentrations of compound 7 (321 ± 121 μM and 3.4 ± 1.1 μM, respectively). Compound 10 was also considered MB specific and with plasma levels of 2.9 ± 0.5 μM did not achieve significant inhibition in the ex vivo blood assay since only S-COMT activity is monitored. MB-COMT specific compounds 7 and 10 achieved significant effects on CSF DOPAC and/or HVA thus highlighting the possibility that despite an S-COMT majority ex vivo, the localization of COMT and affinity of dopamine for MB versus S-COMT may also be contributing factors.17,32
Mitochondrial Membrane Potential Assessment
To distinguish novel leads from the hepatotoxic liability associated with tolcapone, novel COMT inhibitors were tested alongside tolcapone and nonhepatotoxic entacapone in MMP assays.28 Rat heart mitochondria were isolated and incubated with multiple concentrations of tolcapone, entacapone, or novel COMT inhibitors to determine the influence on the MMP. Evaluated novel compounds were approximately 1000- or 500-fold less potent than tolcapone and entacapone, respectively. Figure 5 demonstrates that 35–40 nM tolcapone leads to a 50% reduction in membrane potential. In contrast, all of the novel COMT inhibitors tested were considerably less potent uncouplers of MMP compared to tolcapone. Moreover, all novel compounds tested were differentiated from entacapone, which is considered nonhepatotoxic. In fact, concentrations of the new compounds varying from 35 μM to greater than 300 μM were required to reduce the MMP by 50%.
Figure 5.

Effect of tolcapone, entacapone, or novel COMT inhibitors on rat heart mitochondrial membrane potential (MMP). Values listed next to compounds are concentrations that resulted in a 50% reduction of the mitochondrial membrane potential based on a vehicle treatment control representing maximum membrane potential and 3 mM carbonylcyanide m-chlorophenyl hydrazone (CCCP) treatment representing minimum membrane potential.
Therefore, these in vitro data support the idea that non-nitrocatechol COMT inhibitors may be developed with reduced risk for hepatotoxicity. In pilot studies, we compared the tolcapone and entacapone MMP response in mitochondria isolated from rat liver with mitochondria isolated from rat heart (data not shown). In both liver and heart mitochondria, tolcapone was ∼2–5 fold more potent MMP inhibitor when compared to entacapone, although MMP effects were observed at ∼100-fold lower concentrations in heart mitochondria as compared to liver mitochondria. These differences in the response could possibly result due to a greater basal level of coupled mitochondria isolated from heart as compared to liver. The tolcapone MMP response observed in the liver mitochondria was analogous to the response observed by Haasio and co-workers, although for entacapone a more potent response was observed as compared to what was previously reported.36
Conclusions
In summary, our current research led to the discovery of novel, non-nitrocatechol COMT inhibitors with improved safety profiles. These compounds may provide a starting point for the development of a therapeutic that targets negative symptoms and cognitive deficit associated with schizophrenia. We recognize the limitation of using Wistar rats in our preclinical studies due to the predominance of brain S-COMT versus MB-COMT and that this ratio does not accurately reflect the evolutionary species change moving to human where MB-COMT is preferentially expressed in brain.16 Future studies utilizing transgenic rats expressing human MB-COMT on a rat COMT knockout background, S-COMT deficient mice,37 or mice overexpressing human MB-COMT (Val158) (16) are expected to improve correlations between biomarker and ex vivo data when evaluating MB-COMT specific and preferring compounds. Thus, along with the identification and evaluation of additional compounds, future exploratory work with the novel compounds utilizing a preclinical model with preferential MB-COMT brain expression will help determine whether MB-COMT specific or preferring compounds have similar biomarker and MB-COMT brain inhibition effects versus tolcapone.
Methods
Compounds
Tolcapone and entacapone were synthesized by Synfine (Richmond Hill, ON, Canada). 3-Methoxy-tolcapone and 4-methoxy-tolcapone along with novel compounds 1–14 were synthesized at Merck Research Laboratories (West Point, PA).
Preparation of Recombinant COMT
Bacmid expressing human MB-COMT (M51A, V158) with a C-terminal 6 histidine (HIS) tag was utilized to infect Sf-9 insect cells with a multiplicity of infection of 3 for 48 h (pFastBac vector, Invitrogen, Carlsbad, CA). For the preparation of rat MB-COMT (M44A) with a C-terminal 10 HIS tag/pET29b(+) (pET expression vectors, EMD Chemicals Inc., Gibbstown, NJ), human S-COMT (V158) with a C-terminal enterokinase cleavage site and 6-HIS/pET29b(+) and rat S-COMT with C-terminal enterokinase cleavage site followed by a 6-HIS tag/pET29b(+), approximately 50 ng DNA were transformed into BL21-Gold (DE3) or BL21-CodonPlus(DE3)-RIPL (for rat MB-COMT only) according to manufacturer protocols (Stratagene, La Jolla, CA). Colonies were selected for growth in YT media (Teknova, Hollister, CA) supplemented with 10 mM MgCl2, 10 mM MgSO4, 3 mM KCl, and 50 ug/mL kanamycin, or Optigrow Superbroth (Fisher Scientific, Pittsburgh, PA) plus 50 ug/mL kanamycin (for rat MB-COMT only) and incubated at 37 °C until an OD600 of approximately 0.7. Temperature was then reduced to 18 °C (20 °C for rat S-COMT only). When the OD600 reached approximately 0.7, cultures were induced with 0.4 mM IPTG and incubated at 18–20 °C for 16–18 h. Cells were then collected according to standard protocol and supernatant discarded.
Cell pellets were resuspended in Buffer A, pH 7.4 without detergent (50 mM Tris-HCl, pH 7.4, 150–200 mM NaCl, Roche complete, EDTA-free, protease inhibitor tablets (2 tablets/50 mL; Roche Applied Science, Indianapolis, IN), 1–2 mM tris[2-carboxyethyl] phosphine hydrochloride (TCEP, Pierce Biotechnology, Rockford, IL), 30–40 mM imidazole (Fisher Scientific), 2 mM MgCl2 for MB-COMT constructs or with Buffer A plus detergent (1% Fos-choline 12 (FC-12, Anatrace, Maumee, OH, #F308)) for S-COMT constructs. Buffer A contained 25 units of benzonase nuclease (EMD Chemicals Inc.) per milliliter upon resuspension of cells. Pellets were homogenized by hand and then sonicated (power 5) with 10 s pulses for 3–4 min. In some cases, pellets were fluidized once at 14 000 psi. Suspension was collected at 23 800g with an SLA 1500 rotor at 4 °C for 1 h. Supernatant from S-COMT preparations was loaded onto a HIS Trap FF, 5 mL column (GE Healthcare, Piscataway, NJ) prewashed with Buffer A containing detergent using an Äktaxpress (GE Healthcare). Supernatant from MB-COMT preparations was discarded and pellet was resuspended in Buffer A without detergent, sonicated and then collected as above. Supernatant was again discarded and pellet was resuspended with detergent (1% FC-12) and sonicated as detailed above. Insoluble cell debris was removed by centrifugation at 32 000 or 48 000g for 1 h at 4 °C using a JA-20 rotor and then loaded onto a HIS Trap FF, 5 mL column as detailed above. After loading, column was washed with 3 column volumes (CVs) 100% Buffer A with detergent. After a system wash, elution of purified recombinant COMT occurred using a step elution with 50% Buffer B (Buffer A with detergent and 300–500 mM imidazole) for 10 CVs. Start and end level was 40 and 35 milli absorbance units, respectively. Aliquots of Äktaxpress fractions were separated using 4–12% Bis-Tris gels with 1× MES buffer (Invitrogen) as detailed below, stained with Instant Blue (Expedeon, San Diego, CA) and fractions pooled for dialysis. Dialysis on pooled fractions containing recombinant COMT was completed at least 100 fold in Dialysis Buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.125 mM TCEP, 2 mM MgCl2, 5% glycerol and 0.4% FC-12) to remove imidazole. For rat MB-COMT only, Buffer A and B, pH 7.5, contained 50 mM HEPES instead of 50 mM Tris-HCl. Also instead of dialysis, rat MB-COMT containing fractions were pooled and desalted with Buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 2 mM TCEP, 0.5% FC-12) using a 20 mL HiPrep 16/10 desalting column (GE Healthcare).
Western Blotting
Proteins were separated on 4–12% Bis-Tris gel with 1× MES buffer (Invitrogen) according to manufacturer protocol. Samples were heated 10 min at 70 °C prior to loading, except standards. Gels were transferred using an iBlot as directed by the manufacturer (Invitrogen). PVDF membranes were blocked for approximately 1 h at room temperature in blocking solution (5% milk (Bio-Rad, Hercules, CA), 0.1% Tween-20, in 1× TBS (Mediatech, Manassas, VA)) with agitation. Blots were incubated overnight at 4 °C with agitation in 10 mL primary antibody #sc-135872 recognizing COMT at a dilution of 1:200 (Santa Cruz Biotechnology, Santa Cruz, CA) or COMT antibody #AB5873 at a dilution of 1:10 000 (Millipore, Billerica, MA) prepared in blocking solution. Blots were washed three times for 5 min in wash buffer (1× TBS plus 0.1% Tween-20) at room temperature with agitation. Blots were then incubated for 45 min to 1 h at room temperature with agitation in secondary antibody prepared in blocking solution at a 1:2000 dilution (anti-mouse IgG, HRP-linked antibody (#7076) or anti-rabbit IgG, HRP-linked antibody (#7074) from Cell Signaling Technology Danvers, MA. Prior to developing with ECL reagents (Pierce Biotechnology, #32106), blots were washed three times for 5 min in wash buffer with agitation. Blots were then developed using a VersaDoc imager (Bio-Rad) using the chemiluminescent setting.
COMT FP Assay
The activity of the compounds as COMT inhibitors may be readily determined using an FP methodology.30 Compounds had activity in reference assays by exhibiting the ability to inhibit the production of SAH. Any compound exhibiting an IC50 below 1 μM was considered a COMT inhibitor.
To determine IC50 values in the fluorescence polarization assay, 10 mM compound stock in DMSO was used to prepare 10 point 3-fold dilution series and 1 μL of appropriate dilution was plated into assay wells (black 96-well, round-bottom, polystyrene plates from Corning Inc., Corning, NY, Costar # 3792). Recombinant enzyme was diluted in Assay Buffer (100 mM Na2HPO4 pH 7.4, 1 mM DTT, 0.005% Tween-20), and 35 μL of diluted enzyme was added to the assay wells containing 1 μL of diluted compound. The final amount of enzyme in the assay varied between 1 and 6 ng depending on the preparation of COMT utilized. Preincubation of COMT enzyme and compound progressed for 2 h at room temperature. Enzyme reactions were initiated upon the addition of 5 μL of an 8× mix prepared in Assay Buffer containing 8 μM SAM (USB Corporation, Cleveland, OH, # US10601), 16 μM dopamine (Sigma-Aldrich, St. Louis, MO, # H8502), and 40 mM MgCl2. After a 25 min incubation at room temperature, reactions were quenched with 5 μL 250 mM EDTA, pH 8.2. To quenched reactions, 20 μL of a preformed complex containing a 1:80 000 dilution of SAC TAMRA tracer (2 mM from AnaSpec, Fremont, CA) and a 1:20 dilution of anti-S-adenosyl-l-homocysteine antibody (mouse monoclonal from IMX Homocysteine Reagent Pack, Abbott Laboratories, Abbott Park, IL, #7D29-20) was prepared in Assay Buffer B (Na2HPO4 pH 7.2). Prior to combining with quenched enzyme assays, the SAH antibody/SAC TAMRA tracer complex was preformed at room temperature for 30 min while protected from light. Therefore, the final dilution of the SAH antibody/SAC TAMRA mix was 1:60 and 1:240 000, respectively. After a 2.5 h incubation at room temperature, protected from light, fluorescence polarization was measured using a Tecan Safire2 plate reader using an excitation of 530 nm and an emission of 595 nm (Tecan US, Durham, NC). Titration curves and IC50 values were calculated using standard protocols.
Animals
All experimental protocols described in this study were approved by the Merck and Co., Inc. Institutional Animal Care and Use Committee and conducted in accordance with the Guide for Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996). Adult male Wistar rats were used for all in vivo experiments, while Sprague–Dawley rats were utilized as heart donors for mitochondrial membrane potential assays. Animals were maintained on a 12-h light/dark cycle (lights on 7:00 a.m.), and temperature and relative humidity were maintained at 22 to 24 °C and 50 to 55%, respectively.
COMT Biomarker
CSF concentrations of DOPAC and HVA were evaluated in male Wistar rats 7 to 10 weeks of age (220–250 g). Animals were administered test compounds or vehicle via intraperitoneal injection or, in the case of tolcapone and compound 10, by oral gavage and returned to home cages for a specified time determined from historical literature for tolcapone9 or in-house in vitro and pharmacokinetic data (e.g., 1 h, 100 mg/kg for novel compounds). Animals were anesthetized with an inhalant anesthetic (isoflurane delivered at 5% with oxygen at 2 L/min) in an induction chamber. To ensure proper anesthesia, the animals were monitored and assessed by the lack of response to a toe pinch. CSF sampling was completed from the cisterna magna by needle puncture and aspiration using a 25–27 gauge butterfly needle with tubing attached to a syringe. While under anesthesia, rats were euthanized by decapitation using a small animal guillotine. The brain and liver were dissected and flash frozen on dry ice. Blood samples were obtained from the trunk and collected into tubes containing 0.5 M EDTA. CSF (15 μL) was diluted with 25 μL of 0.1 M perchloric acid containing 0.1 μM ascorbic acid and 4 μL of internal standard (100 ng/mL 3,4-dihydroxybenzylamine hydrobromide (DHBA, Sigma-Aldrich) or 3-methoxytyramine hydrochloride (3-MT, Sigma-Aldrich)). Brain monoamine levels were quantitated using high performance liquid chromatography with electrochemical detection (HPLC-ED). Briefly, 20 μL of the diluted sample was injected onto a C18 reverse phase column (MD150, 3 × 100 mm, 3 μm, ESA; Chelmsford, MA) and the dopamine metabolites detected by an ESA Coulochem III controller (model 5200A, ESA) using a model 5020 guard cell (+350 mV) and a model 5014B microdialysis analytical cell (E1, −150 mV; E2, +220 mV). A commercially available mobile phase MD-TM (ESA) was used at a rate of 0.25 mL/min under isocratic conditions and DHBA was added to samples as an internal standard and sample run time was 35 min. The remaining CSF volume was diluted 1:1 with 100% acetonitrile and frozen at −80 °C for pharmacokinetic analysis. Whole blood was centrifuged (3000g for 10 min at 4 °C) and plasma removed and frozen at −80 °C for pharmacokinetic analysis. Red blood cells (following removal of plasma), brain and peripheral organs were processed in the COMT ex vivo assay.
Pharmacokinetics
For the determination of compound levels in CSF or plasma, samples were prepared and analyzed using ultrahigh performance liquid chromatography (UHPLC) combined with triple quadrupole mass spectrometry. Aliquots (50 μL) of control matrix (species specific EDTA plasma or CSF) were added into 0.75 mL tubes in a 96-well format. A 10 μL aliquot of the appropriate intermediate solution of tolcapone (or other related compound) was added to each tube to generate a standard curve and replicate quality control samples. Each standard, quality control and unknown sample was precipitated with (150 μL for plasma and 75 μL for CSF samples) acetonitrile (HPLC grade; Sigma-Aldrich) + 0.1% formic acid (HPLC grade; Sigma-Aldrich) containing labetalol (US Pharmacopeia, Rockville, MD) as the analytical internal standard. The samples were sealed, vortexed for 2 min and collected at 2760g for 10 min. Following centrifugation, a 75 μL aliquot of the extract was transferred to a plate containing 75 μL of water +0.1% formic acid.
The samples were injected onto the UHPLC (Aria LX-2, Thermo Fisher Scientific, Waltham, MA) to determine linearity and reproducibility. Chromatographic separation was performed on a Supelco Ascentis Express (Sigma-Aldrich), C18–2.7 μm, 5 × 3 mm column. The mobile phase consisted of acetonitrile +0.1% formic acid and water +0.1% formic acid using a gradient at a flow rate of 1.0 mL/min. The gradient started at 90% aqueous and held for 60 s, ramped to 100% organic in 60 s, held at that rate for 75 s, followed by a ramp back to 90% aqueous and held for 75 s.
Following compound separation by UHPLC, samples were analyzed using an API 5000 triple quadrupole mass spectrometer (AB Sciex, Foster City, CA) that was operated in the negative/positive ionization mode (MRM). The TurboV electrospray interface was set at an ionspray voltage of ±4000 V. The temperature for the interface was set at 450 °C.
COMT ex Vivo Assay
COMT activity in blood, brain, and liver was evaluated by the ability to methylate norepinephrine (Sigma-Aldrich) to normetanephrine essentially as described38,39 with modifications. Total COMT was obtained from liver or brain by homogenizing with FastPrep 24 Teen D Lysing Matrix tubes (MP Biomedicals, Solon, OH) 1:16 for liver and 1:4 for brain (w/v) in buffer 1 (100 mM sodium phosphate pH 7.4, 1 mM DTT and 5 mM MgCl2) followed by sonication on power 5, 10 s on and 10 s off for a total of five times for brain or eight times for liver. Homogenate was collected at 20 000g for 30 min at 4 °C, and the resulting supernatant used to evaluate COMT activity. Red blood cells were obtained following the removal of plasma and stored at −80 °C without washing and hemolyzed as described previously.38 Aliquots of 100 μL of enzyme were then preincubated for 20 min at 37 °C with 92 μL 1× assay buffer (100 mM sodium phosphate pH 7.4, 1 mM DTT, 5 mM MgCl2 and 100 μM pargyline) for brain and liver or 2× assay buffer for blood. The reaction was brought to a final volume of 200 μL with 0.4 mM norepinephrine and 25 μM SAM for brain and blood or with 0.05 mM norepinephrine and 100 μM SAM for liver. Reactions were incubated at 37 °C for a further 15 min (liver), 15–35 min (brain), or 40 min (blood) and then quenched on ice with 50 μL of perchloric acid. Prior to HPLC-ED, reactions were clarified by filtration (0.22 μm pore, Corning Inc., Costar #8169). Norepinephrine metabolites were separated on a C18 reverse phase column and detected as detailed above except detector conditions were as follows: E1, −150 mV; E2, +360 mV.
MMP Assay
MMP was measured by modification of a previously described fluorometric method40 and rationale.36 Eight week old Sprague–Dawley rats (Charles River Laboratories, Wilmington, MA) served as heart donors. Hearts were removed from the animals rapidly following anesthesia (isoflurane delivered at 5% with oxygen at 2 L/min) and exsanguinations and minced and homogenized in Buffer C (120 mM KCl, 20 mM HEPES, 2 mM MgCl2, 1 mM EGTA and 5 mg/mL BSA). Cellular debris were removed by centrifugation at 600g followed by mitochondria isolation through differential centrifugation at sequential speeds of 17 000g, 6000g, and 3500g. Isolated mitochondria were resuspended in Buffer D (225 mM mannitol, 75 mM sucrose, 10 mM Tris-HCl and 0.1 mM EDTA), and protein concentration was determined by DC Protein Assay (Bio-Rad). Isolated rat heart mitochondria (35 μL; protein concentration of 5 mg/mL) were incubated in Buffer D with 12.5 mM glutamate, 3.125 mM malate, 15 μM safranine O, and selected COMT inhibitors. Additional assays included a vehicle treatment which served as a maximum membrane potential control, while treatment with 3 mM carbonylcyanide m-chlorophenyl hydrazone (CCCP) served as a minimum membrane potential control. The reaction temperature was maintained at 30 °C and fluorescence with excitation of 495 nm and emission of 584 nm was measured for 20 min. A single point of the plateau was chosen to calculate MMP as a percent of total membrane potential using the formula: [(CCCP signal – COMT inhibitor signal)/(CCCP signal – vehicle signal) × 100].
Acknowledgments
The authors would like to thank Paul D. Zuck, Liam Bonnette, Delphine Collin, Justin Murray, Kartik Narayan, Daniel Riley, Sylvie M. Sur, Matthew C. Kuhls, Gary E. Adamson, Kenneth D. Anderson, Susan J. Crathern, Rena Zhang, Dannette M. Pascarella, Faith A. Mullen, Betsy Martin, Deborah L. Dooney, Scott Fauty, Kristin Geddes, and James W. Monahan for their invaluable analytical and/or screening support. We deeply appreciate intellectual and/or COMT purification support from Pravien D. Abeywickrema, Tim Allison, Noel Byrne, Ronald E. Diehl, Rachael E. Ford, Keith Rickert, Jennifer Shipman, and Joan Zugay-Murphy. We thank Scott Ceglia for timely delivery of synthetic intermediates on scale. We would also like to thank Jay A. Grobler, Ming-Tain Lai, and Marlene A. Jacobson for helpful discussions.
Glossary
Abbreviations
- COMT
catechol-O-methyltransferase
- MB-COMT
membrane bound COMT
- S-COMT
soluble COMT
- CSF
cerebrospinal fluid
- HTS
high throughput screen
- IC50
half maximal inhibitory concentration
- MMP
mitochondrial membrane potential
- PFC
prefrontal cortex
- SNP
single nucleotide polymorphism
- DOPAC
dihydroxyphenylacetic acid
- HVA
homovanillic acid
- FP
fluorescence polarization
- SAH
S-adenosyl-l-homocysteine
- SAM
S-adenosyl-l-methionine
Author Contributions
R.G.R., N.A.S., S.M.S., L.Y., M.K., R.F.S., S.E.W., S.T.H., B.W.T., J.C.B., J.M.S., S.S., S.P., C.R.G., N.B., J.L.K., P.J.M., J.J.M., K.K.N., J.W.S., Z.Z., D.M., and P.H.H. participated in research design. R.G.R., N.A.S., S.M.S., L.Y., M.K., R.F.S., S.E.W., S.T.H., B.W.T., J.M.S., S.S., J.L.K., P.J.M., J.J.M., K.K.N., J.W.S., Z.Z., D.M., and D.L.H. contributed new reagents or analytic tools. R.G.R., N.A.S., L.Y., M.K., R.F.S., S.P., C.R.G., J.L.K., T.L.W., S.S., P.J.M., J.J.M., K.K.N., J.W.S., Z.Z., S.T.H., and D.L.H. conducted experiments. R.G.R., N.A.S., S.M.S., L.Y., M.K., R.F.S., S.P., C.R.G., N.B., J.L.K., T.L.W., S.S., S.E.W., B.W.T., J.C.B., J.M.S., S.T.H., and D.L.H. performed data analysis. R.G.R., N.A.S., S.M.S., N.B., J.L.K., J.C.B., C.R.G., D.M., S.E.W., S.T.H., and J.M.S. wrote or contributed to writing the manuscript.
The authors declare the following competing financial interest(s): Upon completion of the work, authors were employees of Merck Sharp & Dohme Corporation.
References
- Saha S.; Chant D.; Welham J.; McGrath J. (2005) A systematic review of the prevalence of schizophrenia. PLoS Med 2, e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratta P.; Daneluzzo E.; Bustini M.; Prosperini P.; Rossi A. (2000) Processing of context information in schizophrenia: relation to clinical symptoms and WCST performance. Schizophr. Res. 44, 57–67. [DOI] [PubMed] [Google Scholar]
- Addington J.; Addington D. (1993) Premorbid functioning, cognitive functioning, symptoms and outcome in schizophrenia. J. Psychiatry Neurosci. 18, 18–23. [PMC free article] [PubMed] [Google Scholar]
- Green M. F. (1996) What are the functional consequences of neurocognitive deficits in schizophrenia?. Am. J. Psychiatry 153, 321–330. [DOI] [PubMed] [Google Scholar]
- Davis K. L.; Kahn R. S.; Ko G.; Davidson M. (1991) Dopamine in schizophrenia: a review and reconceptualization. Am. J. Psychiatry 148, 1474–1486. [DOI] [PubMed] [Google Scholar]
- Seamans J. K.; Gorelova N.; Durstewitz D.; Yang C. R. (2001) Bidirectional dopamine modulation of GABAergic inhibition in prefrontal cortical pyramidal neurons. J. Neurosci. 21, 3628–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Fang Y.; Shen Y.; Xu Q. (2010) Analysis of association between the catechol-O-methyltransferase (COMT) gene and negative symptoms in chronic schizophrenia. Psychiatry Res. 179, 147–150. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A.; Weinberger D. R. (2006) Intermediate phenotypes and genetic mechanisms of psychiatric disorders. Nat. Rev. Neurosci. 7, 818–827. [DOI] [PubMed] [Google Scholar]
- Tunbridge E. M.; Bannerman D. M.; Sharp T.; Harrison P. J. (2004) Catechol-o-methyltransferase inhibition improves set-shifting performance and elevates stimulated dopamine release in the rat prefrontal cortex. J. Neurosci. 24, 5331–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavich L.; Forsberg M. M.; Karayiorgou M.; Gogos J. A.; Männistö P. T. (2007) Site-specific role of catechol-O-methyltransferase in dopamine overflow within prefrontal cortex and dorsal striatum. J. Neurosci. 27, 10196–10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käenmäki M.; Tammimäki A.; Myöhänen T.; Pakarinen K.; Amberg C.; Karayiorgou M.; Gogos J. A.; Männistö P. T. (2010) Quantitative role of COMT in dopamine clearance in the prefrontal cortex of freely moving mice. J. Neurochem. 114, 1745–1755. [DOI] [PubMed] [Google Scholar]
- Tenhunen J.; Salminen M.; Lundström K.; Kiviluoto T.; Savolainen R.; Ulmanen I. (1994) Genomic organization of the human catechol O-methyltransferase gene and its expression from two distinct promoters. Eur. J. Biochem. 223, 1049–1059. [DOI] [PubMed] [Google Scholar]
- Reenilä I.; Männistö P. T. (2001) Catecholamine metabolism in the brain by membrane-bound and soluble catechol-o-methyltransferase (COMT) estimated by enzyme kinetic values. Med. Hypotheses 57, 628–632. [DOI] [PubMed] [Google Scholar]
- Tunbridge E. M.; Harrison P. J.; Weinberger D. R. (2006) Catechol-o-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol. Psychiatry 60, 141–151. [DOI] [PubMed] [Google Scholar]
- Myöhänen T. T.; Schendzielorz N.; Männistö P. T. (2010) Distribution of catechol-O-methyltransferase (COMT) proteins and enzymatic activities in wild-type and soluble COMT deficient mice. J. Neurochem. 113, 1632–1643. [DOI] [PubMed] [Google Scholar]
- Papaleo F.; Crawley J. N.; Song J.; Lipska B. K.; Pickel J.; Weinberger D. R.; Chen J. (2008) Genetic dissection of the role of catechol-O-methyltransferase in cognition and stress reactivity in mice. J. Neurosci. 28, 8709–8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Song J.; Yuan P.; Tian Q.; Ji Y.; Ren-Patterson R.; Liu G.; Sei Y.; Weinberger D. R. (2011) Orientation and cellular distribution of membrane-bound catechol-o-methyltransferase in cortical neurons: implications for drug development. J. Biol. Chem. 286, 34752–34760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachman H. M.; Papolos D. F.; Saito T.; Yu Y. M.; Szumlanski C. L.; Weinshilboum R. M. (1996) Human catechol-O-methyltransferase pharmacogenetics: description of a functional polymorphism and its potential application to neuropsychiatric disorders. Pharmacogenetics 6, 243–250. [DOI] [PubMed] [Google Scholar]
- Chen J.; Lipska B. K.; Halim N.; Ma Q. D.; Matsumoto M.; Melhem S.; Kolachana B. S.; Hyde T. M.; Herman M. M.; Apud J.; Egan M. F.; Kleinman J. E.; Weinberger D. R. (2004) Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am. J. Hum. Genet. 75, 807–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett J. H.; Jones P. B.; Robbins T. W.; Müller U. (2007) Effects of the catechol-O-methyltransferase Val158Met polymorphism on executive function: a meta-analysis of the Wisconsin Card Sort Test in schizophrenia and healthy controls. Mol. Psychiatry 12, 502–509. [DOI] [PubMed] [Google Scholar]
- Egan M. F.; Goldberg T. E.; Kolachana B. S.; Callicott J. H.; Mazzanti C. M.; Straub R. E.; Goldman D.; Weinberger D. R. (2001) Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 98, 6917–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi G.; Mattay V. S.; Bertolino A.; Elvevåg B.; Callicott J. H.; Das S.; Kolachana B. S.; Egan M. F.; Goldberg T. E.; Weinberger D. R. (2005) Effect of catechol-O-methyltransferase val158met genotype on attentional control. J. Neurosci. 25, 5038–5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett J. H.; Scoriels L.; Munafò M. R. (2008) Meta-analysis of the cognitive effects of the catechol-O-methyltransferase gene Val158/108Met polymorphism. Biol. Psychiatry 64, 137–144. [DOI] [PubMed] [Google Scholar]
- Apud J. A.; Mattay V.; Chen J.; Kolachana B. S.; Callicott J. H.; Rasetti R.; Alce G.; Iudicello J. E.; Akbar N.; Egan M. F.; Goldberg T. E.; Weinberger D. R. (2007) Tolcapone improves cognition and cortical information processing in normal human subjects. Neuropsychopharmacology 32, 1011–1020. [DOI] [PubMed] [Google Scholar]
- Giakoumaki S. G.; Roussos P.; Bitsios P. (2008) Improvement of prepulse inhibition and executive function by the COMT inhibitor tolcapone depends on COMT Val158Met polymorphism. Neuropsychopharmacology 33, 3058–3068. [DOI] [PubMed] [Google Scholar]
- Nackley A. G.; Shabalina S. A.; Tchivileva I. E.; Satterfield K.; Korchynskyi O.; Makarov S. S.; Maixner W.; Diatchenko L. (2006) Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science 314, 1930–1933. [DOI] [PubMed] [Google Scholar]
- Assal F.; Spahr L.; Hadengue A.; Rubbia-Brandt L.; Burkhard P. R. (1998) Tolcapone and fulminant hepatitis. Lancet 352, 958. [DOI] [PubMed] [Google Scholar]
- Parashos S. A.; Wielinski C. L.; Kern J. A. (2004) Frequency, reasons, and risk factors of entacapone discontinuation in Parkinson disease. Clin. Neuropharmacol. 27, 119–123. [DOI] [PubMed] [Google Scholar]
- Apud J. A.; Weinberger D. R. (2007) Treatment of cognitive deficits associated with schizophrenia: potential role of catechol-O-methyltransferase inhibitors. CNS Drugs 21, 535–557. [DOI] [PubMed] [Google Scholar]
- Graves T. L.; Zhang Y.; Scott J. E. (2008) A universal competitive fluorescence polarization activity assay for S-adenosylmethionine utilizing methyltransferases. Anal. Biochem. 373, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurkela M.; Siiskonen A.; Finel M.; Tammela P.; Taskinen J.; Vuorela P. (2004) Microplate screening assay to identify inhibitors of human catechol-O-methyltransferase. Anal. Biochem. 331, 198–200. [DOI] [PubMed] [Google Scholar]
- Borges N.; Vieira-Coelho M. A.; Parada A.; Soares-da-Silva P. (1997) Studies on the tight-binding nature of tolcapone inhibition of soluble and membrane-bound rat brain catechol-O-methyltransferase. J. Pharmacol. Exp. Ther. 282, 812–817. [PubMed] [Google Scholar]
- Sun J.; Von Tungeln L. S.; Hines W.; Beger R. D. (2009) Identification of metabolite profiles of the catechol-O-methyl transferase inhibitor tolcapone in rat urine using LC/MS-based metabonomics analysis. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 877, 2557–2565. [DOI] [PubMed] [Google Scholar]
- LeWitt P. A. (1993) Assessment of the dopaminergic lesion in Parkinson’s disease by CSF markers. Adv. Neurol. 60, 544–547. [PubMed] [Google Scholar]
- Thiffault C.; Langston J. W.; Di Monte D. A. (2003) Cerebrospinal fluid 3,4-dihydroxyphenylacetic acid level after tolcapone administration as an indicator of nigrostriatal degeneration. Exp. Neurol. 183, 173–179. [DOI] [PubMed] [Google Scholar]
- Haasio K.; Koponen A.; Penttilä K. E.; Nissinen E. (2002) Effects of entacapone and tolcapone on mitochondrial membrane potential. Eur. J. Pharmacol. 453, 21–26. [DOI] [PubMed] [Google Scholar]
- Käenmäki M.; Tammimäki A.; Garcia-Horsman J. A.; Myöhänen T.; Schendzielorz N.; Karayiorgou M.; Gogos J. A.; Männistö P. T. (2009) Importance of membrane-bound catechol-O-methyltransferase in L-DOPA metabolism: a pharmacokinetic study in two types of Comt gene modified mice. Br. J. Pharmacol. 158, 1884–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida L.; Vaz-da-Silva M.; Silveira P.; Falcão A.; Maia J.; Loureiro A.; Torrão L.; Machado R.; Wright L.; Soares-da-Silva P. (2004) Pharmacokinetic-pharmacodynamic interaction between BIA 3–202, a novel COMT inhibitor, and levodopa/carbidopa. Clin. Neuropharmacol. 27, 17–24. [DOI] [PubMed] [Google Scholar]
- Vieira-Coelho M. A.; Soares-da-Silva P. (1999) Effects of tolcapone upon soluble and membrane-bound brain and liver catechol-O-methyltransferase. Brain Res. 821, 69–78. [DOI] [PubMed] [Google Scholar]
- Åkerman K. E.; Wikström M. K. (1976) Safranine as a probe of the mitochondrial membrane potential. FEBS Lett. 68, 191–197. [DOI] [PubMed] [Google Scholar]