

Abstract
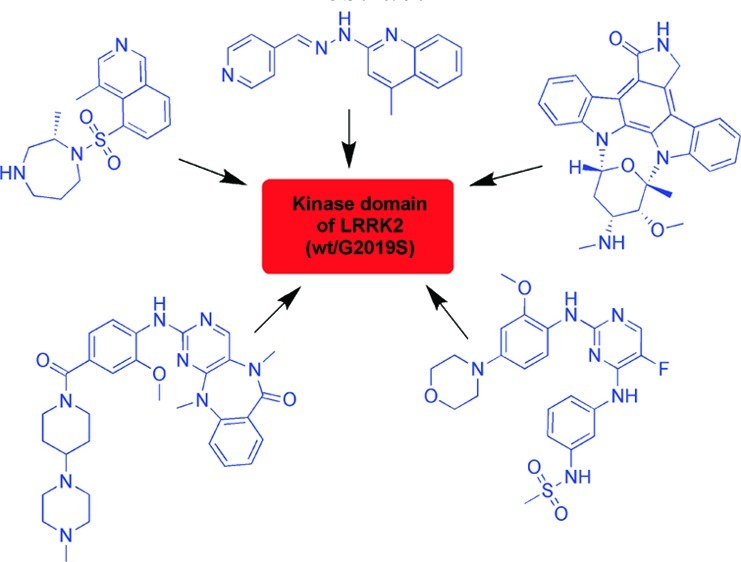
Parkinson's disease (PD) is the second most common neurodegenerative disorder. Several single gene mutations have been linked to this disease. Mutations in the gene encoding leucine-rich repeat kinase 2 (LRRK2) indicate LRRK2 as promising therapeutic target for the treatment of PD. LRRK2 mutations were observed in sporadic as well as familial PD patients and have been investigated intensively. LRRK2 is a large and complex protein, with multiple enzymatic and protein-interaction domains, each of which is effected by mutations. The most common mutation in PD patients is G2019S. Several LRRK2 inhibitors have been reported already, although the crystal structure of LRRK2 has not yet been determined. This review provides a summary of known LRRK2 inhibitors and will discuss recent in vitro and in vivo results of these inhibitors.
Keywords: Parkinson's disease, leucine-rich repeat kinase 2 (LRRK2), LRRK2 inhibitors, mutations, animal models
Parkinson's disease (PD) is the second most prevalent neurodegenerative disorder after Alzheimer's disease, effecting up to ∼4% of the population over 80.1,2 PD is characterized by a large number of motoric and non-motoric symptoms. Four of them are designated as cardinal features: tremor at rest, rigidity, akinesia, and postural instability.3 PD patients usually develop dementia during the course of the disease and gradually develop depression. The National Institute for Health and Clinical Excellence (NICE, U.K.) published guidelines for the diagnosis and management of patients with PD, which is used by most experts.2,4 The typical hallmarks of PD in post-mortem brain tissue are loss of dopaminergic neurons of the substantia nigra, associated with the formation of fibrillar aggregates composed of α-synuclein and other proteins (e.g. Lewy bodies).5,6 Presently, there is no cure for PD and the mainstay therapy is still the drug levodopa (L-Dopa).2,7 L-Dopa is highly effective in reducing motor symptoms; nevertheless, there are two major problems, the side-effects and that patients become therapy resistant.7,8 Adenosine A2a receptor antagonists have been shown to reduce PD-like features in animal studies through interaction with the specific dopamine receptor subtype D2 in the basal ganglia, making it more sensitive to dopamine. The adenosine A2a receptor antagonists SYN-115 from Synosia Therapeutics is currently in a phase IIb trial.7 Other approaches are the metabotropic glutamate receptors (mGluRs), which are members of the G-protein-coupled receptor (GPCR) superfamily. They participate in the modulation of synaptic transmission and neuronal excitability throughout the central nervous system. Several studies indicate the therapeutic utility of mGluR ligands in neurological and psychiatric disorders and make mGluRs promising targets for non-dopaminergic drug discovery in PD. mGluR5 is the target of drug development programmes at major pharmaceutical companies, e.g. Roche and Novartis.7,9,10
Several single gene mutations have been identified and linked to PD, for example, DJ-1, UCH-L1, SNCA, PRKN, and LRRK2.1,2,5,11−14 However, mutations in the LRRK2 gene are the most common cause of familial and sporadic late-onset PD.15 Since the LRRK2 gene mutations have been linked to PD, several inhibitors were reported to inhibit this kinase. We reviewed the literature and summarized the relevant data.
LRRK2: Structure and Mutations
The leucine-rich repeat kinase 2 (LRRK2) encodes a large multidomain protein with 2527 amino acids. Several independent domains have been established or predicted for the LRRK2 protein, including an ankyrin-like (ANK) domain, a leucine-rich repeat (LRR) domain, a Ras (renin-angiotensin system) of complex (Roc) domain, which belongs to the Ras GTPase family, followed by a C-terminal of ROC (COR) domain, a kinase (Kinase) domain, and a C-terminal WD40 domain (Figure 1).16−18
Figure 1.

Schematic illustration of domains and most common PD-linked point mutations of LRRK2. Red marked mutations have been linked to altered kinase activity. ANK, ankyrin-like domain; LRR, leucin-rich repeat domain; Roc, Ras of complex domain, which belongs to the Ras GTPase family; COR, C-terminal of Roc domain; Kinase, kinase domain; WD40, C-terminal WD40 domain. The five putatively pathogenic mutations are enlarged.
Beside the structural homology to the MAP kinase kinase kinases (MAPKKK) LRRK2 shares other biochemical features with MAPKKK, like autophosphorylation and interaction with kinase-specific chaperones.19 LRRK2 is expressed in various brain regions and in several other organs, for example, in the lung, kidney, and heart.20−22 LRRK2 efficiently phosphorylates moesin at Thr558 in vitro, raising the idea that moesin may be a physiological substrate of LRRK2. Moreover, ezrin and radixin are phosphorylated by LRRK2, which are involved in moesin binding actin.23,24 LRRK2 predominantly exists as a dimer under native conditions. The wild-type (wt) LRRK2 dimer displays increased kinase activity versus its monomeric counterpart.25,26 More than 20 LRRK2 mutations have been linked to autosomal-dominant parkinsonism, and five of them are considered definitely pathogenic (R1441C, R1441G, Y1699C, G2019S, I2020T).17,18,27 The most common mutation, which is present in more than 85% of PD patients carrying LRRK2 mutations, is G2019S.28 In some ethnic groups the frequency of the LRRK2-G2019S mutation has been found to be even higher. For example, 13–40% of all PD patients in the Ashkenazi Jewish and northern African Arab population have this mutation, whereas in the Asian population this mutation is much less common.17,28 The G2019S, R1441C, and R1441G mutations increase the LRRK2 kinase activity and both the kinase as well as the GTPase activities of LRRK2 are required to induce cell death.6,29−32 The GTPase domain-associated R1441C mutation in combination with the G2019S kinase domain mutation increased the kinase activity up to 7-fold relative to wild-type protein.33 It was reported that expression of G2019S mutant in Drosophila dendritic arborization neurons causes several dendrite defects, including tau mislocalization in dendrites, tau hyperphosphorylation at the T212/S214 sites, dendrite degeneration, and microtubule fragmentation.34 Furthermore, the LRRK2-G2019S mutant caused a progressive degeneration of nigral dopaminergic neurons in rats.35 In addition, the autokinase activity of the LRRK2 mutant I2020T was found to be increased in comparison to wild-type.19 Mutations within or near the GTPase domain including R1514Q, Y1699C, and I1371V increase kinase activity, while the alteration of the lysine residue K1347A leads to an ablation of this. The I1122V mutation in the LRR domain nominally increases kinase activity, whereas the D1994A and K1906M mutations in the kinase domain are able to diminish respectively to inhibit kinase activity.31 Replacing the kinase domain with a “kinase-dead” version blocks inclusion body formation and delays cell death.36 In addition, LRRK2 is able to phosphorylate MAPKKK 3, 4, 6, and 7 in vitro. This indicates that MAPKKK are molecular targets of LRRK2 mutants, whereby LRRK2 could be linked to neurotoxicity, cellular stress, and apoptosis.37,38 LRRK2 provides a potential therapeutic target utilizing the knowledge gained in neuroprotective kinase inhibition.
In Vivo Models and Studies
Several studies using in vivo models of LRRK2 fostered the understanding of neurobiology, pathogenesis, and utility of potential therapeutics. The Drosophila model revealed that a LRRK2 loss-of-function mutant leads to significantly impaired locomotive activity and that LRRK2 is critical for the integrity of dopaminergic neurons (DA).39 Another study showed that transgenic Drosophila harboring G2019S, Y1699C, or G2385 LRRK2 variants exhibit late-onset loss of DA and reduced lifespan.40 Liu et al. used the GAL4/UAS system to generate transgenic Drosophila expressing either wild-type human LRRK2 or LRRK2-G2019S. They reported that expression of either wild-type human LRRK2 or LRRK2-G2019S in the photoreceptor cells caused retinal degeneration. Furthermore, they observed that expression of LRRK2 or LRRK2-G2019S in neurons produced adult-onset selective loss of dopaminergic neurons and locomotor dysfunction.41,42 Overexpression of human LRRK2 wild-type, R1441C, and G2019S in DA of transgenic C. elegans models was sufficient to induce neurodegeneration and behavioral deficits, whereas knockout of the C. elegans LRRK2 homologue, LRK-1, prevents the LRRK2-induced neurodegeneration.43 The blockage of zebrafish LRRK2 protein by morpholinos caused embryonic lethality and severe development defects such as growth retardation and loss of neurons. In addition, the deletion of the WD40 domain of zebrafish LRRK2 by morpholinos revealed Parkinsonism-like phenotypes, including loss of dopaminergic neurons in the diencephalon and locomotion defects.44 Remarkably, another research group failed to reproduce the phenotypic loss of dopaminergic neurons in zebrafish.45 Nevertheless, the zebrafish model may be a useful vertebrate model. The presence of a LRRK2 protein excess in LRRK2 wild-type and G2019S mice showed exacerbated α-synuclein A53T-mediated cytotoxicity. This result raised the idea that inhibition of LRRK2 expression may provide an applicable strategy to ameliorate α-synuclein-induced neurodegeneration in PD.46 Expression of full-length LRRK2 wild-type did not induce any significant neuronal loss in the nigrostriatal system of adult rats, whereas expression of human LRRK2-G2019S mutant causes progressive degeneration of nigral dopaminergic neurons.35 Bacterial artificial chromosome (BAC) transgenic mice expressing LRRK2 wild-type, LRRK2-R1441G, and LRRK2-G2019S have shown evidence of neurodegeneration.24,47,48 Furthermore, the LRRK2-R1441G BAC transgenic mice revealed tau to be hyperphosphorylated in brain tissues.48 However, LRRK2 knockout mice lacking the kinase domain of LRRK2 are viable and live a normal life span. Thus, LRRK2 is not essential for mouse development and maintenance of DA.49 However, expression of the human LRRK2-G2019S mutation in transgenic mice is sufficient to recreate the slowly progressive degeneration of dopaminergic neurons that forms the hallmark pathology of familial and sporadic PD.50 Several mice studies investigated the potential of LRRK2 as therapeutic strategy for the treatment of PD.51−57 Two independent lines of LRRK2 germ-line deletion mice indicated that LRRK2 plays an essential role in the regulation of protein homeostasis during aging. Therefore, the authors concluded that LRRK2 inhibition may not represent a suitable therapeutic strategy for the treatment of PD.54 Another research group created inducible transgenic rats expressing LRRK2 with G2019S substitution and recapitulated the initiation process of dopaminergic dysfunction. However, the mutation was not sufficient to develop dopaminergic neurodegeneration or to induce neuron death in transgenic rats.57 Data obtained from a R1441C knockin mouse suggested that this mutation impairs stimulated dopamine neurotransmission and D2 receptor function. The R1441C mutation could represent pathogenic precursors preceding dopaminergic degeneration in PD brains.53 A novel herpes simplex virus (HSV) amplicon-based mouse model of LRRK2 dopaminergic neurotoxicity was developed to determine the efficacy of several LRRK2 kinase inhibitors. Nonetheless, a significant loss of tyrosine hydroxylase-positive neurons was induced due to HSV amplicon-mediated delivery of LRRK2-G2019S, whereas the HSV amplicon-mediated delivery of LRRK2-D1994A caused no neuronal loss. The injection of the LRRK2 kinase inhibitors can attenuate the loss of tyrosine hydroxylase-positive neurons induced by HSV-G2019S. Thus, the inhibition of LRRK2 kinase activity may hold potential to protect against LRRK2 toxicity and consequently for the treatment of neurodegeneration in PD.58 Hence, LRRK2 kinase inhibition holds potential for the treatment of PD. In the following, we will give a summary of small molecule LRRK2 kinase inhibitors. The inhibition effect of ROCOLRRK2 fragments will not be discussed.59
Small Molecule Kinase Inhibitors for LRRK2
LRRK2 is a large protein with several discrete domains. It surfaced as a therapeutic target when the kinase activity and the most common LRRK2 mutation, G2019S, were associated with neurotoxicity and PD. The first LRRK2 inhibitors derived from library screening efforts were mostly ATP-competitive. There are only few inhibitors, which were specifically developed to inhibit LRRK2. Thus, the majority of the compounds inhibits more than one kinase at the concentration indicated in the tables. The data in Table 1 derived from a limited number of in vitro assays using wild-type LRRK2 and G2019S-LRRK2. These assays vary in the concentration of LRRK2-constructs, substrate, and ATP; thus, the mere comparison of IC50 is misleading. The high sensitive assays utilize radioisotopes, which allow detection of both autophosphorylation and substrate phosphorylation, but are less suitable for high-throughput screening (HTS). High-throughput capability was achieved by time-resolved fluorescence resonance energy transfer (TF-FRET) and the amplified luminescent proximity homogeneous (AlphaScreen) assays.62 Although truncated LRRK2 and its full-length analog display similar phosphorylation activity, differences have been noticed. This may be a result from the utilization of different substrates, for example, LRRKtide and myelin basic protein (MBP).60,63
Table 1. Staurosporine and Derivatives as LRRK2 Inhibitors.

| IC50 |
|||||||
|---|---|---|---|---|---|---|---|
| no. | name | wild-type LRRK2 | LRRK2-G2019S | substrate | selectivitya | in vivo | lit. |
| 1 | Staurosporine | ∼1 nM;b 2 nM;b 8.2 nM;d 40 nMe | 0.2 nM;f 1.8 nM;c 40 nMe | GST-moesin; LRRKtide; MBPg | 2 | (58, 60−62) | |
| 2 | K-252a | ∼25 nM;b 3.6 nMc | 2.8 nMc | LRRKtide | (60, 61) | ||
| 3 | K-252b | ∼50 nMb | LRRKtide | (61) | |||
| 4 | Gö6976 | ∼250 nMb | LRRKtide | (61) | |||
Number of all kinases, including LRRK2.
Goat GST-LRRK2.
GST-LRRK2 (970–2527; wt/G2019S).
full-length LRRK2.
GST-LRRK2 (wt/G2019S).
Full-length Strep-tag LRRK2 (G2019S).
Myelin basic protein (MBP).
Staurosporine (1) is one of the widely used kinase inhibitors. This unselective compound equipotently inhibited both wild-type LRRK2 and LRRK2-G2019S (truncated and full-length) with an IC50 ranging from 0.2 to 40 nM (Table 1).58,60−62 Its inhibitory effect concerning LRRK2 was determined in different in vitro assays, for example, radioactive, TF-FRET, and AlphaScreen assay. These assays utilized different substrates such as synthetic peptides, for example, LRRKtide and potential physiological substrates: GST-Moesin. Staurosporine (1) had a similar inhibitory profile against LRRK1/LRRK2 autophosphorylation and MBP phosphorylation.58 Its isoindolinone derivatives K-252a/b (2/3) and Gö6976 (4) also inhibited wild-type LRRK2 and LRRK2-G2019S in the nanomolar range, whereas the maleimide analoga GF109203X (5) and Ro31-8220 (6) inhibited wild-type and LRRK2-G2019S in the low micromolar range only (Table 2).58,60,61 Ro31-8220 (6) is remarkable for the potent inhibition of MBP phosphorylation with an IC50 of 50 nM in the TF-FRET assay.
Table 2. Maleimide Derivatives as LRRK2 Inhibitors.

| IC50 |
|||||||
|---|---|---|---|---|---|---|---|
| no. | name | wild-type LRRK2 | LRRK2-G2019S | substrate | selectivitya | in vivo | lit. |
| 5 | GF109203X | 2190 nMb | 2620 nMb | MBPc | 2 | (58) | |
| 6 | Ro31-8220 | 2671 nM;d 50 nMb | 1922 nM;d 5160 nMb | LRRKtide; MBPc | 2 | (58, 60) | |
Number of all kinases, including LRRK2.
GST-LRRK2 (wt/G2019S).
Myelin basic protein (MBP).
GST-LRRK2 (970-2527; wt/G2019S).
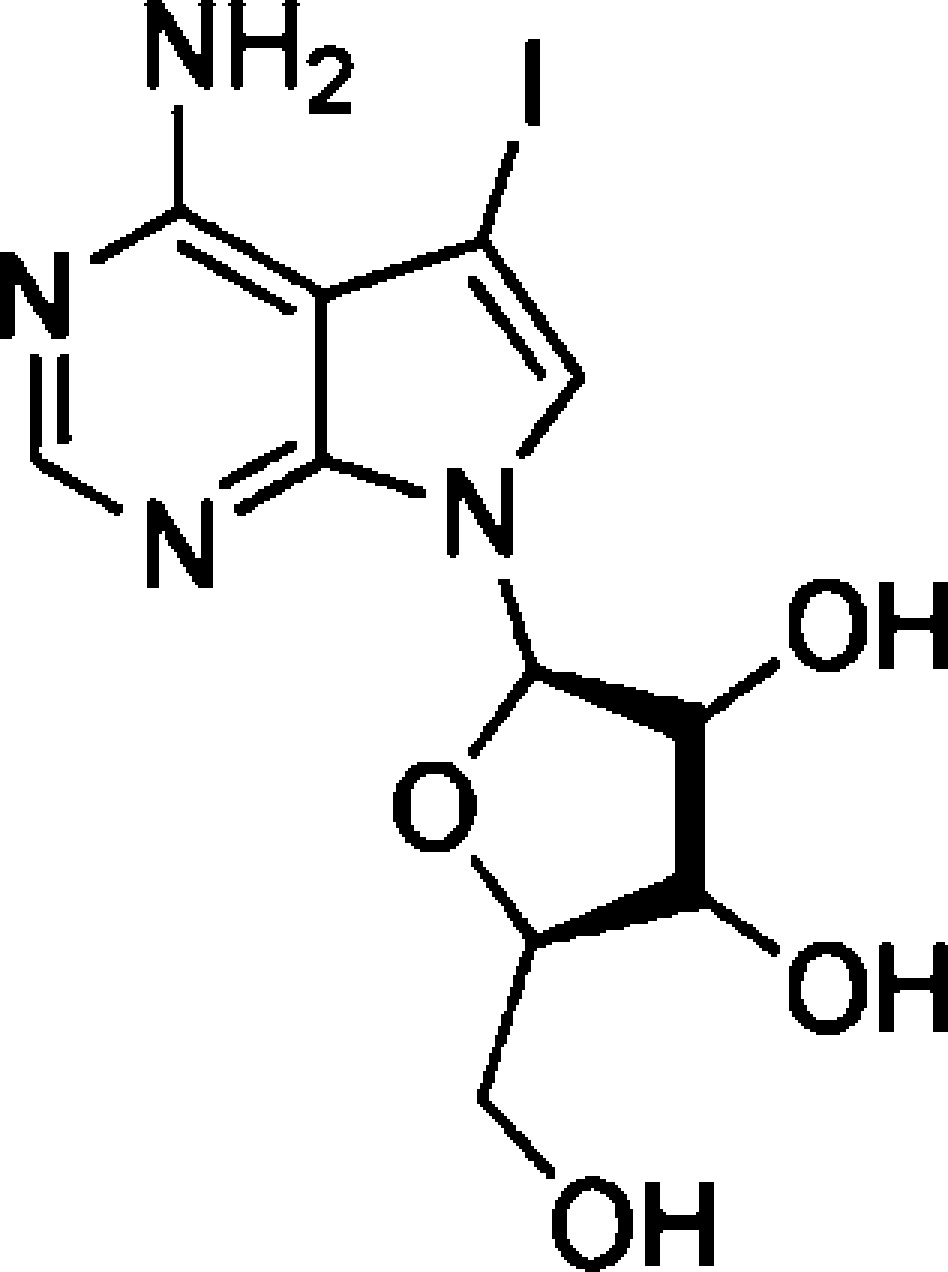
The inhibitory potency of 5-Iodotubericidin (7) (Table 3) and Sorafenib (8) (Table 4) was more than 4-fold higher for LRRK2-G2019S compared to wild-type LRRK2.58 Sorafenib (8) was up to 50% more selective for wild-type LRRK2 than for wild-type LRRK1. LRRK2-G2019S induced toxicity in rat primary cortical neuronal cultures (TUNEL assay) was completely protected by 5 μM of Sorafenib (8). Furthermore, it protected against LRRK2-G2019S-induced neurodegeneration in C. elegans and in Drosophila.64
Table 3. 5-Iodotubericidin as LRRK2 Inhibitor.
| IC50 |
|||||||
|---|---|---|---|---|---|---|---|
| no. | name | wild-type LRRK2 | LRRK2-G2019S | substrate | selectivitya | in vivo | lit. |
| 7 | 5-iodo-tubericidin | 14780 nMb | 3410 nMb | MBPc | 2 | (58) | |
Number of all kinases, including LRRK2.
GST-LRRK2 (wt/G2019S).
Myelin basic protein (MBP).
Table 4. Sorafenib as LRRK2 Inhibitor.

| IC50 |
|||||||
|---|---|---|---|---|---|---|---|
| no. | name | wild-type LRRK2 | LRRK2-G2019S | substrate | selectivitya | in vivo | lit. |
| 8 | sorafenib | 5580 nMb | 1230 nMb | MBPc | 2 | C. elegans; Drosophila | (58) |
Number of all kinases, including LRRK2.
GST-LRRK2 (wt/G2019S).
Myelin basic protein (MBP).
The indolinones GW5074 (9) and Indirubin-3'-monoxime (10) were ∼3-fold more active concerning LRRK2-G2019S than its corresponding wild-type. GW5074 (9) and Indirubin-3'-monoxime (10) inhibited LRRK2-G2019S with IC50's of 880 nM and ∼1.3 μM, respectively.58,61 They inhibited the closely related LRRK1 and LRRK2 in a similar manner. GW5074 (9) and Indirubin-3'-monoxime (10) inhibited LRRK2-mediated phosphorylation of LRRKtide and MBP, and additionally the phosphorylation of the eukaryotic translation initiation factor 4E-binding protein (4E-BPI), a putative physiological LRRK2 substrate. Furthermore, they attenuated LRRK2-G2019S induced cell injury and cell death. GW5074 (9) rescued the reduction of the density of tyrosine hydroxylase-positive fibers in a herpes simplex virus (HSV) amplicon-based mouse model of LRRK2 dopaminergic neurotoxicity. In addition, GW5074 (9) prevented LRRK2-G2019S induced inflammation, increase in isolectin B4 (ILB4)-positive cells in the striatum and substantia nigra pairs compacta.58 Both Sorafenib (8) and GW5074 (9) were found to protect against LRRK2-G2019S-induced neurodegeneration in C. elegans and in Drosophila.64 The well-known kinase inhibitor Sunitinib (11) was also investigated concerning its ability to inhibit LRRK2 (Table 5). It inhibited the LRRK2-mediated phosphorylation of Nictide and LRRKtide with an IC50 of 15–79 nM (truncated wild-type and LRRK2-G2019S).60,63,65 The selectivity of Sunitinib (11) was profiled in a panel of 85 kinases. Sunitinib (11) inhibits LRRK2-G2019S, its wild-type, and 12 further kinases at 1 μM by more than 80%.60,63,65 However, the less selective Sunitinib (11) is capable to suppress the activity of full-length LRRK2 expressed from Swiss-3T3 fibroblast cells.63 The apparent potency of Sunitinib (11) to inhibit wild-type LRRK2 drops to an IC50 of 370 nM at cellular ATP concentration (1 mM).66 In addition, studies of endogenous LRRK2 activity and phosphorylation in EBV-transformed lymphoblastoid cells, derived from a PD patient harboring a homozygous LRRK2-G2019S mutation, revealed that Sunitinib (11) inhibited the phosphorylation of Ser910 and Ser935 more potently than in wild-type cells.65 A comparable result was observed for the inhibitor H-1152 (12) (Table 6). H-1152 (12) is a known ROCK2 inhibitor. This compound was profiled in a panel of 85 kinases and found to inhibit Aurora B kinase, BRSK2, wild-type LRRK2, and LRRK2-G2019S at the relevant concentration. H-1152 (12) displayed IC50's ranging from 150 to 600 nM in radioisotope or AlphaScreen phosphorylation assays of wild-type LRRK2 and LRRK2-G2019S. Unfortunately, the structure determination of the LRRK2 kinase domain by X-ray crystallography was not reported yet. A docking analysis of H-1152 (12) utilized homology modeling of LRRK2, by superimposing the protein Cα atoms of the LRRK2 model with the reported ROCK1-H-1152 complex. This analysis indicated a backbone interaction with the NH atom of Ala1950 (PDB code of ROCK1-H-1152 not published). Furthermore, the two methyl groups of H-1152 (12) were observed to make lipophilic contacts with the ATP binding site. The amino acid Ala2016 was found to be close to H-1152 (12). This can contribute to crucial drug resistance, because the IC50 of H-1152 (12) for the LRRK2 mutant A2016T increased up to ∼30-fold.63 Another commercially available quinoline derivative, Compound 4 (13), inhibited the autophosphorylation of LRRK2-G2019S with an IC50 of 4.1 μM (Table 6). It was screened in a small kinase panel of 13 kinases at 10 μM concentration. There was no other inhibitory effect, except for MLK1.67 This observation can be rationalized by the similarity of the kinase domains of MLK and LRRK2. Compound 4 (13) was analyzed in a homology model of LRRK2 using the structure of the transforming growth factor-beta (TGF-β) activated kinase 1 (TAK1; PDB code: 2EVA). This homology model of the LRRK2 kinase domain with inhibitor Compound 4 (13) indicated hydrogen bond interactions between Compound 4 (13) and Ala1950. It was found that the 4-pyridine ring was located in the solvent exposed region of LRRK2. Moreover, a comparison of ATP and Compound 4 (13) docking revealed that the 4-methyl quinoline moiety overlapped with the adenine rings of ATP. The treatment of murine dopaminergic SN4741 cells with 10 μM of Compound 4 (13) restored their cell survival rates in an oxidative stress-induced test to the level of the control cells. The neurotoxicity test with primary rat cortical neuronal cells revealed cell survival rates of 85% at 10 μM and a significantly increased toxicity at 100 μM.67 In a kinase panel of 85 kinases the known ROCK inhibitor Y-27632 (14) was found to additionally inhibit PRK2, MNK1, wild-type LRRK2 and LRRK2-G2019S at 10 μM (Table 7). Y-27632 (14) inhibited truncated LRRK2-G2019S with an IC50 of 1 μM in a radioisotope assay. Furthermore, the full-length Strep-tag LRRK2-G2019S was inhibited with an IC50 of 1.8 μM in an AlphaScreen assay.62,63 The anthracene and phenanthrene derivatives SP60012 (15), Damnacanthal (16), LDN-73794 (17), and LDN-22684 (18) inhibited LRRK2-G2019S and wild-type LRRK2 in the low micromolar range (Table 8).58,68,69 LDN-73794 (17) was confirmed to be ATP competitive, whereas LDN-22684 (18) was found to be a non-ATP competitive inhibitor. Further studies revealed LDN-22684 (18) to be neither GTP competitive nor substrate competitive. Hence, it was deduced that LDN-22684 (18) is an allosteric LRRK2 inhibitor.68,69 Three compounds were especially developed to inhibit LRRK2, namely, CZC-25146 (19), CZC-54252 (20), and LRRK2-IN-1 (21) (Table 9).70,71 CZC-25146 (19) and CZC-54252 (20) inhibited the activity of recombinant human wild-type LRRK2 with an IC50 ranging from ∼1 to ∼5 nM. The G2019S mutant was inhibited with an IC50 ranging from ∼2 to ∼7 nM in a TF-FRET assay. In addition, they were screened against a kinase panel of 185 kinases and exhibited good selectivity. CZC-25146 (19) inhibited five other kinases, PLK4, GAK, TNK1, CAMKK2, and PIP4K2C, with high potency only, but none of them have been classified as predictors of genotoxicity or hematopoietic toxicity.72,73 Furthermore, it prevents mutant LRRK2-induced injury of cultured rodent and human neurons with mid-nanomolar potency. In vivo pharmacology established a volume of distribution of 5.4 L/kg and a clearance of 2.3 L/h/kg for CZC-25146 (19). Unfortunately, it exhibited a poor brain penetration of just 4% (Table 10).71
Table 5. Indolinone Dervivatives as LRRK2 Inhibitors.

| IC50 |
|||||||
|---|---|---|---|---|---|---|---|
| no. | name | wild-type LRRK2 | LRRK2-G2019S | substrate | selectivitya | in vivo | lit. |
| 9 | Raf-1 kinase inhibitor I (GW5074) | ∼500 nM;b3150 nMc | 880 nMc | LRRKtide; MBPd | 2 | HSVg amplicon-based mouse model | (58, 61) |
| 10 | Indirubin-3′-monooxime | 4830 nMc | 1310 nMc | MBPd | 2 | HSVg amplicon-based mouse model | (58) |
| 11 | Sunitinib | 79 nM;e 15 nMf | 19 nM;e 26 nMf | Nictide; LRRKtide | 85 | (60, 63, 65) | |
Number of all kinases, including LRRK2.
Goat GST-LRRK2.
GST-LRRK2 (wt/G2019S).
Myelin basic protein (MBP).
GST-LRRK2 (1326–2527; wt/G2019S).
GST-LRRK2 (970–2527; wt/G2019S).
Herpes simplex virus.
Table 6. Quinoline Derivatives as LRRK2 Inhibitors.

| IC50 |
|||||||
|---|---|---|---|---|---|---|---|
| no. | name | wild-type LRRK2 | LRRK2-G2019S | substrate | selectivitya | in vivo | lit. |
| 12 | H-1152 | 244 nMb | 600 nM;c 150 nMb | GST-Moesin; Nictide | 85 | (62, 63, 65) | |
| 13 | Compound 4 | 4100 nMd | 13 | (67) | |||
Number of all kinases, including LRRK2.
GST-LRRK2 (1326-2527; wt/G2019S).
full-length Strep-tag LRRK2 (G2019S).
GST-LRRK2 (G2019S).
Table 7. Y-27632 as LRRK2 Inhibitor.
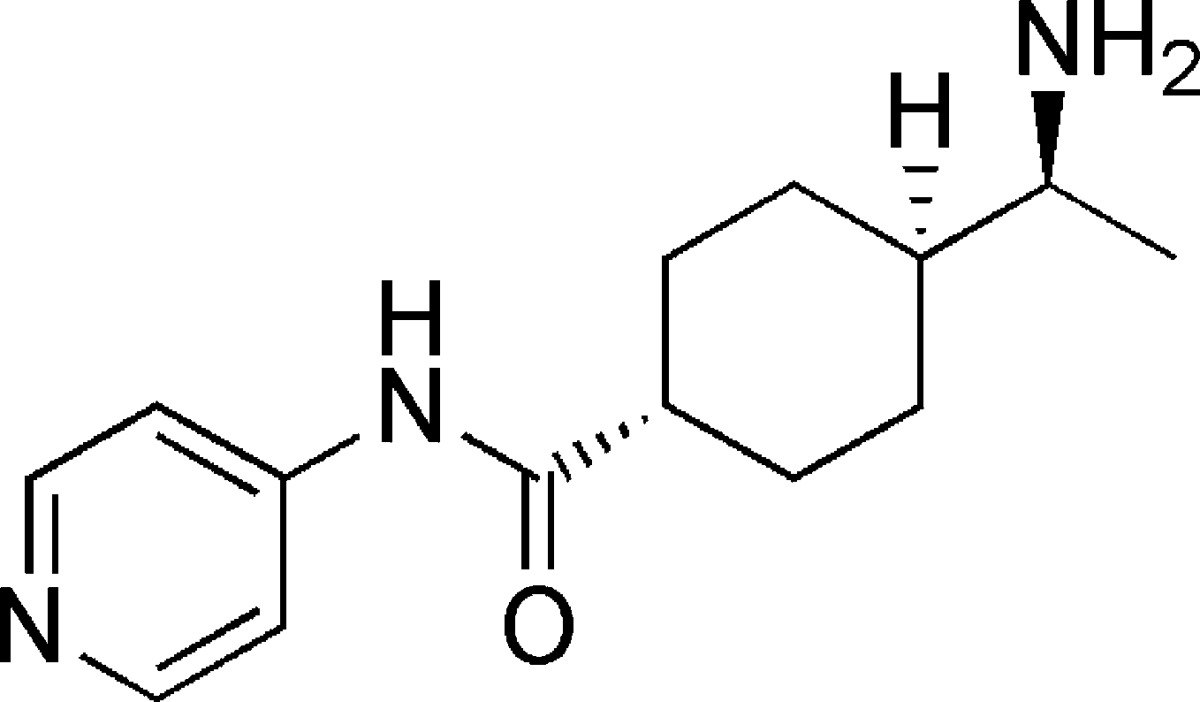
| IC50 |
|||||||
|---|---|---|---|---|---|---|---|
| no. | name | wild-type LRRK2 | LRRK2-G2019S | substrate | selectivitya | in vivo | lit. |
| 14 | Y-27632 | 2300 nMb | 1800 nM;c 1000 nMb | GST-Moesin; Nictide | 85 | (62, 63) | |
Number of all kinases, including LRRK2.
GST-LRRK2 (1326-2527; wt/G2019S).
Full-length Strep-tag LRRK2 (G2019S).
Table 8. Anthracene and Phenanthrene Derivatives as LRRK2 Inhibitors.

| IC50 |
|||||||
|---|---|---|---|---|---|---|---|
| no. | name | wild-type LRRK2 | LRRK2-G2019S | substrate | substrate selectivitya | in vivo | lit. |
| 15 | SP600125 | 3100 nMb | 5000 nMb | MBPc | 2 | (58) | |
| 16 | Damnacanthal | 7810 nMb | 9450 nMb | MBPc | 2 | (58) | |
| 17 | LDN-73794 | 3500 nMd | PLK-peptide | 2 | (68) | ||
| 18 | LDN-22684e | 6900 nMf | 6100 nMf | PLK-peptide | (69) | ||
Number of all kinases, including LRRK2.
GST-LRRK2 (wt/G2019S).
Myelin basic protein (MBP).
Purified from BAC-transgenic mouse brain.
Non-ATP competitive.
Human LRRK2 (970-2527; wt/G2019S).
Table 9. Pyrimidine Derivatives as LRRK2 Inhibitors.

| IC50 |
|||||||
|---|---|---|---|---|---|---|---|
| no. | name | wild-type LRRK2 | LRRK2-G2019S | substrate | selectivitya | in vivo | lit. |
| 19 | CZC-25146 | 4.76 nMb | 6.87 nMb | LRRKtide | LRRKtide | male CD-1 mice | (71) |
| 20 | CZC-54252 | 1.28 nMb | 1.85 nMb | LRRKtide | LRRKtide | male CD-1 mice | (71) |
| 21 | LRRK2-IN-1 | 13 nMc | 6 nMc | Nictide | Nictide | Male C57BL/6 mice | (70) |
Number of all kinases, including LRRK2.
Human LRRK2 (wt/G2019S).
GST-LRRK2 (1326–2527; wt/G2019S).
Table 10. Pharmocokinetic Profile of the Currently Best Published LRRK2 inhibitors CZC-25146 and LRRK2-IN-1a.
| name | route | dose (mg/kg) | Tmax (h) | Cmax(ng/mL) | AUC0–∞(h·ng/mL) | T1/2 (h) | CL (mL/min/kg) | Vss(L/kg) | F (%) | BBB penetration (%) | lit. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| CZC-25146 (male CD-1 mice) | IV | 1 | 0 | 154 | 419–434 | 1.6 | 2.3 | 5.4 | 4 | (71) | |
| PO | 5 | 0.25 | 1357 | 2878–2894 | 1 | 133 | |||||
| LRRK2-IN-1 (male C57BL/6 mice) | IV | 1 | 2974 | 4.47 | 5.6 | 1.71 | not efficiently | (70) | |||
| PO | 10 | 1.0 | 1618 | 14758 | 49.3 |
IV = intravenous injection; PO = oral delivery; Tmax = time of maximum plasma concentration; Cmax = maximum plasma concentration; AUC0–∞ = area under the curve (measure of exposure); T1/2 = half life; CL = plasma clearance; Vss = volume of distribution; F = oral bioavailability; BBB = blood-brain barrier.
A HTS and subsequent lead optimization provided the LRRK2 inhibitor LRRK2-IN-1 (21). It inhibited both truncated wild-type LRRK2 and LRRK2-G2019S with IC50 values of 13 and 6 nM, but LRRK2-A2016T and LRRK2-A2016T+G2019S mutants were found to be ∼400-fold more resistant to LRRK2-IN-1 (21) (Table 9).70 This was explained by a molecular docking study of LRRK2-IN-1 (21) bound to a homology model of LRRK2 (A2016T), which revealed an unfavorable steric interaction as observed for H-1152 (12). The confirmed reversible ATP competitive inhibitor LRRK2-IN-1 (21) was selective in kinase panels containing more than 470 kinases. Surprisingly, under the same conditions as employed for LRRK2, LRRK2-IN-1 lacked inhibition of LRRK1. The kinase panels revealed additional inhibition of DCLK1, DCLK2, as well as MAPK7 and supported IC50's of greater than 1 μM for AURKB, CHEK2, MKNK2, MYLK (smMLCK), NUAK1, PLK1, and RPS6KA2. LRRK2-IN-1 (21) induced a similar dose-dependent Ser910 and Ser935 dephosphorylation and loss of 14–3–3 binding to endogenous LRRK2 in human-derived neuroblastoma SHSY5Y cells and mouse Swiss3T3 cells. Pharmacokinetic studies of LRRK2-IN-1 (21) revealed a half-life of 4.5 h and a bioavailability of 49.3% in mice (Table 10). An insufficient blood-brain barrier (BBB) permeation was concluded from the LRRK2 phosphorylation status in the kidney versus brain, which imposes limits on this “useful first-generation 'tool'”.70
So far, just a small number of LRRK2 inhibitors have been synthesized and profiled in kinase panels. The best of the reported compounds display both high activity and selectivity. However, these reported best-in-class compounds do not pass the BBB efficiently, which limits their potential in animal models of PD. Maybe the patent literature holds additional treasures, waiting to be released.74−79
Conclusion and Perspectives
The importance of the G2019S mutation in the kinase domain of LRRK2 derives from the association with the second most common neurodegenerative disease: Parkinson’s disease. The late-onset sporadic classical PD affects almost 2% of the world population over 65 years of age.80 The availability of suitable HTS assay formats such as TF-FRET or AlphaScreen provided first inhibitors of LRRK2 activity with moderate selectivity. Lead optimization resulted in first generation tools and second-generation inhibitors, which displayed promising pharmacokinetic properties, but are limited by insufficient brain uptake or brain activity. The increase in patent applications (e.g., Glaxo Group Limited and Cellzome Limited) indicates a target on the rise. TTT-3002, a drug candidate of TauTaTis, exhibited good results in LRRK2 inhibition. A phase I clinical trial of TTT-3002 is expected to start in 2011.74−79,81
The clinical development of LRRK2 inhibitors is impaired by the lack of public data of relevant pharmacology, biology, and even biomarkers. Moreover, the gain of function in LRRK2 mutations may require fundamentally different dosing regimes for mutation carriers versus other PD patients resulting in personalized medicine. This dosage may very up to 100-fold, implying genomic profiling, patient stratification and a wide therapeutic window. Several in vivo invertebrate models indicate neurotoxic hyperactivity of LRRK2 kinase, but the number of LRRK2 kinase activity studies in mouse models is rather limited. New LRRK2 animal models may provide essential information for target validation and suitable biomarkers for end point identification in drug development. Passage of the blood-brain barrier remains a challenge, yet LRRK2 function may be important outside the CNS. Moreover, the consequences of LRRK2 inhibition are not sufficiently established in animal models or humans to conclude a safe inhibition rate in mutation carriers or normal PD patients nor to rule out therapy resistance in gain of function mutations. Furthermore, just a small number of substrates has been reported to date and this may increase. Hence, an effective and safe inhibition of LRRK2 kinase activity has yet to be confirmed in vivo, in vitro. Any novel therapy must be evaluated against established PD therapies, which results in either extended or large clinical trials. A robust and approved biomarker may enable shorter or smaller trials; however, this may take years to develop and to gain approval.82−84 Recently, the LRRK-2 phosphorylation sites Ser910, Ser935, Ser955, and Ser973 were suggested as biomarkers for LRRK2 because the treatment of LRRK2 expressing cells with LRRK2-IN-1 (21) revealed these phosphorylations to be disrupted. However, the kinases and phosphatases responsible for the regulation of these phosphorylation sites have yet to be identified.85
Type-I-kinase inhibitors are notorious for their selectivity problems, which frequently cause adverse events in humans. In addition, drug resistance or diminished activity has been observed, which may be caused by kinase domain mutations. LRRK2 features many mutations, which imposes problems to find a useful drug candidate.63,70 A new generation of kinase inhibitors, called type-II-inhibitors, may reduce some of these problems.86,87 They bind to the ATP site and extend into an adjacent allosteric site. This allosteric site is not as highly conserved as the ATP binding site and may provide a strategy to obtain improved selectivity. LRRK2 is a particularly challenging target: the G2019S gain of function mutation requires very efficient inhibition to reduce the activity to the wild-type level. The lack of brain permeable inhibitors leaves an open fundamental question: how much G2019S LRRK2 inhibition is required in vivo? In summary, it can be stated that LRRK2 inhibition provides potential to treat PD, albeit further research and clarification is inevitable.
This work was supported by the Technische Universität Darmstadt.
The authors declare no competing financial interest.
Author Contributions
† These authors contributed equally to this work.
References
- Ross O. A.; Farrer M. J. (2005) Pathophysiology, pleiotrophy and paradigm shifts: genetic lessons from Parkinson’s disease. Biochem. Soc. Trans. 33, 586–590. [DOI] [PubMed] [Google Scholar]
- Davie C. A. (2008) A review of Parkinson’s disease. Br. Med. Bull. 86, 109–127. [DOI] [PubMed] [Google Scholar]
- Jankovic J. (2008) Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376. [DOI] [PubMed] [Google Scholar]
- Clarke C. E. (2007) Parkinson’s disease. BMJ - Clin. Rev. 335, 441–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A.; Biskup S.; Leitner P.; Lichtner P.; Farrer M.; Lincoln S.; Kachergus J.; Hulihan M.; Uitti R. J.; Calne D. B.; Stoessl A. J.; Pfeiffer R. F.; Patenge N.; Carbajal I. C.; Vieregge P.; Asmus F.; Müller-Myhsok B.; Dickson D. W.; Meitinger T.; Strom T. M.; Wszolek Z. K.; Gasser T. (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607. [DOI] [PubMed] [Google Scholar]
- Berwick D. C.; Harvey K. (2011) LRRK2 signaling pathways: the key to unlocking neurodegeneration?. Trends Cell Biol. 21, 257–265. [DOI] [PubMed] [Google Scholar]
- Smith K. (2010) Treatment frontiers. Nature - Parkinson's Disease Outlook 466, 15–18. [DOI] [PubMed] [Google Scholar]
- Poewe W. (2006) The natural history of Parkinson’s disease. J. Neurol. 253, VII/2–VII/6. [DOI] [PubMed] [Google Scholar]
- Duty S. (2010) Therapeutic potential of targeting group III metabotropic glutamate receptors in the treatment of Parkinson’s disease. Br. J. Pharmacol. 161, 271–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender C. M.; Conn P. J. (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisán-Ruíz C.; Jain S.; Evans E. W.; Gilks W. P.; Simón J.; van der Brug M.; López de Munain A.; Aparicio S.; Martínez Gil A.; Khan N.; Johnson J.; Martinez J. R.; Nicholl D.; Carrera I. M.; Pena A. S.; de Silva R.; Lees A.; Martí-Massó J. F.; Pérez-Tur J.; Wood N. W.; Singleton A. B. (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600. [DOI] [PubMed] [Google Scholar]
- Valente E. M.; Abou-Sleiman P. M.; Caputo V.; Muqit M. M. K.; Harvey K.; Gispert S.; Ali Z.; Del Turco D.; Bentivoglio A. R.; Healy D. G.; Albanese A.; Nussbaum R.; González-Maldonado R.; Deller T.; Salvi S.; Cortelli P.; Gilks W. P.; Latchman D. S.; Harvey R. J.; Dallapiccola B.; Auburger G.; Wood N. W. (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. [DOI] [PubMed] [Google Scholar]
- Bonifati V.; Rizzu P.; van Baren M. J.; Schaap O.; Breedveld G. J.; Krieger E.; Dekker M. C. J.; Squitieri F.; Ibanez P.; Joosse M.; van Dongen J. W.; Vanacore N.; van Swieten J. C.; Brice A.; Meco G.; van Duijn C. M.; Oostra B. A.; Heutink P. (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. [DOI] [PubMed] [Google Scholar]
- Kitada T.; Asakawa S.; Hattori N.; Matsumine H.; Yamamura Y.; Minoshima S.; Yokochi M.; Mizuno Y.; Shimizu N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. [DOI] [PubMed] [Google Scholar]
- Tan E. K.; Schapire A. H. (2011) LRRK2 as a therapeutic target in Parkinson’s disease. Eur. J. Neurol. 18, 545–546. [DOI] [PubMed] [Google Scholar]
- Deng J.; Lewis P. A.; Greggio E.; Sluch E.; Beilina A.; Cookson M. R. (2008) Structure of the ROC domain from the Parkinson’s disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc. Natl. Acad. Sci. U.S.A. 105, 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson B. I.; Van Deerlin V. M. (2008) Mutations in LRRK2 as a cause of Parkinson’s disease. Neurosignals 16, 99–105. [DOI] [PubMed] [Google Scholar]
- Mata I. F.; Wedemeyer W. J.; Farrer M. J.; Taylor J. P.; Gallo K. A. (2006) LRRK2 in Parkinson’s disease: protein domains and functional insights. Trends Neurosci. 29, 286–293. [DOI] [PubMed] [Google Scholar]
- Gloeckner C. J.; Kinkl N.; Schumacher A.; Braun R. J.; ÓNeill E.; Meitinger T.; Kolch W.; Prokisch H.; Ueffing M. (2006) The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum. Mol. Genet. 15, 223–232. [DOI] [PubMed] [Google Scholar]
- Li X.; Tan Y.; Poulose S.; Olanow C. W.; Huang X.; Yue Z. (2007) Leucine-rich repeat kinase 2 (LRRK2)/PARK8 possesses GTPase activity that is altered in familial Parkinson’s disease R1441C/G mutants. J. Neurochem. 103, 238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biskup S.; Moore D. J.; Celsi F.; Higashi S.; West A. B.; Andrabi S. A.; Kurkinen K.; Yu S.; Savitt J. M.; Waldvogel H. J.; Faull R. L. M.; Emson P. C.; Torp R.; Ottersen O. P.; Dawson T. M.; Dawson V. L. (2006) Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 60, 557–569. [DOI] [PubMed] [Google Scholar]
- Giasson B. I.; Covy J. P.; Bonini N. M.; Hurtig H. I.; Farrer M. J.; Trojanowski J. Q.; Van Deerlin V. M. (2006) Biochemical and pathological characterization of Lrrk2. Ann. Neurol. 59, 315–322. [DOI] [PubMed] [Google Scholar]
- Jaleel M.; Nichols R. J.; Deak M.; Campbell D. G.; Gillardon F.; Knebel A.; Alessi D. R. (2007) LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem. J. 405, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T.; Yang D.; Sushchky S.; Liu Z.; Smith W. (2011) Models for LRRK2-Linked Parkinsonism. Parkinson's Dis. 2011, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Z.; Smith K. A.; La Voie M. J. (2010) Membrane localization of LRRK2 is associated with increased formation of the highly active LRRK2 dimer and changes in its phosphorylation. Biochemistry 49, 5511–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E.; Zambrano I.; Kaganovich A.; Beilina A.; Taymans J.; Daniels V.; Lewis P.; Jain S.; Ding J.; Syed A.; Thomas K. J.; Baekelandt V.; Cookson M. R. (2008) The Parkinson disease-associated leucine-rich repeat kinase 2 (LRRK2) is a dimer that undergoes intramolecular autophosphorylation. J. Biol. Chem. 283, 16906–16914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J. P.; Mata I. F.; Farrer M., J. (2006) LRRK2: a common pathway for parkinsonism, pathogenesis and prevention?. Trends Mol. Med. 12, 76–82. [DOI] [PubMed] [Google Scholar]
- Seol W. (2010) Biochemical and molecular features of LRRK2 and its pathophysiological roles in Parkinson’s disease. BMB Rep. 43, 233–244. [DOI] [PubMed] [Google Scholar]
- Cookson M. R. (2010) The role of leucine-rich repeat kinase 2 (LRRK2) in Parkinson’s disease. Nat. Rev. Neurosci. 11, 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A. B.; Moore D. J.; Biskup S.; Bugayenko A.; Smith W. W.; Ross C. A.; Dawson V. L.; Dawson T. M. (2005) Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc. Natl. Acad. Sci. U.S.A. 102, 16842–16847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A. B.; Moore D. J.; Choi C.; Andrabi S. A.; Li X.; Dikeman D.; Biskup S.; Zhang Z.; Lim K.; Dawson V. L.; Dawson T. M. (2007) Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum. Mol. Genet. 16, 223–232. [DOI] [PubMed] [Google Scholar]
- Xiong Y.; Coombes C. E.; Kilaru A.; Li X.; Gitler A. D.; Bowers W. J.; Dawson V. L.; Dawson T. M.; Moore D. J. (2010) GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet. 6, e1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber P. J.; Smith A. D.; Sen S.; Renfrow M. B.; Mobley J. A.; West A. B. (2011) Autophosphorylation in the Leucine-Rich Repeat Kinase 2 (LRRK2) GTPase Domain Modifies Kinase and GTP-Binding Activities. J. Mol. Biol. 412, 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.; Tsai P.; Wu R.; Chien C. (2010) LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ß. J. Neurosci. 30, 13138–13149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusonchet J.; Kochubey O.; Stafa K.; Yound S. M. Jr; Zufferey R.; Moore D. J.; Schneider B. L.; Aebischer P. (2011) A rat model of progressive nigral neurodegeneration induced by the Parkinson’s disease-associated G2019S mutation in LRRK2. J. Neurosci. 31, 907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greggio E.; Jain S.; Kingsbury A.; Bandopadhyay R.; Lewis P.; Kaganovich A.; van der Brug M. P.; Belina A.; Blackinton J.; Thomas K. J.; Ahmad R.; Miller D. W.; Kesavapany S.; Singleton A.; Lees A.; Harvey R. J.; Harvey K.; Cookson M. R. (2006) Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 23, 329–341. [DOI] [PubMed] [Google Scholar]
- Hsu C. H.; Chan D.; Greggio E.; Saha S.; Guillily M. D.; Ferree A.; Raghavan K.; Shen G. C.; Segal L.; Ryu H.; Cookson M. R.; Wolozin B. (2010) MKK6 binds and regulates expression of Parkinson’s disease-related protein LRRK2. J. Neurochem. 112, 1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloeckner C. J.; Schumacher A.; Boldt K.; Uefffing M. (2009) The Parkinson disease-associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. J. Neurochem. 109, 959–968. [DOI] [PubMed] [Google Scholar]
- Lee S. B.; Kim W.; Lee S.; Chung J. (2007) Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem. Biophys. Res. Commun. 358, 534–539. [DOI] [PubMed] [Google Scholar]
- Ng C.; Mok S. Z. S.; Koh C.; Ouyang X.; Fivaz M. L.; Tan E.; Dawson V. L.; Dawson T. M.; Yu F.; Lim K. (2009) Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J. Neurosci. 29, 11257–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Wang X.; Yu Y.; Li X.; Wang T.; Jiang H.; Ren Q.; Jiao Y.; Sawa A.; Moran T.; Ross C. A.; Montell C.; Smith W. W. (2008) A Drosophila model for LRRK2-linked parkinsonism. Proc. Natl. Acad. Sci. U.S.A. 105, 2693–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith W. W. (2010) Leucine-rich kinase 2 (LRRK2) Drosophila Model For Parkinsońs Disease: Wildtype1 (WT1) and G2019S Mutant Flies. U.S. 2010/0175140 A1.
- Yao C.; El Khoury R.; Wang W.; Byrd T. A.; Pehek E. A.; Thacker C.; Zhu X.; Smith M. A.; Wilson-Delfosse A. L.; Chen S. G. (2010) LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol. Dis. 40, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng D.; Qu D.; Kwok K. H. H.; Ng S. S.; Lim A. Y. M.; Aw S. S.; Lee C. W. H.; Sung W. K.; Lufkin T.; Jesuthasan S.; Sinnakaruppan M.; Liu J. (2010) Deletion of the WD40 domain of LRRK2 in Zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS Genet. 6, e1000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G.; Xin S.; Zhong H.; Lin S (2011) Disruption of LRRK2 does not cause specific loss of dopaminergic neurons in zebrafish. PLoS Genet. 6, e20630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X.; Parisiadou L.; Gu X.; Wang L.; Shim H.; Sun L.; Xie C.; Long C.; Yang W.; Ding J.; Chen Z. Z.; Gallant P. E.; Tao-Cheng J.; Rudow G.; Troncoso J. C.; Liu Z.; Li Z.; Cai H. (2009) Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 64, 807–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Patel J. C.; Wang J.; Avshalumov M. V.; Nicholson C.; Buxbaum J. D.; Elder G. A.; Rice M. E.; Yue Z. (2010) Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J. Neurosci. 30, 1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Liu W.; Oo T. F.; Wang L.; Tang Y.; Jackson-Lewis V.; Zhou C.; Geghman K.; Bogdanov M.; Przedborski S.; Beal M. F.; Burke R. E.; Li C. (2009) Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat. Neurosci. 12, 826–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreas-Mateos E.; Mejias R.; Sasaki M.; Li X.; Lin B. M.; Biskup S.; Zhang L.; Banerjee R.; Thomas B.; Yang L.; Liu G.; Beal M. F.; Huso D. L.; Dawson T. M.; Dawson V. L. (2009) Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine). J. Neurosci. 29, 15846–15850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramonet D.; Daher J. P. L; Lin B. M.; Stafa K.; Kim J.; Banerjee R.; Westerlund M.; Pletnikova O.; Glauser L.; Yang L.; Liu Y.; Swing D. A.; Beal M. F.; Troncoso J. C.; McCaffery J. M.; Jenkins N. A.; Copeland N. G.; Galter D.; Thomas B.; Lee M. K.; Dawson T. M.; Dawson V. L.; Moore D. J. (2011) Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PloS ONE 6, e18568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melrose H. L.; Dächsel J. C.; Behrouz B.; Lincoln S. J.; Yue M.; Hinkle K. M.; Kent C. B.; Korvatska E.; Taylor J. P.; Witten L.; Liang Y. Q.; Beevers J. E.; Boules M.; Dugger B. N.; Serna V. A.; Gaukhman A.; Yu X.; Castanedes-Casey M.; Braithwaite A. T.; Ogholikhan S.; Yu N.; Bass D.; Tyndall G.; Schellenberg G. D.; Dickson D. W.; Janus C.; Farrer M. J. (2010) Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol. Dis. 40, 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melrose H. L.; Kent C. B.; Taylor J. P.; Dachsel J. C.; Hinkle K. M.; Lincoln S. J.; Mok S. S.; Culvenor J. G.; Masters C. L.; Tyndall G. M.; Bass D. I.; Ahmed Z.; Andorfer C. A.; Ross O. A.; Wszolek Z. K.; Delldonne A.; Dickson D. W.; Farrer M. J. (2007) A comparative analysis of leucine-rich repeat kinase 2 (Lrrk2) expression in mouse brain and Lewy body disease. Neuroscience 147, 1047–1058. [DOI] [PubMed] [Google Scholar]
- Tong Y.; Pisani A.; Martella G.; Karouani M.; Yamaguchi H.; Pothos E. N.; Shen J. (2009) R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc. Natl. Acad. Sci. U.S.A. 106, 14622–14627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y.; Yamaguchi H.; Giaime E.; Boyle S.; Kopan R.; Kelleher R. J. III; Shen J. (2010) Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. U.S.A. 107, 9879–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Xie C.; Greggio E.; Parisiadou L.; Shim H.; Sun L.; Chandran J.; Lin X.; Lai C.; Yang W.; Moore D. J.; Dawson T. M.; Dawson V. L.; Chiosis G.; Cookson M. R.; Cai H. (2008) The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J. Neurosci. 28, 3384–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B.; Melrose H. L.; Zhao C.; Hinkle K. M.; Yue M.; Kent C.; Braithwaite A. T.; Ogholikhan S.; Aigner R.; Winkler J.; Farrer M. J.; Gage F. H. (2011) Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol. Dis. 41, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H.; Huang C.; Tong J.; Hong W. C.; Liu Y.; Xia X. (2011) Temporal expression of mutant LRRK2 in adult rats impairs dopamine reuptake. Int. J. Biol. Sci. 7, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. D.; Shin J.; VanKampen J.; Petrucelli L.; West A. B.; Ko H. S.; Lee Y.; Maguire-Zeiss K. A.; Bowers W. J.; Federoff H. J.; Dawson V. L.; Dawson T. M. (2010) Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat. Med. 16, 998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C. L.; Rovelli G.; Springer W.; Schall C.; Gasser T.; Kahle P. J. (2009) Homo- and heterodimerization of ROCO kinases: LRRK2 kinase inhibition by the LRRK2 ROCO fragment. J. Neurochem. 111, 703–715. [DOI] [PubMed] [Google Scholar]
- Anand V. S.; Reichling L. J.; Lipinski K.; Stochaj W.; Duan W.; Kelleher K.; Pungaliya P.; Brown E. L.; Reinhart P. H.; Somber R.; Hirst W.; Riddle S. M.; Braithwaite S. P. (2009) Investigation of leucine-rich repeat kinase 2: enzymological properties and novel assays. FEBS J. 276, 466–478. [DOI] [PubMed] [Google Scholar]
- Covy J. P.; Giasson B. I. (2009) Identification of compounds that inhibit the kinase activity of leucine-rich repeat kinase 2. Biochem. Biophys. Res. Commun. 378, 473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedro L.; Padrós J.; Beaudet L.; Schubert H.-D.; Gillardon F.; Dahan S. (2010) Development of a high-throughput AlphaScreen assay measuring full-length LRRK2(G2019S) kinase activity using moesin protein substrate. Anal. Biochem. 404, 45–51. [DOI] [PubMed] [Google Scholar]
- Nichols R. J.; Dzamko N.; Hutti J. E.; Cantley L. C.; Deak M.; Moran J.; Bamborough P.; Reith A. D.; Alessi D. R. (2009) Substrate specificity and inhibitors of LRRK2, a protein kinase mutated in Parkinson’s disease. Biochem. J. 424, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Hamamichi S.; Lee B. D.; Yang D.; Ray A.; Caldwell G. A.; Caldwell K. A.; Dawson T. M.; Smith W. W.; Dawson V. L. (2011) Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s disease models. Hum. Mol. Genet. 20, 3933–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamko N.; Deak M.; Hentati F.; Reith A. D.; Prescott A. R.; Alessi D. R.; Nichols R. J. (2010) Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14–3-3 binding and altered cytoplasmic localization. Biochem. J. 430, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichling L. J.; Riddle S. M. (2009) Leucine-rich repeat kinase 2 mutants I2020T and G2019S exhibit altered kinase inhibitor sensitivity. Biochem. Biophys. Res. Commun. 384, 255–258. [DOI] [PubMed] [Google Scholar]
- Yun H.; Heo H. Y.; Kim H. H.; DooKim N.; Seol W. (2011) Identification of chemicals to inhibit the kinase activity of leucine-rich repeat kinase 2 (LRRK2), a Parkinson’s disease-associated protein. Bioorg. Med. Chem. Lett. 21, 2953–2957. [DOI] [PubMed] [Google Scholar]
- Liu M.; Dobson B.; Glicksman M. A.; Yue Z.; Stein R. L. (2010) Kinetic mechanistic studies of wild-type leucine-rich repeat kinase 2: characterization of the kinase and GTPase activities. Biochemistry 49, 2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.; Poulose S.; Schuman E.; Zaitsev A. D.; Dobson B.; Auerbach K.; Seyb K.; Cuny G. D.; Glicksman M. A.; Stein R. L.; Yue Z. (2010) Development of a mechanism-based high-throughput screen assay for leucine-rich repeat kinase 2--discovery of LRRK2 inhibitors. Anal. Biochem. 404, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X.; Dzamko N.; Prescott A.; Davies P.; Liu Q.; Yang Q.; Lee J.-D.; Patricelli M. P.; Nomanbhoy T. N.; Alessi D. R.; Gray N. S. (2011) Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat. Chem. Biol. 7, 203–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden N.; Perrin J.; Ren Z.; Lee B. D.; Zinn N.; Dawson V. L.; Tam D.; Bova M.; Lang M.; Drewes G.; Bantscheff M.; Bard F.; Dawson T. M.; Hopf C. (2011) Chemoproteomics-Based Design of Potent LRRK2-Selective Lead Compounds That Attenuate Parkinson’s Disease-Related Toxicity in Human Neurons. ACS Chem. Biol. 6, 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaharski A. J.; Bitter H.; Gonzaludo N.; Kondru R.; Goldstein D. M.; Zabka T. S.; Lin H.; Singer T.; Kolaja K. (2010) Modeling bone marrow toxicity using kinase structural motifs and the inhibition profiles of small molecular kinase inhibitors. Toxicol. Sci. 118, 266–275. [DOI] [PubMed] [Google Scholar]
- Olaharski A. J.; Gonzaludo N.; Bitter H.; Goldstein D.; Kirchner S.; Uppal H.; Kolaja K. (2009) Identification of a kinase profile that predicts chromosome damage induced by small molecule kinase inhibitors. PLoS Comput. Biol. 5, e1000446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan B., Estrada A., Sweeney Z., and Mciver E. G. (2011) Pyrazolopyridines as inhibitors of the kinase LRRK2. WO 2011/141756 A1.
- Kim J. W., Lee J., Song H.-J., Kim Y., Lee H. K., Choi J.-S., Lim S.-H., and Chang S. (2011) Kinase Inhibitors. WO 2011/053861 A1.
- Lee J., Song H.-J., Koh J. S., Lee L. K., Kim Y., Chang S., Kim H. W., Chang S., Lim S.-H., Choi J.-S., Kim J.-H., and Kim S.-W. (2011) Kinase Inhibitors. WO 2011/060295 A1.
- Mciver E. G., Smiljanic E., Harding D. J., and Hough J. (2010) Compounds (I). WO 2010/106333 A1.
- Nichols P. L., Eatherton A. J., Bamborough P., Jandu K. S., Philips O. J., and Andreotti D. (2011) WO 2011/038872 A1.
- Ramsden N. (2009) Use Of LRRK2 Inhibitors For Neurodegenerative Disease. WO 2009/127642 A2.
- Correia Guedes L.; Ferreira J. J.; Rosa M. M.; Coelho M.; Bonifati V.; Sampaio C. (2010) Worldwide frequency of G2019S LRRK2 mutation in Parkinson’s disease: A systematic review. J. Prak. Rel. Dis. 16, 237–242. [DOI] [PubMed] [Google Scholar]
- http://www.tautatis.com/home.html (2011).
- Yue Z. (2012) Genetic mouse models for understanding LRRK2 biology, pathology and pre-clinical application. Parkinsonism Relat. Disord. 18(Suppl 1), S180–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z.; Lachenmayer M. L. (2011) Genetic LRRK2Models of Parkinson’s Disease: Dissecting the Pathogenic Pathway and Exploring Clinical Applications. Movement Disord. 26, 1386–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://www.pdonlineresearch.org/ (2012).
- Doggett E. A.; Zhao J.; Mork C. N.; Hu D.; Nichols R. J. (2012) Phosphorylation of LRRK2 serines 955 and 973 is disrupted by Parkinsońs disease mutations and LRRK2 pharmacological inhibition. J. Neurochem. 120, 37–45. [DOI] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Rauh D. (2010) Inaktive Kinasekonformationen stabilisieren. Nachr. Chem. 58, 118–121. [Google Scholar]