

Abstract
Neuropeptide Y (NPY) is a 36 amino acid peptide, widely distributed within central nervous system neurons. More recently, it has been shown that NPY is involved in Alzheimer's disease (AD), a disorder characterized by accumulation of amyloid β-peptide (Aβ) in neurons. In a previous study, we investigated the effect of NPY on neuronal damage by exposing SH-SY5Y cells (an established human derived neuroblastoma cell line) to Aβ’s pathogenic fragment 25–35 (Aβ25–35). We found a NPY-neuroprotective action associated with changes in intracellular production of nerve growth factor (NGF), a member of the neurotrophin family. Since our results were encouraging, we decided to replicate our data using primary cortical neurons cultured in presence of Aβ25–35, and investigated whether NPY had similar neuroprotective action. Moreover, since cortical neurons are able to produce and release NGF, we investigated whether the synthesis and release of NGF were modified in such experimental conditions. Our results showed that a preincubation with NPY counteracted the toxic effect of Aβ, as measured by increased cell viability. Moreover, NPY pretreatment had an effect on NGF since its intracellular synthesis was increased, release was normalized, and mRNA expression was downregulated. Notably, these effects on NGF were in the opposite direction of those produced by incubating the cells with Aβ alone. This study in primary cortical neurons supports the hypothesis that NPY may be a neuroprotective agent against β-amyloid neurotoxicity. These data also suggest that NPY may influence the synthesis and the release of NGF by cortical neurons.
Keywords: NPY, β-amyloid, primary cells, cerebral cortex, NGF, cortical neurons
Neuropeptide Y (NPY) is a 36 amino acid peptide, widely distributed within neurons of the central and peripheral nervous systems, and it is found in mammalian brain in higher concentrations than all other peptides studied to date.1 It acts as a neurotransmitter and/or a modulator of several neuroendocrine functions including cardiovascular physiology and feeding behavior.2 Studies performed in laboratory animals have shown that, in the central nervous system (CNS), NPY produces behavioral effects that are close to those induced by anxiolytics,3−5 antiepileptics,6 and antidepressants.7−9
More recently, it has been shown that NPY is involved in Alzheimer's disease (AD), a disorder characterized by the accumulation of amyloid plaques, built up mainly of the amyloid β-peptide (Aβ), and neurofibrillary tangles, made by the microtubule-binding protein tau.10 A significant NPY reduction in the hippocampal regions of AD brains has been shown11 in conjunction with an altered number of NPY receptors.12 Also, reduced plasma13 and cerebrospinal fluid NPY levels14 have been observed during AD progression, thus confirming a close correlation between NPY and AD pathogenesis and/or pathophysiology.
Interestingly, a neuroprotective effect of NPY against AD neuronal damages in the CNS has been reported in AD transgenic mouse models,15 although the mechanism of action of this NPY-mediated neuroprotection is still under investigation. In the attempt to elucidate this mechanism, in a previous study, we investigated the effect of NPY on neuronal damage by exposing SH-SY5Y cells (a well established human derived neuroblastoma cell line) to Aβ’s pathogenic fragment 25–35 (Aβ25–35).16 We found a NPY-neuroprotective action on these cells associated with changes in intracellular production of nerve growth factor (NGF), a member of the neurotrophin family. NGF is relevant to AD because it supports survival and function of cholinergic neurons of the basal forebrain (BFCNs)17 which undergo degeneration early and contribute significantly to cognitive decline in AD.18 Accordingly, it has been shown that decreased supply of NGF at the level of BFCN cell bodies leads to loss of neuronal markers and shrinkage, mimicking what is observed in AD.19 In addition, NPY shares with NGF the ability to counteract excitotoxicity20 and modulate neurogenesis.21−23
Nevertheless, although our results were encouraging, the neuroblastoma cells used in that study were tumor cells with protein expression patterns rather different from nontumor cells. Thus, being aware of these issues, we decided to replicate our data using primary cells which are more close to normal physiologic conditions. We used the rat primary cortical neurons cultured in presence of Aβ25–35, and investigated whether NPY had similar neuroprotective action. Moreover, since cortical neurons are able to produce and release NGF, which is in turn retrogradely transported to BFCNs,24 we investigated whether the synthesis and release of NGF would be modified in such experimental conditions.
Results and Discussion
In this study, we investigated the effect of preincubation with NPY on primary cortical neurons of fetal rats exposed to amyloid beta peptide fragment 25–35. Our aim was to verify the possibility that NPY may exert neuroprotective effects on cortical neurons and regulate the expression of nerve growth factor by these cells.
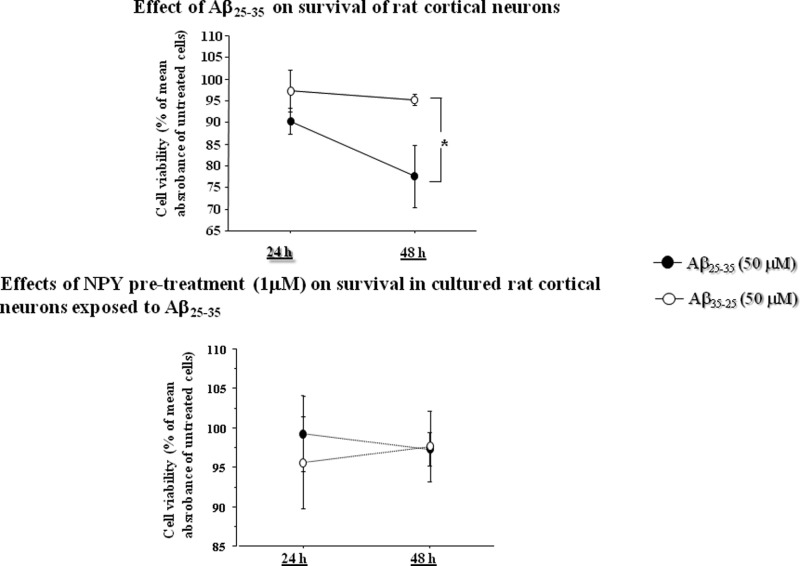
Aβ25–35 Reduces Rat Cortical Neuron Viability and Pretreatment with NPY Protects Neurons from Aβ25–35 Toxicity
The neuroprotective effect of NPY on neuronal survival was investigated. Primary cortical neurons were preincubated either alone (positive control) or with three concentrations of NPY (0.5, 1, and 2 μM) for 24 h and then exposed to Aβ25–35 (50 μM) or Aβ35–25 (50 μM) for 48 h (Figure 1). At 24 and 48 h, neuronal survival was evaluated with MTS assay.
Figure 1.

NPY effects on cell viability in cultured rat cortical neurons exposed to Aβ25–35. Cortical neurons were preincubated alone (a) or with three concentrations of NPY [(b) 2 mM, (c) 1 mM, and (d) 0.5 mM] for 24 h and then exposed to Aβ25–35 (50 μM) or Aβ35–25 (50 μM) for 48 h (number of experiments performed = 4). After 24 and 48 h, cells were tested with MTS assay. NPY treatment restored normal cell survival. Cell viability is expressed as percent of the mean absorbance value of untreated cells at each time point. Data represent means ± SEM
As shown in Figure 1a, ANOVA showed that there was a significant effect of the treatment in positive control (p < 0.05). Posthoc comparisons revealed that after 48 h of incubation Aβ25–35 significantly reduced cell viability as compared to Aβ35–25 (50 μM) (p < 0.05).
The results also showed that NPY was able to protect cortical neurons from Aβ25–35 toxicity. We found that 2 μM NPY abolished the toxic effects of Aβ25–35 (p = 0.27 for the effect of the treatment) at 24 and 48 h (Figure 1b). The same effect on neuronal survival was observed in neurons exposed to 1 μM (Figure 1c) (p = 0.71; treatment effect) and 0.5 μM (Figure 1d) (p = 0.27; treatment effect) NPY pretreatments.
Aβ25–35 Reduces NGF Synthesis and Release and Increases NGF mRNA
In order to evaluate the effect of Aβ treatment on NGF synthesis and release, we incubated for 24 h cortical neurons with Aβ25–35 (50 μM) or Aβ35–25 (50 μM) and measured NGF protein levels in neurons and the release in culture medium (Figure 2a,b). We found an effect of the treatment on NGF synthesis (p < 0.05) and an interaction between treatment and time (p < 0.05) (Figure 2a). Post hoc analysis showed that Aβ25–35 reduced NGF synthesis at 24 h (p < 0.01) and 48 h (p < 0.05). The release of NGF was also affected by Aβ25–35 treatment (p < 0.01) and reduced NGF was observed in the culture medium of Aβ25–35 exposed cortical neurons at 24 h (p < 0.05) and 48 h (p < 0.05) (Figure 2b).
Figure 2.

NGF protein [(a) synthesis, (b) release] and mRNA (c) in cortical neurons incubated with Aβ25–35 (50 μM) or its inactive control Aβ35–25 (50 μM). Cell pellets were collected at 24 and 48 h. NGF protein levels are expressed in pg/g total proteins. NGF mRNA was measured by real time PCR and expressed as amount of target gene normalized to an endogenous reference (β-actin) and relative to a calibrator (ΔΔCt) Data represent means ± SEM. Asterisks indicate statistical level of significance (*p < 0.05; **p < 0 01).
NGF mRNA was evaluated by real time PCR. An effect of Aβ treatment was observed (p < 0.05). While NGF synthesis was reduced, we found that NGF mRNA was increased by Aβ25–35 at 24 h (p < 0.05). It is possible that the increase in NGF mRNA at 24 h may represent an early attempt to counteract Aβ25–35 neurotoxic insult. An effect of time was also noted (p < 0.01) because NGF mRNA increased with time in both groups (Figure 2c).
Pretreatment with NPY Increases NGF Synthesis, Reduces NGF mRNA, and Restores NGF Release in Cortical Neurons Exposed to Aβ25–35
To investigate whether the effects of NPY on cell survival were associated with changes in NGF production, NPY (1 μM) pretreated cortical neurons were exposed to Aβ25–35 (50 μM) or its inactive control Aβ35–25 (50 μM) (Figure 3).
Figure 3.

NGF protein [(a) synthesis, (b) release] and mRNA (c) in cortical neurons pretreated with NPY (1 mM) for 24 h and then exposed to toxic concentration (50 μM) of Aβ25–35 or its inactive control Aβ35–25 (50 μM). Cell pellets were collected at 24 and 48 h. NGF protein levels are expressed in pg/g total protons. NGF mRNA was measured by real time PCR and expressed as amount of target gene normalized to an endogenous reference (β-actin) and relative to a calibrator (ΔΔCt). Data represent means ± SEM. Asterisks indicate statistical level of significance (*p < 0.05).
For intracellular NGF production, we found an effect of the treatment (p < 0.05) and an interaction between treatment and time (p < 0.01). Post hoc analysis showed that NGF synthesis was increased in Aβ25–35 exposed neurons (p < 0.05) at 24 h (Figure 3a). Release of NGF in the culture medium was normalized by NPY pretreatment (Figure 3b). No differences in NGF release between Aβ25–35 and Aβ35–25 were noted. An effect of time was present, since release of NGF significantly decreased at 48 h in both groups (p < 0.01).
NGF mRNA was also evaluated in cortical neurons pretreated with NPY (1 μM) and exposed to Aβ25–35 (50 μM) or its inactive control Aβ35–25 (50 μM) (Figure 3c). There was a significant effect of the treatment (p < 0.05) and an interaction between treatment and time (p < 0.05). We found that NGF mRNA level after Aβ25–35 treatment was not different from that of Aβ35–25 at 24 h and even decreased at 48 h suggesting that NPY pretreatment had a counteracting effect on NGF mRNA on neurons subsequently exposed to toxic levels of Aβ25–35.
This study was the logical extension of a previous experiment performed in our laboratory using a human neuroblastoma cell line.16 In that study, we observed a neuroprotective effect of NPY against Aβ neurotoxicity associated with increased synthesis of neurotrophins by neuroblastoma cells. Thus, in order to verify these data in primary cells (which are taken directly from living tissue) and further investigate the mechanism of action of NPY on neurotrophins, we exposed rat cortical neurons to the biologically active region of Aβ (fragment Aβ25–35),25 pretreated them with NPY, and measured synthesis, release, and gene expression of nerve growth factor.
Our results showed that a preincubation with NPY counteracted the toxic effect of Aβ, as measured by increased cell viability. Moreover, NPY pretreatment also had an effect on NGF since its intracellular synthesis was increased, release was normalized, and mRNA expression was downregulated. Notably, these effects on NGF were in the opposite direction of those produced by incubating the cells with Aβ alone.
This study represents an additional step to further investigate the possibility that NPY is a potential neuroprotective agent in Alzheimer's disease. The fact that NPY exerted neuroprotection against Aβ toxicity in both neuroblastoma (an immortalized cell line) and primary cells is encouraging. However, the mechanism of this NPY-induced neuroprotection is not completely clear. In our previous paper,16 we hypothesized that NPY exerts its neuroprotective action by inhibiting neuronal excitability through a mechanism involving the inhibition of the voltage-dependent Ca2+ influx in presynaptic nerve terminals,26−28 an effect probably mediated by presynaptic activation of NPY Y2 receptors.20,29,30 This idea is supported by some studies showing that NPY is able to inhibit neuronal excitability in in vivo and in vitro models.31 In addition, transgenic mice for human amyloid precursor protein (hAPP) have spontaneous nonconvulsive seizure activity in cortical and hippocampal networks, associated with GABAergic sprouting, enhanced synaptic inhibition, and synaptic plasticity deficits in the dentate gyrus supporting the idea that agents that prevent neuronal excitability may be neuroprotective to Aβ -induced neurological deficits.32
Further pharmacological characterization of NPY-receptor subtypes will assess the possibility that NPY-receptor agonists or antagonists can be considered as valuable compounds to prevent neurodegeneration. In addition, it is of interest that NPY may cross the blood-brain barrier (BBB) by passive diffusion,33 while other proteins of potential therapeutic interest in AD, including NGF, possess poor permeability to the BBB.
Another important finding is the effect of NPY on synthesis and release of NGF. It is well documented that NGF is a critical survival factor for cholinergic neurons which undergo neurodegeneration in late stage Alzheimer's disease.18,19 We found that Aβ reduced the synthesis and release of this neurotrophins. Since NGF is synthesized in the cortex and retrogradely transported to basal forebrain cholinergic neurons,19 it is possible that basal forebrain neurons may be damaged not only by the accumulation of Aβ and neurofibrillary tangles but also by reduced NGF support from cerebral cortex neurons. Thus, there is the possibility that NPY by increasing NGF synthesis and release in cortical neurons may have beneficial effects to the basal forebrain. However, this hypothesis needs to be further investigated. A necessary step to validate this hypothesis could be to administer NPY in appropriate AD animal models and verify its potential neuroprotective effects to the cholinergic system. This idea is also supported by an immunocytochemistry study demonstrating that loss of cholinergic neurons in the basal forebrain of AD patients is selective and, notably, NPY-positive neurons are preserved.34 Another issue to be studied is the potential side effects of NPY in vivo, consisting in peripheral hemodynamic effects which may limit its use in human therapy.
In conclusion, this study in primary cortical neurons supports the hypothesis that NPY may be a neuroprotective agent against beta amyloid neurotoxicity. These data also suggest that NPY may influence the synthesis and the release of NGF by cortical neurons. The possible implications of these findings in AD pathophysiology need to be explored.
Methods
Primary Cell Cultures
Cortical neuron cultures were prepared from embryonic day 17–19 fetal Wistar rats. The brains were removed and cortices were freed of meninges, washed with 6 mL of Earl’s balanced salt solution, and centrifuged for 2 min at 150g. The tissue was resuspended and incubated for 30 min at 37 °C with 0.02% trypsin followed by addition of DNase I (80 μg/mL final) and trypsin inhibitor (0.52 mg/mL). Digested tissues were mechanically dissociated twice and centrifuged at 150g for 10 min. The dissociated cells were plated at a density of 2 × 105 cells/cm2 on poly-l-lysine (Sigma-Aldrich) wells in Neurobasal media (Gibco, cat. no. 21103-049) supplemented with 2% B27 (Gibco, cat. no. 17504-044),35 0.5 mM l-glutammine (Euroclone, cat. no. ECB3000D), and 50 U/mL penicillin/streptomycin solution (Euroclone, cat. no. ECB3001D). Cells were grown at 37 °C and 5% CO2, changing half medium every 3 days from the seeding to the execution of the experiments, when we halved the concentration of B-27 supplement. Cells from the 11th day on were used for experiments.
All animal experiments were performed in accordance to the Italian law on the use and care of laboratory animals (DL 116\92).
Drugs
NPY (human, rat; cat. no. H-6375, Bachem AG, Switzerland) was resuspended in distilled H2O at a stock concentration of 0.1 mM, filtered/sterilized, and stored at −20 °C.
Aβ protein (fragment 25–35; cat. no. H-1192, Bachem AG, Switzerland) and its inactive form (Aβ protein 35–25; cat. no. H-2964, Bachem AG, Switzerland) were both reconstituted in sterile distilled H2O at a concentration of 1 mM, filtered/sterilized, incubated for 72 h at 37 °C to induce aggregation, and immediately stored at −20 °C. Aggregation was confirmed by electron microscope. The reason why we used Aβ25–35 is that this short fragment retains the toxicity of Aβ1–4236−39 but, unlike the full-length peptide, it forms fibrils immediately after solution.40
Cell Treatments for Survival Study and NGF Analysis
To evaluate the neuroprotective effects of NPY, neurons were preincubated alone or with different NPY concentrations (0.5, 1, and 2 μM) for 24 h and then the same concentration of active/inactive Aβ-peptide (50 μM) was added to the medium for 48 h. In such conditions, we have evidence in primary cell cultures that NPY may possess biological activity at least for 48 h.41−43 After incubation, neuronal survival was evaluated with MTS assay. Three independent experiments were performed for each condition.
Based on the data obtained from survival study, for NGF studies, peptide concentrations were fixed at 50 μM for Aβ25–35 and at 1 μM for NPY pretreatment. Neurons were first exposed to Aβ25–35 or its inactive control Aβ35–25 for 48 h. In the second experiment, the cells were pretreated with NPY (1 μM) and then exposed to Aβ peptides at the same conditions. At the end of each experiment, cortical neurons were gathered with QIAzol Lysis Reagent (Qiagen) and stored at −80 °C for analyses using the RNA, while cells (gathered in RIPA buffer) and media were collected and stored at −80 °C for NGF immunoassay (ELISA) and for quantification of total proteins with Bradford assay (BioRad Protein Assay, BioRad Laboratories GmbH). The suitable times of cell collection were fixed at 24 and 48 h. Three independent experiments were performed for each experimental treatment.
MTS Assay
At 24 and 48 h, we performed a MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)] assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay, Promega) in order to study cell survival and select the concentration of β-amyloid and NPY to be used in successive experiments. MTS assay was performed according to manufacturer instructions. Briefly, cortical neurons were cultured in a 96-well microtiter plate. After treatment, 20 μL of MTS solution was added to each well. Then, after an incubation of 1–2 h, the absorbance was measured at 492 nm by a multiplate reader (Benchmark Plus, Biorad).
Real-Time PCR
Total RNA was extracted and purified using the miRNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. The quality and quantity of total RNA was assessed by using the NanoDrop ND-1000 spectrophotometer (A260/280 > 2.00; A260/230 > 1.9) and its integrity was checked by 1% agarose gel electrophoresis. For total cDNA reverse transcription (RT), we used the protocol and reagents of the ImProm-II Reverse Transcription System (Promega) kit. The final volume of 20 μL contained 500 ng RNA, 500 ng of random examers, 0.5 mM dNTPs mix, 1× ImProm-II Reaction Buffer, 5 mM MgCl2, 20 U Rnasin, and 1 μL of ImProm-II reverse transcriptase.
Real-time PCR for NGF quantification was performed in the Mx3000P Stratagene (La Jolla, CA) instrument, using the DNA binding dye SYBR Green (SYBR Green PCR Master Mix; Applied Biosystems). Twenty microliters of PCR mix containing 10 μL of SYBR Green mix, forward and reverse primers, and 2 μL of cDNA obtained from the RT procedure were used for comparative quantification. We used 300 nM of forward and reverse primers for NGF and 150 nM of both primers for the housekeeping gene β-actin. Primers were designed using Primer Blast (NCBI) and synthesized by Primm srl (Italy). All primers were designed to be specific for a unique sequence within the total rat genome as well as to be located on different exons to avoid DNA contamination. The sequences of each primer are listed in table 1.
Table 1. Real-Time PCR Primer Sequences.
| NGF forward | 5′-GGACGCAGCTTTCTATCCTG-3′ | product length (bp): 368 |
| NGF reverse | 5′-CATGGGCCTGGAAGTCTAAA-3′ | |
| β-actin forward | 5′-CTTCCTGGGTATGGAATCCTGT-3′ | product length (bp): 364 |
| β-actin reverse | 5′-GGTGTAAAACGCAGCTCAGTAAC-3′ |
Negative controls with no cDNA sample were run to exclude PCR mix contamination. Samples for NGF and β-actin transcripts were run in triplicate.
The thermal profile was as follows: 5 min at 95 °C followed by 50 cycles of 95 °C for 15 s, 59 °C for 30 s, and 72 °C for 30 s. The amplification specificity was confirmed by melting-curve analysis of the PCR products. Standard curves for NGF and β-actin were also performed to verify that the amplification efficiencies of both the target and control genes were comparable within a range of 5% from each other. The 2-DDCt method was used to calculate relative changes in gene expression determined from real-time quantitative PCR experiments.44
Protein Extraction
Cell pellets were resuspended and homogenized in 0.3 mL of ice-cold RIPA buffer, containing 150 mM NaCl, 50 mM Tris–HCl (pH 7.5), 1% NP40, 0.1% SDS, 1 mM AEBSF (freshly prepared), 2 mM DTT (freshly prepared), and 0.5% deoxycholate. The tissue homogenate solutions were centrifuged at 14 000g for 5 min at 4 °C. The supernatants were collected and used for quantification of NGF and total proteins.
NGF Determination with Immunoassay (ELISA)
NGF concentrations in cell pellet and culture medium were determined in sandwich ELISAs according to the manufacturer’s instructions (Promega). NGF concentrations were determined from the regression line for the standard (ranging from 3.9 to 500 pg/mL, purified mouse NGF) incubated under similar conditions in each assay. The detection limit was <4 pg/mL. NGF concentration was expressed as pg NGF/g total proteins. Cross-reactivity to other related neurotrophins, such as neurotrophin-3 (NT-3) and neurotrophin-4/5 (NT-4/5), was less than 3%. All assays were performed in duplicate.
Bradford Assay
We performed a Bradford assay on pellets and media used for ELISA assays to quantify the total protein amount. We used Bradford solution (Biorad) and BSA 1 mg/mL (Sigma) to do the standard curve in a Varian spectrophotometer.
Statistical Analysis
Differences between groups of treatment were analyzed by analysis of variance (ANOVA) followed by Fisher’s protected least significant difference (PLSD) posthoc test. Interaction between treatment and time was evaluated by two-way ANOVA. A p-value of <0.05 was considered statistically significant. Statistical analysis was performed using the Statview software from SAS Institute.
Glossary
Abbreviations
- NPY
neuropeptide Y
- AD
Alzheimer's disease
- CNS
central nervous system
- Aβ
amyloid β-peptide
- NGF
nerve growth factor
- BBB
blood-brain barrier
- BFCNs
cholinergic neurons of the basal forebrain
Author Contributions
F.A., N.C., and F.G. designed the study and wrote the protocol. T.C. provided primary rat cortical neurons and protocols. N.C., F.G., and S.C. performed experimental procedures. G.F., C.C., and S.B. managed the literature searches and analyses. F.A. undertook the statistical analysis, and F.A. and N.C. wrote the first draft of the manuscript. All authors contributed to and have approved the final manuscript.
Grant nEUROsyn Italian Ministry of Health (RC2008) in the frame of ERA-Net Neuron.
The authors declare no competing financial interest.
References
- Gray T. S.; Morley J. E. (1986) Neuropeptide Y: Anatomical distribution and possible function in mammalian nervous system. Life Sci. 38, 389–401. [DOI] [PubMed] [Google Scholar]
- Colton C. A.; Vitek M. P. (2006) NPY and chronic neurodegenerative disease: NPY family of peptides in neurobiology, cardiovascular and metabolic disorders from genes to therapeutics. J. Neural Transm. 113, 231–238.15959845 [Google Scholar]
- Gutman A. R.; Yang Y.; Ressler K. J.; Davis M. (2008) The role of neuropeptide Y in the expression and extinction of fear-potentiated startle. J. Neurosci. 28, 12682–12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.; Feder A.; Wegener G.; Bailey C.; Saxena S.; Charney D.; Mathé A. A. (2011) Central functions of neuropeptide Y in mood and anxiety disorders. Expert Opin. Ther. Targets 15, 1317–1331. [DOI] [PubMed] [Google Scholar]
- Cohen H.; Liu T.; Kozlovsky N.; Kaplan Z.; Zohar J.; Mathé A. A. (2012) The Neuropeptide Y (NPY)-ergic System is Associated with Behavioral Resilience to Stress Exposure in an Animal Model of Post-Traumatic Stress Disorder. Neuropsychopharmacology 37, 350–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bahh B.; Balosso S.; Hamilton T.; Herzog H.; Beck-Sickinger A. G.; Sperk G.; Gehlert D. R.; Vezzani A.; Colmers W. F. (2005) The anti-epileptic actions of neuropeptide Y in the hippocampus are mediated by Y and not Y receptors. Eur. J. Neurosci. 22, 1417–1430. [DOI] [PubMed] [Google Scholar]
- Goyal S. N.; Upadhya M. A.; Kokare D. M.; Bhisikar S. M.; Subhedar N. K. (2009) Neuropeptide Y modulates the antidepressant activity of imipramine in olfactory bulbectomized rats: involvement of NPY Y1 receptors. Brain Res. 1266, 45–53. [DOI] [PubMed] [Google Scholar]
- Bjørnebekk A.; Mathé A. A.; Brené S. (2010) The antidepressant effects of running and escitalopram are associated with levels of hippocampal NPY and Y1 receptor but not cell proliferation in a rat model of depression. Hippocampus 20, 820–828. [DOI] [PubMed] [Google Scholar]
- Jiménez-Vasquez P. A.; Diaz-Cabiale Z.; Caberlotto L.; Bellido I.; Overstreet D.; Fuxe K.; Mathé A. A. (2007) Electroconvulsive stimuli selectively affect behavior and neuropeptide Y (NPY) and NPY Y(1) receptor gene expressions in hippocampus and hypothalamus of Flinders Sensitive Line rat model of depression. Eur. Neuropsychopharmacol. 17, 298–308. [DOI] [PubMed] [Google Scholar]
- Querfurth H. W.; LaFerla F. M. (2010) Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. [DOI] [PubMed] [Google Scholar]
- Kowall N. W.; Beal M. F. (1988) Cortical somatostatin, neuropeptide Y, and NADPH diaphorase neurons: normal anatomy and alterations in Alzheimer’s disease. Ann. Neurol. 23, 105–114. [DOI] [PubMed] [Google Scholar]
- Martel J. C.; Alagar R.; Robitaille Y.; Quirion R. (1990) Neuropeptide Y receptor binding sites in human brain: possible alteration in Alzheimer’s disease. Brain Res. 519, 228–235. [DOI] [PubMed] [Google Scholar]
- Koide S.; Onishi H.; Hashimoto H.; Kai T.; Yamagami S. (1995) Plasma neuropeptide Y is reduced in patients with Alzheimer’s disease. Neurosci. Lett. 198, 149–151. [DOI] [PubMed] [Google Scholar]
- Alom J.; Galard R.; Catalan R.; Castellanos J. M.; Schwartz S.; Tolosa E. (1990) Cerebrospinal fluid neuropeptide Y in Alzheimer’s disease. Eur. Neurol. 30, 207–210. [DOI] [PubMed] [Google Scholar]
- Rose J. B.; Crews L.; Rockenstein E.; Adame A.; Mante M.; Hersh L. B.; Gage F. H.; Spencer B.; Potkar R.; Marr R. A.; Masliah E. (2009) Neuropeptide Y fragments derived from neprilysin processing are neuroprotective in a transgenic model of Alzheimer’s disease. J. Neurosci. 29, 1115–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce N.; Dinallo V.; Ricci V.; Federici G.; Caltagirone C.; Bernardini S.; Angelucci F. (2011) Neuroprotective effect of neuropeptide Y against beta-amyloid 25–35 toxicity in SH-SY5Y neuroblastoma cells is associated with increased neurotrophin production. Neurodegener. Dis. 8, 300–309. [DOI] [PubMed] [Google Scholar]
- Schliebs R.; Arendt T. (2011) The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 221, 555–563. [DOI] [PubMed] [Google Scholar]
- Allen S. J.; MacGowan S. H.; Treanor J. J.; Feeney R.; Wilcock G. K.; Dawbarn D. (1991) Normal beta-NGF content in Alzheimer’s disease cerebral cortex and hippocampus. Neurosci. Lett. 131, 135–139. [DOI] [PubMed] [Google Scholar]
- Salesi A.; Delcroix J. D.; Swaab D. F. (2004) Alzheimer’s disease and NGF signaling. J. Neural Transm. 111, 323–345. [DOI] [PubMed] [Google Scholar]
- Silva A. P.; Pinheiro P. S.; Carvalho A. P.; Carvalho C. M.; Jakobsen B.; Zimmer J.; Malva J. O. (2003) Activation of neuropeptide Y receptors is neuroprotective against excitotoxicity in organotypic hippocampal slice cultures. FASEB J. 17, 1118–1120. [DOI] [PubMed] [Google Scholar]
- Decressac M.; Wright B.; David B.; Tyers P.; Jaber M.; Barker R. A.; Gaillard A. (2010) Exogenous neuropeptide Y promotes in vivo hippocampal neurogenesis. Hippocampus 21, 233–238. [DOI] [PubMed] [Google Scholar]
- Howell O. W.; Doyle K.; Goodman J. H.; Scharfman H. E.; Herzog H.; Pringle A.; Beck-Sickinger A. G.; Gray W. P. (2005) Neuropeptide Y stimulates neuronal precursor proliferation in the post-natal and adult dentate gyrus. J. Neurochem. 93, 560–570. [DOI] [PubMed] [Google Scholar]
- Howell O. W.; Silva S.; Scharfman H. E.; Sosunov A. A.; Zaben M.; Shatya A.; McKhann G.; Herzog H.; Laskowski A.; Gray W. P. (2007) Neuropeptide Y is important for basal and seizure-induced precursor cell proliferation in the hippocampus. Neurobiol. Dis. 26, 174–188. [DOI] [PubMed] [Google Scholar]
- Niewiadomska G.; Mietelska-Porowska A.; Mazurkiewicz M. (2011) The cholinergic system, nerve growth factor and the cytoskeleton. Behav. Brain Res. 221, 515–526. [DOI] [PubMed] [Google Scholar]
- Del Mar Martinez-Senac M.; Villalain J.; Gomez-Fernandez J. C. (1999) Structure of the Alzheimer beta-amyloid peptide (25–35) and its interaction with negatively charged phospholipid vesicles. Eur. J. Biochem. 265, 744–753. [DOI] [PubMed] [Google Scholar]
- Qian J.; Colmers W. F.; Saggau P. (1997) Inhibition of synaptic transmission by neuropeptide Y in rat hippocampal area CA1: modulation of presynaptic Ca2+ entry. J. Neurosci. 17, 8169–8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A. P.; Carvalho A. P.; Carvalho C. M.; Malva J. O. (2001) Modulation of intracellular calcium changes and glutamate release by neuropeptide Y1 and Y2 receptors in the rat hippocampus: differential effects in CA1, CA3 and dentate gyrus. J. Neurochem. 79, 286–296. [DOI] [PubMed] [Google Scholar]
- Silva A. P.; Carvalho A. P.; Carvalho C. M.; Malva J. O. (2003) Functional interaction between neuropeptide Y receptors and modulation of calcium channels in the rat hippocampus. Neuropharmacology 44, 282–292. [DOI] [PubMed] [Google Scholar]
- Smiałowska M.; Domin H.; Zieba B.; Koźniewska E.; Michalik R.; Piotrowski P.; Kajta M. (2009) Neuroprotective effects of neuropeptide Y-Y2 and Y5 receptor agonists in vitro and in vivo. Neuropeptides 43, 235–249. [DOI] [PubMed] [Google Scholar]
- Xapelli S.; Silva A. P.; Ferreira R.; Malva J. O. (2007) Neuropeptide Y can rescue neurons from cell death following the application of an excitotoxic insult with kainate in rat organotypic hippocampal slice cultures. Peptides 28, 288–294. [DOI] [PubMed] [Google Scholar]
- Colmers W. F.; Bleakman D. (1994) Effects of neuropeptide Y on the electrical properties of neurons. Trends Neurosci. 17, 373–379. [DOI] [PubMed] [Google Scholar]
- Palop J. J.; Chin J.; Roberson E. D.; Wang J.; Thwin M. T.; Bien-Ly N.; Yoo J.; Ho K. O.; Yu G. Q.; Kreitzer A.; Finkbeiner S.; Noebels J. L.; Mucke L. (2007) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastin A. J.; Akerstrom V. (1999) Nonsaturable entry of neuropeptide Y into brain. Am. J. Physiol. 276, 479–482. [DOI] [PubMed] [Google Scholar]
- Lehéricy S.; Hirsch E. C.; Cervera-Piérot P.; Hersh L. B.; Bakchine S.; Piette F.; Duyckaerts C.; Hauw J. J.; Javoy-Agid F.; Agid Y. (1993) Heterogeneity and selectivity of the degeneration of cholinergic neurons in the basal forebrain of patients with Alzheimer’s disease. J. Comp. Neurol. 330, 15–31. [DOI] [PubMed] [Google Scholar]
- Di Penta A.; Mercaldo V.; Florenzano F.; Munck S.; Ciotti M. T.; Zalfa F.; Mercanti D.; Molinari M.; Bagni C.; Achsel T. (2009) Dendritic LSm1/CBP80-mRNPs mark the early steps of transport commitment and translational control. J. Cell. Biol. 184, 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen L. L.; Mortishire-Smith R. J.; Pollack S. J.; Shearman M. S. (1995) The toxicity in vitro of beta-amyloid protein. Biochem. J. 311, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike C. J.; Walencewicz-Wasserman A. J.; Kosmoski J.; Cribbs D. H.; Glabe C. G.; Cotman C. W. (1995) Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25–35 region to aggregation and neurotoxicity. J. Neurochem. 64, 253–265. [DOI] [PubMed] [Google Scholar]
- Shearman M. S.; Ragan C. I.; Iversen L. L. (1994) Inhibition of PC12 cell redox activity is a specific, early indicator of the mechanism of beta-amyloid-mediated cell death. Proc. Natl. Acad. Sci. U.S.A. 91, 1470–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzi E.; Holzemann G.; Seelig J. (1994) Reversible random coil-beta-sheet transition of the Alzheimer beta-amyloid fragment (25–35). Biochemistry 33, 1345–1350. [DOI] [PubMed] [Google Scholar]
- Hensley K.; Carney J. M.; Mattson M. P.; Aksenova M.; Harris M.; Wu J. F.; Floyd R. A.; Butterfield D. A. (1994) A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 91, 3270–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel D. E.; Eipper B. A.; Ronnett G. V. (2001) NeuropeptideY functions as a neuroproliferative factor. Nature 410, 940–944. [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z.; Pruszczyk P.; Colton C.; Yao J.; Shen G. H.; Myers A. K.; Wahlestedt C. (1993) Mitogenic effect of neuropeptide Y in rat vascular smooth muscle cells. Peptides 14, 263–268. [DOI] [PubMed] [Google Scholar]
- Protas L.; Qu J.; Robinson R. B. (2003) Neuropeptide Y: neurotransmitter or trophic factor in the heart?. News Physiol. Sci. 18, 181–185. [DOI] [PubMed] [Google Scholar]
- Zhao S.; Fernald R. D. (2005) Comprehensive algorhythm for quantitative real-time polymerase chain reaction. J. Comput. Biol. 12, 1045–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]