

Abstract
Histamine is recognized as a neurotransmitter or neuromodulator in the brain, and it plays a major role in the pathogenic progression after cerebral ischemia. Extracellular histamine increases gradually after ischemia, and this may come from histaminergic neurons or mast cells. Histamine alleviates neuronal damage and infarct volume, and it promotes recovery of neurological function after ischemia; the H1, H2, and H3 receptors are all involved. Further studies suggest that histamine alleviates excitotoxicity, suppresses the release of glutamate and dopamine, and inhibits inflammation and glial scar formation. Histamine may also affect cerebral blood flow by targeting to vascular smooth muscle cells, and promote neurogenesis. Moreover, endogenous histamine is an essential mediator in the cerebral ischemic tolerance. Due to its multiple actions, affecting neurons, glia, vascular cells, and inflammatory cells, histamine is likely to be an important target in cerebral ischemia. But due to its low penetration of the blood-brain barrier and its wide actions in the periphery, histamine-related agents, like H3 antagonists and carnosine, show potential for cerebral ischemia therapy. However, important questions about the molecular aspects and pathophysiology of histamine and related agents in cerebral ischemia remain to be answered to form a solid scientific basis for therapeutic application.
Keywords: Cerebral ischemia, histamine, histamine receptor, carnosine
Cerebral ischemia is defined as reduction of cerebral blood flow (CBF) to a critical threshold that propagates brain damage involving the entire brain or a selective region. Global cerebral ischemia entails reduction in CBF over the entire brain, which occurs commonly in patients who have a variety of clinical conditions such as cardiac arrest, shock, and asphyxia and in patients undergoing complex cardiac surgery.1 Ischemic stroke results from a transient or permanent reduction in CBF that is restricted to the territory of a major brain artery. The reduction in flow is, in most cases, caused by the occlusion of a cerebral artery either by an embolus or by local thrombosis.2 Every year, 15 million people worldwide suffer a stroke, among whom nearly 6 million die and another 5 million are left permanently disabled. So, stroke remains the third leading cause of death and the leading neurologic cause of long-term disability. So far, over 100 neuroprotective drugs shown effective in animals have been found to be virtually ineffective in the treatment of human ischemia and clinical trials of many compounds ended prematurely due to disruption of normal brain function and adverse effects, except for reperfusion with recombinant tissue plasminogen activator (rtPA) for stroke.3−5 rtPA is approved by the FDA for acute ischemic stroke treatment but with certain limitations such as a narrow therapeutic time window of 4.5 h and the risk of hemorrhage.6 No doubt, translating experimental results into effective drug therapies is extremely difficult. But the complex sequence of pathophysiological events that evolve over time and space may be the reason for the failure to find a viable neuroprotective agent. The major pathogenic mechanisms after ischemia include excitotoxicity, programmed cell death, inflammation, and cerebral reconstruction.2 Moreover, beside neurons, the other components of the neurovascular unit, glia (astrocytes, microglia, and oligodendroglia) and vascular cells (endothelia, pericytes, and vascular smooth muscle cells), also participate in the neurological injury. Therefore, it is necessary to search for protective agents directed at multiple targets.
Histamine is an important neurotransmitter or neuromodulator in the central nervous system. The soma of histaminergic neurons are located in the tuberomamillary nucleus in the hypothalamus, and their fibers are widely distributed throughout the brain.7 It is now thought that the histaminergic system commands general states of metabolism, consciousness, and memory.8 The non-neuronal pools in the brain include mast cells, which contain almost half of the histamine.9 Mast cells have been identified in the brain parenchyma, mainly in the thalamus and hypothalamus, and their function is still unclear. Histamine in the brain is not transported from plasma since it cannot penetrate the blood-brain barrier, but is formed from l-histidine by a specific enzyme, l-histidine decarboxylase (HDC).10 There are two major pathways of histamine metabolism: ring methylation and oxidative deamination by diamine oxidase.11 In the brain, most of the histamine is catalyzed by histamine-N-methyltransferase to form tele-methylhistamine, which is converted by monoamine oxidase B to tele-methylimidazole acid. Three of the four histamine receptors that have been identified (H1–H3) are prominently expressed in the brain, whereas the fourth receptor (H4), which was discovered recently,12 is found predominantly in the periphery, for example, in bone marrow and leukocytes. The H1 receptor is a 486–491 amino acid protein, and is coupled to the Gq/11 protein and phospholipase C, while the H2 receptor consists of 358–359 amino acids and is coupled to Gs and protein kinase A.13 Unlike the H1 and H2 receptors, the H3 receptor is mainly located presynaptically, signals though Gi/Go proteins, displays significant constitutive activity, and negatively controls histamine release and synthesis.14 Recently, numerous studies have suggested that histamine and its receptors play important roles in cerebral ischemia by acting on multiple targets,15 which will be highlighted in this Review by concentrating on data related to the complex pathogenic mechanisms after cerebral ischemia.
Alteration of Histamine Content and Its Receptors after Cerebral Ischemia
In the experimental model of focal cerebral ischemia induced by occlusion of the middle cerebral artery (MCAO) in rats, the levels of histamine and tele-methylhistamine in the striatum and surrounding cerebral cortex gradually increase and these changes become pronounced and statistically significant 6–12 h after induction of ischemia.16 The striatal histamine and tele-methylhistamine reach levels three- and 2-fold higher, respectively, than those of the contralateral side. By microdialysis, the release of histamine was found to increase gradually over several hours after MCAO, whereas the release of glutamate and dopamine occurs immediately after induction of cerebral ischemia, and their values recover to basal levels after reperfusion.17 In the primate Macaca radiata, a rise in cerebral histamine level was also found following experimental infarction induced in the basal ganglia by coagulation of the middle cerebral artery.18
Adachi et al. reported that histamine release is markedly reversed by preischemic administration of alpha-fluoromethylhistidine (α-FMH, 100 mg/kg), an irreversible inhibitor of HDC, which generates histamine from l-histidine.17 α-FMH depletes histaminergic neurons of their histamine within the first few hours after treatment, since the turnover rate of neuronal histamine (half-life, <1 h) is more rapid than that of mast cell histamine (half-life, several days).9,19,20 So, Adachi et al. proposed that activation of histaminergic neurons is the predominant factor in the increase of histamine release after cerebral ischemia.17 However, in previous studies, we found that the number of mast cells decreases markedly after global ischemia, and this may be caused by degranulation.21 Meanwhile, the histamine content increases significantly in the thalamus and striatum. Moreover, Subramanian et al. also reported that the elevation of histamine level in the basal ganglia after infarction in this region is probably due to the proliferation of mast cells.18 Therefore, where the histamine comes from may depend on the ischemic region.
The expression and the binding density of histamine receptors also change after cerebral ischemia.22 At 48 h after transient global ischemia, H1 receptor mRNA expression increases but H2 receptor binding density and protein expression decrease in the caudate-putamen. H3 receptor mRNA expression increases in the caudate-putamen, but decreases in the globus pallidus and the thalamus. In association with this, H3 receptor binding density increases in the cortex, caudate-putamen, globus pallidus, and hippocampus; this receptor may be involved in regulating continuous neuronal histamine release. The increase of histamine release and the alteration of central histamine receptor expression or ligand binding suggest that histamine participates in the pathophysiological processes after cerebral ischemia.
Protective Effect of Histamine and Its Receptors on Cerebral Ischemia
Considerable evidence shows that histamine has a protective effect on neurological injury after cerebral ischemia. Intraperitoneal administration of histidine, a precursor of histamine, immediately and 6 h after reperfusion, markedly alleviates the infarction after MCAO.23 Intracerebroventricular administration of histamine (10–100 nmol) improves the delayed ischemic damage in hippocampal CA1 pyramidal cells induced by 3 min of transient forebrain ischemia.24 Our recent study found that histidine markedly protects against long-term injury in terms of neurological score, learning and memory, and infarct area, even 56 days after cerebral ischemia (unpublished data). Rats pretreated with α-FMH show significantly more necrotic hippocampal CA1 pyramidal cells than controls after cerebral ischemia caused by 10 min of 4-vessel occlusion.25 Moreover, in gerbils, α-FMH also aggravates the death of hippocampal CA2 neurons after 3 min ischemia, while the histamine content of the brain is significantly reduced.26 In addition, we found that the histamine from mast cells protects against neuronal death induced by oxygen-glucose deprivation, which is an in vitro model of ischemia.27
To further investigate the role of histamine receptors in cerebral ischemia, various receptor antagonists and agonists have been used. Intracerebroventricular administration of cimetidine and ranitidine, H2 antagonists, aggravate the neuronal damage after MCAO, which is alleviated by the H2 agonist dimaprit.28 But the H1 agonist HTMT and the H1 antagonist mepyramine do not affect the outcome. However, in a previous study, the H1 receptor was found to participate in delayed neuronal death caused by cerebral ischemia.25 Excitotoxicity is a predominant reason for neuronal death in the early phase after cerebral ischemia, due to the accumulation of extracellular glutamate and the activation of N-methyl-d-aspartate (NMDA) receptors. Blockade of H1 receptors by terfenadine, chlorpheniramine, oxatomide, or triprolidine enhances the excitotoxic response of NMDA receptors, which is prevented by histamine.29 We also found that the histamine precursor carnosine increases the viability of differentiated PC12 cells (a neuronal cell line) after NMDA administration through the carnosine-histidine-histamine-H1/H3 receptor pathway.30,31 Due to the exclusive CNS distribution of H3 compared with H1 and H2 receptors, agents targeted to H3 receptors have more potential, and their functions are the focus of much attention. The H3 agonist α-methylhistamine significantly aggravates the delayed neuronal death following ischemia caused by 10 min of 4-vessel occlusion in rats.25 We recently found that mice treated with the H3 antagonists thioperamide or clobenpropit, and H3 receptor knockout mice showed less impairment of neurological function and a reduced infarct area after MCAO (unpublished data). However, we were interested to find that the protection after H3 receptor blockade also occurs in HDC knockout mice, which suggests that blockade of H3 receptors has a protective mechanism other than up-regulating histamine. This evidence all supports the hypothesis that histamine has a protective effect on cerebral ischemic injury, and all three of its receptors may be involved. But animals with genetically modified histamine receptors, especially conditional genetically modified animals, may be useful in elucidating the detailed roles of histamine receptors in the neurovascular unit after cerebral ischemia.
Possible Mechanisms by Which Histamine and Its Receptors Protect against Cerebral Ischemia
Direct Effect on Neurons
With energy depletion, membrane potential is lost, neurons depolarize, and then excitatory amino-acids are released into the extracellular space. As a result, glutamate receptors are overactivated, and excessive Na+, Cl–, and Ca2+ influx initiate a series of cytoplasmic and nuclear events that result in profound tissue damage. We found that histamine alleviates the excitotoxicity induced by NMDA via H2 receptors, and detailed study indicated that histamine works through the cAMP/PKA pathway.32 8-Bromo-cAMP mimicks the effect of histamine, while in contrast, both the adenylyl cyclase inhibitor 9-(tetrahydro-2-furanyl)-9H-purine-6-amine and the PKA inhibitor N-[2-(p-bromocinnamylamino) ethyl]-5-isoquinolinesulfonamide reverse the protection conferred by histamine. Our further study suggests that the increased NMDA receptor internalization may be involved in the protection by histamine (Figure 1). Moreover, the H1 antagonist terfenadine is reported to enhance the excitotoxic response to NMDA in cerebellar neurons, by increasing intracellular Ca2+, cGMP synthesis and the production of reactive oxygen species.29 In a recent study, we found the mechanism of protection by H3 receptor antagonist against neuronal apoptosis after MCAO or OGD involves the up-regulation of autophagy. Moreover, H3 receptor knockout mice also show alleviated neurological injury and up-regulated level of autophagy (unpublished data).
Figure 1.

Internalization of NMDA receptor increases after NMDA (100 μM) and/or histamine (0.1 μM) administration determined by cell ELISA assay. Values are expressed as mean ± SEM, n = 7–10. *P < 0.05, **P < 0.01, compared with control group.
Neurotransmitters
The histamine neurons in the tuberomamillary nucleus send out axons to innervate almost the entire brain. Moreover, H3 receptor expression is not confined to histaminergic neurons, and, as a heteroreceptor, it is known to modulate various neurotransmitter systems in the brain, like those of glutamate, dopamine, gamma-aminobutyric acid (GABA) and acetylcholine.33 We found that hippocampal glutamate levels significantly increase in HDC knockout mice after contextual fear conditioning, which indicates that histamine deficiency may improve memory by up-regulating glutamate.34 Therefore, elevated histamine content may affect the release of other transmitters, especially glutamate, to impact cerebral ischemia. It has been reported that preischemic administration of histamine suppresses the increase in glutamate and dopamine levels during ischemia.28 Dopamine also has detrimental effects on ischemic neuronal damage, most probably due to the generation of reactive oxygen species.35 The effect of histamine on neurotransmitter release was considered to be due to activation of H3 receptors, and then inhibition of presynaptic Ca2+ entry, via direct G-protein-mediated inhibition of multiple Ca2+ channels.36,37 In our study, the mechanism of the protection by carnosine against NMDA-induced neurotoxicity in PC12 cells includes transformation to histamine and then effective inhibition of glutamate release through H3 receptors.30 However, it is also suggested that suppression of the ischemic release of glutamate and dopamine by H2 receptor action is a contributing factor in alleviating the histologic outcome, but how the H2 receptor modulates excitatory neurotransmitter release is quite unclear.28
The level of glutamate in the synaptic cleft is not only affected by the release from neurons, but also related to the uptake by astrocytes. Following synaptic release, glutamate in the forebrain is mainly taken up by the astrocytic glutamate transporter GLT-1, and then converted into glutamine by glutamine synthetase (GS).38 After cerebral ischemia, the expression of GLT-1 and GS declines, which may aggravate the accumulation of glutamate in the extracellular space and the excitotoxicity.39,40 We found that histamine up-regulates the current of glutamate transporter and the expression of GS, which is abrogated by the H1 antagonists (Figures 2, 3). But whether such an effect is involved in the protection by histamine against cerebral ischemia is still under study. Moreover, we showed that carnosine suppresses the generation of mitochondrial reactive oxygen species to recover the expression of GLT-1 and then reduces the glutamate excitotoxicity; however, this effect is not through the carnosine-histamine pathway.41
Figure 2.

Histamine increases glutamate transporter currents through H1 receptor in astrocytes. Astrocytes were treated with histamine (HA) as the indicated concerntration, or cotreated with H1 antagonist diphenhydramine (Diphen) 100 μM for 24 h. Whole-cell glutamate transporter currents in astrocyte were activated by exogenous application of l-glutamate, APV, and CNQX. Representative traces of glutamate transporter currents are shown (A). Peak glutamate transporter current of astrocyte increased after histamine treatment, but was reversed by Diphen (B). Values are expressed as mean ± SEM, n = 9–13. *P < 0.05, compared with control group; $$P < 0.01 compared with HA 100 μM group.
Figure 3.

Histamine reverses the decrease of GS protein expression through H1 receptor after 6 h OGD. Astrocytes were given histamine as the indicated concerntration and exposed to 6 h OGD, and then the GS protein level was analyzed by Western blot. H1 receptor antagonist pyrilamine (Py, 10 μM) and H2 receptor antagonist cimetidine (Cim, 10 μM) were given 15 min before OGD. Values are expressed as mean ± SEM, n = 3–7. **P < 0.01, ***P < 0.001.
Modulation of GABA release may also be involved the protection by H3 antagonists. We found that clobenpropit reverses the neurotoxicity induced by NMDA in a concentration-dependent manner.42 The protection by clobenpropit is inhibited by the GABAA receptor antagonists picrotoxin and bicuculline. Further study demonstrated that the protection by clobenpropit is due to enhanced GABA release. The inducible GABA release is also inhibited by the H3 agonist α-methylhistamine, but not by pyrilamine or cimetidine. Furthermore, the elevation of GABA release induced by clobenpropit is through the cAMP/PKA pathway and increase of intracellular Ca2+ level, which protects against NMDA-induced excitotoxicity.
Cerebral Blood Flow and Edema
H1 and H2 receptors are distributed in the endothelium or smooth muscle cells in the cerebral vascular wall,43,44 and histaminergic fibers have been shown to innervate cerebral blood vessels.45,46 Nevertheless, it remains an undeniable fact that histamine has inconsistent effects on regional CBF (rCBF). Intracerebroventricular injection of histamine causes a dose-dependent increase in rCBF in the hippocampus, and similar findings are found with not only the H1 agonist 2-thiazolylethylamine, but also the H2 agonist dimaprit.47 Intraperitoneal injection of l-histidine also results in an increase in rCBF in the hippocampus, in parallel with elevation of histamine content in the brain. The increase in rCBF in the hippocampus induced by l-histidine is antagonized by both H1 and H2 antagonists (diphenhydramine, pyrilamine, and zolantidine). We found that intracerebroventricular injection of clobenpropit, a representative H3 antagonist, dose-dependently and significantly increases rCBF in the hippocampus of rats, which is enhanced by metoprine, a selective histamine N-methyltransferase inhibitor; furthermore, this is antagonized by an H3 agonist, α-methylhistamine, an H1 antagonist, mepyramine, and an H2 antagonist, zolantidine.48 However, it was also reported that histamine induces cortical vessel contraction via H1 receptors in rats.49 Recently, Yang et al. also reported that histamine induces a reduction of rCBF in the rat parietal lobe and this effect may be partly attributed to its combination with H1 receptors.50 In our study of rCBF after focal cerebral ischemia, we did not find a change of rCBF in the penumbra or core of ischemia after histamine or histidine administration, and there was no difference between HDC knockout and wild-type mice.51 Such discrepancy may be due to the dose of histamine used, the different region detected, or the different effects of histamine under physiological and pathological conditions. More detailed evaluation of rCBF is necessary for the study and development of histamine-related agents to treat cerebral ischemia.
With endothelial cell damage and tight junction breakdown, vasogenic edema is characterized by an increase in extracellular fluid volume due to increased permeability of brain capillary endothelial cells to serum proteins such as albumin.52 Vasogenic edema then causes an increase in tissue pressure and, thereby, aggravates the primary reduction in blood flow.53 With respect to capillary permeability, several reports indicate that blood-brain barrier permeability is regulated by brain histamine. Infusion of histamine into the carotid artery enhances the penetration of albumin through the capillary and increases the cortical water content by activating H2 receptors.54,55 Electron microscopic examination revealed that perivascular astrocytic processes are swollen. Blocking H2 receptors prevents both the transport of albumin and ultrastructural changes, whereas H1 blockade fails to do this. Further, the H2 antagonist ranitidine attenuates the brain edema induced by the systemic cerebral administration of kainic acid.56 So, histamine is suggested to promote brain edema formation, but what is its action on the edema induced by cerebral ischemia? Recently, Irisawa et al. found that repeated administration of l-histidine, immediately and 6 h after reperfusion, reduces the increase in water content in the ischemic regions, and simultaneous administration of thioperamide with l-histidine completely prevents edema formation and alleviates the infarction after MCAO, with markedly increased brain histamine concentration.57 On the contrary, either pretreatment or post-treatment with ranitidine is able to attenuate the ischemia-induced water accumulation and maldistribution of ions in brain tissue in a bilateral common carotid artery occlusion model.58 But in the global ischemia model induced by 4-vessel occlusion, the ischemic edema formation is not prevented a intracellular histamine receptor antagonist.59 Despite these paradoxical effects of brain histamine, it contributes to neuroprotection against cerebral ischemia, as described previously.
Inflammation
Cerebral ischemia evokes a strong inflammatory response, which is thought to contribute to the progression of injury. The Ca2+-related activation of intracellular second messenger systems, the increase in oxygen free radicals, as well as hypoxia itself, trigger the expression of a number of inflammatory mediators, such as platelet-activating factor, tumor necrosis factor alpha and interleukin-1beta.2,60 Subsequently, the expression of adhesion molecules on the endothelial cell surface attracts neutrophils, macrophages, and monocytes to cross the vascular wall and enter the brain parenchyma. Resident brain cells, astrocytes and microglia are also activated to participate in the inflammatory response. Postischemic inflammation injures neurons and glia by releasing proteolytic enzymes, reactive oxygen species and toxic prostanoids and cytokines.61 When adhesion molecules and inflammatory mediators or their receptors are blocked by neutralizing antibodies and when mice are deficient in the expression of inflammation-related genes, cerebral ischemic damage is reduced.62−64
Histamine is known to augment allergic reactions, such as H1 receptor's action in bronchial asthma and anaphylaxis. In contrast, H2 receptor's action has been shown to suppress inflammation by reducing the production of proinflammatory cytokines and the chemotactic responsiveness of leukocytes.65−67 Then what is the role of histamine in the inflammation after cerebral ischemia? Administration of l-histidine, immediately and 6 h after MCAO reperfusion, reduces the number of neutrophils to 52%. Simultaneous administration of thioperamide further decreases this number to 32%.68 Likewise, the ischemia-induced increase in the number of CD68-positive cells (including polymorphonuclear leukocytes and macrophages/microglia) after 24 h is suppressed by l-histidine injection. The l-histidine administration decreases the number of CD4+ T lymphocytes on both the ischemic and contralateral sides after 12 h, and concurrent administration of thioperamide prolongs the effect. Although administration of the H1 antagonist mepyramine does not affect the suppression of leukocyte infiltration, ranitidine tends to reverse the effect of l-histidine. These data suggest that facilitation of central histaminergic activity suppresses inflammatory cell recruitment after ischemic events through H2 receptors. The detailed mechanism is still unknown, but H2 receptor stimulation has been reported to inhibit the induction of cytokines like interleukin-12 (a potent Th1-driving cytokine) and then the chemotactic responsiveness of leukocytes.69,70 Further study is needed to investigate whether inhibition of inflammation is involved in the protection by histamine and how histamine impacts the progression of inflammation after ischemia in detail.
Cerebral Reconstruction
After cerebral ischemia, astrocytes undergo a vigorous response termed “astrogliosis” characterized by escalated expression of glial fibrillary acidic protein, hypertrophy, and cell proliferation.71,72 Accompanying these morphological changes is a range of physiological changes, including secretion of a variety of cytokines.73,74 At the late phase of focal cerebral ischemia, astrogliosis results in glial scar formation, which obstructs axonal regeneration, and finally impedes neurological recovery.75 So, mechanisms to limit glial scar formation are the focus of much research. We found that histidine markedly improves neurological score and learning and memory, but reduces infarct area during the long-term progression after cerebral ischemia. Further study showed that inhibition of glial scar formation is one possible reason (unpublished data). Moreover, we for the first time established a glial scar model induced by oxygen-glucose deprivation in vitro (submitted for publication), which can be used to further study the mechanism of action of histamine.
Neurogenesis, an important component of cerebral reconstruction, contributes to the possibility of improved functional recovery.76 The adult brain harbors a population of neural stem cells (NSCs) throughout life in the subgranular zone of the hippocampus and adjacent to the lateral ventricles in the subventricular zone.77,78 These NSCs are self-renewing, with the potential to generate all three basic cell types: neurons, oligodendrocytes, and astrocytes.79 Now, pharmacologically based therapies targeting enhancement of endogenous neurogenesis are attracting much attention.80−82 Recently, the proliferating and differentiated neuroepithelial stem cells from rat cerebral cortex were found to express H1, H2, and H3 receptors by RT-PCR and Western blot analysis.83 Treatment with histamine significantly increases the proliferation of neuroepithelial stem cells through H2 receptors, but without affecting the activation of nestin expression. Moreover, apoptotic cell death during the proliferation is significantly decreased. After differentiation, histamine increases the number of neurons 3-fold, mainly by activation of the H1 receptor, and also significantly decreases the proportion of glial (astrocytic) cells, compared to control conditions. Therefore, histamine might play a role in neurogenesis by promoting the proliferation of neural precursors through activation of H2 receptors, and favoring a neuronal fate by H1-mediated stimulation. In addition, histamine is also reported to promote neuronal differentiation of midbrain neural stem cells.84 However, it is not certain that histamine promotes neurogenesis after cerebral ischemia in vivo.
Now it is considered that only inhibiting glial scar formation or promoting neurogenesis may not result in significant recovery, but combinatorial strategies may be more effective.75,85 Histamine may show benefit in limiting glial scar formation, along with stimulation of the intrinsic growth potential of adult neurons, so histamine-related agents have potential value in cerebral reconstruction.
Ischemic Tolerance and Histamine
In 1986, Murry and colleagues first demonstrated that multiple brief ischemic episodes in the canine heart limits the histologic infarct size from a subsequent sustained ischemic insult.86 This phenomenon is known as ischemic tolerance or preconditioning, and means that subthreshold ischemic or hypoxic damage activates certain cellular pathways that help to reduce the damage caused by subsequent severe ischemia.87 Ischemic tolerance has also been confirmed in global and focal cerebral ischemia in animals.88−90 For example, Kitagawa et al. reported that 2 min ischemic treatments at 1 day intervals 2 days before 5 min ischemia induced by occlusion of both common carotids result in strikingly complete protection against neuronal death in gerbils.88 In patients, it is also found that patients with transient ischemic attacks before cerebral infarction have a better outcome than patients without them.91 This opens a window into endogenous neuroprotection and, potentially, a window of opportunity to use these mechanisms in the clinic to treat patients with stroke and global cerebral ischemia.92 Consequently, a better understanding of the endogenous neuroprotective mechanisms involved in ischemic tolerance has significant clinical implications for preventing neuronal damage in susceptible patients.
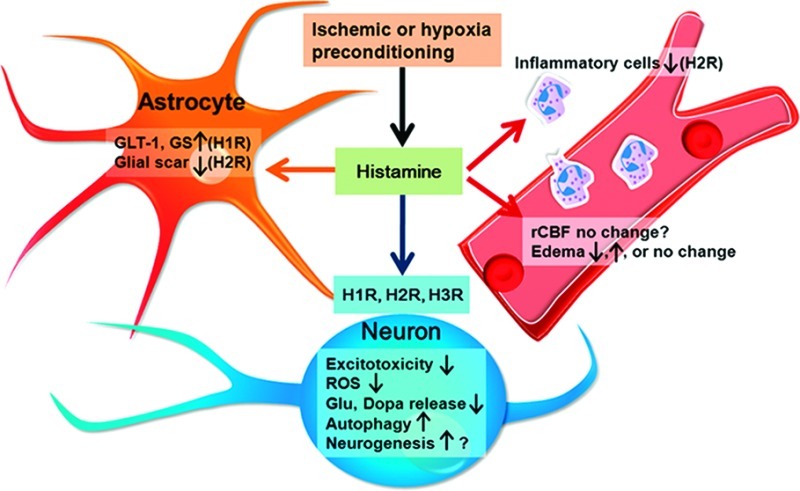
In a previous study, we were interested to find that activation of the central histaminergic system is required for the neuroprotection induced by ischemic or hypoxic preconditioning.51,93 Hypoxic preconditioning improves neurologic function and decreases infarct volume in wild-type (WT) and HDC knockout mice treated with histamine, but not in HDC knockout and WT mice treated with α-FMH. Laser-Doppler flowmetry showed that hypoxic preconditioning ameliorates cerebral blood flow in the periphery of the MCA territory during ischemia in WT mice but not in HDC knockout mice. Vascular endothelial growth factor (VEGF) mRNA and protein expression show a greater increase after hypoxia than in HDC knockout or α-FMH-treated WT mice. In addition, the VEGF receptor-2 antagonist SU1498 prevents the protective effect of hypoxic preconditioning on infarct volume and reverses the increased peripheral cerebral blood flow in WT mice. Therefore, endogenous histamine is an essential mediator of hypoxic preconditioning. It may function by enhancing hypoxia-induced VEGF expression and then increasing the rCBF. Moreover, 10 min ischemic preconditioning significantly prolongs the survival time of WT mice subjected to permanent bilateral carotid artery occlusion. However, in HDC knockout mice, the ischemic tolerance is not induced with 10 min preconditioning. The histamine levels at 0.5 and 48 h increase after 10 min preconditioning, but not at 5 h. Therefore, endogenous histamine in brain may be an essential mediator in hypoxic or ischemic preconditioning-induced cerebral ischemic tolerance, which supports histamine as an important target in cerebral ischemia.
Prospects for Histamine-Related Agents in Cerebral Ischemia
Although histamine shows marked protection against cerebral ischemia, its low penetration of the blood-brain barrier and its wide actions in the peripheral system (in the allergic reaction and gastric acid secretion) limit its direct use, and that of its H1 and H2 receptor agonists. Since the H3 receptor is a presynaptic negative modulator of histamine release, it is thought that an H3 receptor antagonist would enhance histamine release. By virtue of its extensive CNS localization (cortex, hippocampus, amygdala, nucleus accumbens, globus pallidus, striatum, thalamus, and hypothalamus), it is hypothesized that H3 receptor antagonists produce a unique profile of CNS activation, although it is also located presynaptically on the postganglionic sympathetic nerve fibers in the cardiovascular system.94−96 The H3 receptor is recognized as a drug target for neuropathic pain, sleep-wake disorders (narcolepsy), and the cognitive impairment associated with attention deficit hyperactivity disorder, schizophrenia, Alzheimer, and Parkinson disease, while the first H3 receptor ligands have already entered phase I–III clinical trials.97 As described previously, blocking H3 receptors reduces the infarct area and improves neurological function, while activating H3 receptors significantly aggravates the delayed neuronal death after cerebral ischemia. Therefore, H3 antagonists have therapeutic potential in cerebral ischemia, although the detailed mechanism still needs to be understood. Besides regulating neurotransmitter release by regulation of Ca2+ channel, additional interactions with other effector signaling cascades may also contribute to their action, including the activation of adenylyl cyclase, phospholipase A2, mitogen-activated protein kinase, and phosphatidylinositol 3-kinase pathways, which activate extracellular signal-regulated kinases and Akt and subsequently inhibit the action of glycogen synthase kinase 3β.33
Besides the histamine receptor ligands, histamine precursors, like carnosine, are another choice for therapeutic use. Carnosine (β-alanyl-l-histidine) is a naturally occurring dipeptide, widely distributed in tissues including the animal and human brain, at concentrations up to 20 mM, and can easily enter the central nervous system.98,99 There are many theories about its biological functions, such as anti-inflammatory agent, free radical scavenger, membrane-protective agent, and protein glycosylation inhibitor.100,101 Previous studies have demonstrated that carnosine (via histidine) is metabolically transformed into histamine in muscle and kidney.102,103 We recently reported that the metabolic carnosine-histidine-histamine pathway also exists in the CNS.30,104 Carnosine prevents amygdaloid kindling seizures in rats by partial transformation to histamine. Given the relationship between carnosine and histamine, carnosine is proposed as a new histaminergic drug that can replace histamine and avoid inflammation. It is reported that carnosine shows a pronounced protective effect on neurological symptoms and animal mortality after global ischemia, and on infarct volume and neurological function after focal cerebral ischemia or hypoxic-ischemic brain damage.41,105−108 Antioxidant and membrane-protective properties may contribute to this protection; however, whether the carnosine-histidine-histamine pathway is involved remains in doubt. In a recent study, we found that carnosine significantly improves neurological function and decreases infarct size after permanent MCAO in both HDC knockout and the corresponding WT mice to the same extent, and this cannot be reversed by α-FMH.41 It is suggested that the action of carnosine on permanent focal cerebral ischemia is not mediated by the carnosine-histidine-histamine pathway but by decreasing the glutamate levels and preserving the expression of GLT-1. But carnosine protects against NMDA-induced neurotoxicity in differentiated rat PC12 cells through the carnosine-histidine-histamine pathway and H1/H3 receptors.30,31 The different cells and different models may account for the inconsistent results; however, carnosine itself is considered as a useful agent in cerebral ischemia therapy. Certainly, long-term evaluation of the action of histamine-related agents on cerebral ischemia by various ischemic animal models and the studies of their detailed mechanism are needed for their preclinical investigations for treating cerebral ischemia.
Conclusion
Although histamine is not considered to play a pivotal role in the pathogenic cascade following cerebral ischemia, its multitarget-directed actions, including neurons, glia, vascular cells, and inflammatory cells, indicate potential (Figure 4). However, important questions about the molecular aspects and pathophysiology of histamine and its related agents remain to be answered to form a solid scientific basis for the therapeutic application of histamine-related agents.
Figure 4.

Schematic representation of the multitarget-directed actions of histamine and its receptors against cerebral ischemia, acting on neurons, glia, vascular cells, and inflammatory cells. Histamine can alleviate excitotoxicity and reactive oxygen species (ROS) injury, suppress the release of glutamate (Glu) and dopamine (Dopa), and promote autophagy to protect neurons through its H1, H2, or H3 receptors. Histamine may recover the GLT-1 function and GS expression in astrocytes, and inhibit glial scar formation and inflammation. Histamine may also affect rCBF and edema formation, and promote neurogenesis by H1 and H2 receptors. Moreover, endogenous histamine in the brain may be an essential mediator in hypoxia or ischemic preconditioning induced cerebral ischemic tolerance.
Acknowledgments
We are very grateful to Dr. Iain C. Bruce for reading the manuscript.
Glossary
Abbreviations
- α-FMH
alpha-fluoromethylhistidine
- CBF
cerebral blood flow
- GABA
gamma-aminobutyric acid
- GS
glutamine synthetase
- HDC
l-histidine decarboxylase
- MCAO
occlusion of the middle cerebral artery
- NMDA
N-methyl-d-aspartate
- NSCs
neural stem cells
- rCBF
regional CBF
- rtPA
recombinant tissue plasminogen activator
- VEGF
vascular endothelial growth factor
- WT
wild-type
Author Contributions
W.W.H and Z.C. wrote the manuscript.
This work was funded by the National Basic Research of China 973 Program (2011CB504403), by the National Natural Science Foundation of China (81030061, 81173040, 81102429), and partly by the Youth Foundation of the Innovative Scientific Research of Zhejiang University (2009QNA7007).
The authors declare no competing financial interest.
References
- Harukuni I.; Bhardwaj A. (2006) Mechanisms of brain injury after global cerebral ischemia. Neurol. Clin. 24, 1–21. [DOI] [PubMed] [Google Scholar]
- Dirnagl U.; Iadecola C.; Moskowitz M. A. (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 22, 391–397. [DOI] [PubMed] [Google Scholar]
- Caplan L. R. (1998) Stroke treatment: promising but still struggling. JAMA. 279, 1304–1306. [DOI] [PubMed] [Google Scholar]
- Fisher M.; Bogousslavsky J. (1998) Further evolution toward effective therapy for acute ischemic stroke. JAMA, J. Am. Med. Assoc. 279, 1298–1303. [DOI] [PubMed] [Google Scholar]
- Lo E. H. (2008) A new penumbra: transitioning from injury into repair after stroke. Nat. Med. 14, 497–500. [DOI] [PubMed] [Google Scholar]
- Davis S. M.; Donnan G. A. (2009) 4.5 h: the new time window for tissue plasminogen activator in stroke. Stroke 40, 2266–2267. [DOI] [PubMed] [Google Scholar]
- Wada H.; Inagaki N.; Itowi N.; Yamatodani A. (1991) Histaminergic neuron system in the brain: distribution and possible functions. Brain Res. Bull. 27, 367–370. [DOI] [PubMed] [Google Scholar]
- Brown R. E.; Stevens D. R.; Haas H. L. (2001) The physiology of brain histamine. Prog. Neurobiol. 63, 637–672. [DOI] [PubMed] [Google Scholar]
- Garbarg M.; Barbin G.; Bischoff S.; Pollard H.; Schwartz J. C. (1976) Dual localization of histamine in an ascending neuronal pathway and in non-neuronal cells evidenced by lesions in the lateral hypothalamic area. Brain Res. 106, 333–348. [DOI] [PubMed] [Google Scholar]
- Garbarg M.; Barbin G.; Feger J.; Schwartz J. C. (1974) Histaminergic pathway in rat brain evidenced by lesions of the medial forebrain bundle. Science 186, 833–835. [DOI] [PubMed] [Google Scholar]
- Schwartz J. C.; Arrang J. M.; Garbarg M.; Pollard H.; Ruat M. (1991) Histaminergic transmission in the mammalian brain. Physiol. Rev. 71, 1–51. [DOI] [PubMed] [Google Scholar]
- Nguyen T.; Shapiro D. A.; George S. R.; Setola V.; Lee D. K.; Cheng R.; Rauser L.; Lee S. P.; Lynch K. R.; Roth B. L.; O’Dowd B. F. (2001) Discovery of a novel member of the histamine receptor family. Mol. Pharmacol. 59, 427–433. [DOI] [PubMed] [Google Scholar]
- Haas H.; Panula P. (2003) The role of histamine and the tuberomamillary nucleus in the nervous system. Nat. Rev. Neurosci. 4, 121–130. [DOI] [PubMed] [Google Scholar]
- Morisset S.; Rouleau A.; Ligneau X.; Gbahou F.; Tardivel-Lacombe J.; Stark H.; Schunack W.; Ganellin C. R.; Schwartz J. C.; Arrang J. M. (2000) High constitutive activity of native H3 receptors regulates histamine neurons in brain. Nature 408, 860–864. [DOI] [PubMed] [Google Scholar]
- Adachi N. (2005) Cerebral ischemia and brain histamine. Brain Res. Rev. 50, 275–286. [DOI] [PubMed] [Google Scholar]
- Adachi N.; Oishi R.; Saeki K. (1991) Changes in the metabolism of histamine and monoamines after occlusion of the middle cerebral artery in rats. J. Neurochem. 57, 61–66. [DOI] [PubMed] [Google Scholar]
- Adachi N.; Itoh Y.; Oishi R.; Saeki K. (1992) Direct evidence for increased continuous histamine release in the striatum of conscious freely moving rats produced by middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 12, 477–483. [DOI] [PubMed] [Google Scholar]
- Subramanian N.; Theodore D.; Abraham J. (1981) Experimental cerebral infarction in primates: regional changes in brain histamine content. J. Neural Transm. 50, 225–232. [DOI] [PubMed] [Google Scholar]
- Martres M. P.; Baudry M.; Schwartz J. C. (1975) Histamine synthesis in the developing rat brain: evidence for a multiple compartmentation. Brain Res. 83, 261–275. [DOI] [PubMed] [Google Scholar]
- Kollonitsch J.; Perkins L. M.; Patchett A. A.; Doldouras G. A.; Marburg S.; Duggan D. E.; Maycock A. L.; Aster S. D. (1978) Selective inhibitors of biosynthesis of aminergic neurotransmitters. Nature 274, 906–908. [DOI] [PubMed] [Google Scholar]
- Hu W.; Xu L.; Pan J.; Zheng X.; Chen Z. (2004) Effect of cerebral ischemia on brain mast cells in rats. Brain Res. 1019, 275–280. [DOI] [PubMed] [Google Scholar]
- Lozada A.; Munyao N.; Sallmen T.; Lintunen M.; Leurs R.; Lindsberg P. J.; Panula P. (2005) Postischemic regulation of central histamine receptors. Neuroscience 136, 371–379. [DOI] [PubMed] [Google Scholar]
- Adachi N.; Liu K.; Arai T. (2005) Prevention of brain infarction by postischemic administration of histidine in rats. Brain Res. 1039, 220–223. [DOI] [PubMed] [Google Scholar]
- Fujitani T.; Adachi N.; Nagaro T.; Miyazaki H.; Nakamura Y.; Kataoka K.; Arai T. (1996) Histaminergic H2 action protects hippocampal CA1 neurons by prolonging the onset of the anoxic depolarization in gerbils. J. Neurochem. 67, 2613–2615. [DOI] [PubMed] [Google Scholar]
- Adachi N.; Oishi R.; Itano Y.; Yamada T.; Hirakawa M.; Saeki K. (1993) Aggravation of ischemic neuronal damage in the rat hippocampus by impairment of histaminergic neurotransmission. Brain Res. 602, 165–168. [DOI] [PubMed] [Google Scholar]
- Sugimoto K.; Abe K.; Lee T. H.; Sakurai E.; Yanai K.; Kogure K.; Itoyama Y.; Watanabe T. (1994) Histamine depletion in brain caused by treatment with (S)alpha-fluoromethylhistidine enhances ischemic damage of gerbil hippocampal CA2 neurons. Brain Res. 666, 279–283. [DOI] [PubMed] [Google Scholar]
- Hu W.; Fan Y.; Shen Y.; Yang Y.; Dai H.; Fu Q.; Chen Z. (2007) Mast cell-derived mediators protect against oxygen-glucose deprivation-induced injury in PC12 cells and neurons. Neurosci. Lett. 423, 35–40. [DOI] [PubMed] [Google Scholar]
- Hamami G.; Adachi N.; Liu K.; Arai T. (2004) Alleviation of ischemic neuronal damage by histamine H2 receptor stimulation in the rat striatum. Eur. J. Pharmacol. 484, 167–173. [DOI] [PubMed] [Google Scholar]
- Diaz-Trelles R.; Novelli A.; Vega J. A.; Marini A.; Fernandez-Sanchez M. T. (2000) Antihistamine terfenadine potentiates NMDA receptor-mediated calcium influx, oxygen radical formation, and neuronal death. Brain Res. 880, 17–27. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Hu W. W.; Fan Y. Y.; Dai H. B.; Fu Q. L.; Wei E. Q.; Luo J. H.; Chen Z. (2007) Carnosine protects against NMDA-induced neurotoxicity in differentiated rat PC12 cells through carnosine-histidine-histamine pathway and H(1)/H(3) receptors. Biochem. Pharmacol. 73, 709–717. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Fan Y.; Dai H.; Fu Q.; Hu W.; Chen Z. (2007) Neuroprotective effect of carnosine on necrotic cell death in PC12 cells. Neurosci. Lett. 414, 145–149. [DOI] [PubMed] [Google Scholar]
- Dai H.; Zhang Z.; Zhu Y.; Shen Y.; Hu W.; Huang Y.; Luo J.; Timmerman H.; Leurs R.; Chen Z. (2006) Histamine protects against NMDA-induced necrosis in cultured cortical neurons through H receptor/cyclic AMP/protein kinase A and H receptor/GABA release pathways. J. Neurochem. 96, 1390–1400. [DOI] [PubMed] [Google Scholar]
- Leurs R.; Bakker R. A.; Timmerman H.; de Esch I. J. (2005) The histamine H3 receptor: from gene cloning to H3 receptor drugs. Nat. Rev. Drug Discovery 4, 107–120. [DOI] [PubMed] [Google Scholar]
- Liu L.; Zhang S.; Zhu Y.; Fu Q.; Gong Y.; Ohtsu H.; Luo J.; Wei E.; Chen Z. (2007) Improved learning and memory of contextual fear conditioning and hippocampal CA1 long-term potentiation in histidine decarboxylase knock-out mice. Hippocampus 17, 634–641. [DOI] [PubMed] [Google Scholar]
- Clemens J. A.; Phebus L. A. (1988) Dopamine depletion protects striatal neurons from ischemia-induced cell death. Life Sci. 42, 707–713. [DOI] [PubMed] [Google Scholar]
- Brown R. E.; Haas H. L. (1999) On the mechanism of histaminergic inhibition of glutamate release in the rat dentate gyrus. J. Physiol. 515(Pt 3), 777–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Hernandez A.; Nunez A.; Sierra J. J.; Arias-Montano J. A. (2001) Histamine H3 receptor activation inhibits glutamate release from rat striatal synaptosomes. Neuropharmacology 41, 928–934. [DOI] [PubMed] [Google Scholar]
- Bak L. K.; Schousboe A.; Waagepetersen H. S. (2006) The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 98, 641–653. [DOI] [PubMed] [Google Scholar]
- Chen J. C.; Hsu-Chou H.; Lu J. L.; Chiang Y. C.; Huang H. M.; Wang H. L.; Wu T.; Liao J. J.; Yeh T. S. (2005) Down-regulation of the glial glutamate transporter GLT-1 in rat hippocampus and striatum and its modulation by a group III metabotropic glutamate receptor antagonist following transient global forebrain ischemia. Neuropharmacology 49, 703–714. [DOI] [PubMed] [Google Scholar]
- Oliver C. N.; Starke-Reed P. E.; Stadtman E. R.; Liu G. J.; Carney J. M.; Floyd R. A. (1990) Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc. Natl. Acad. Sci. U.S.A. 87, 5144–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; He P.; Fan Y. Y.; Zhang J. X.; Yan H. J.; Hu W. W.; Ohtsu H.; Chen Z. (2010) Carnosine protects against permanent cerebral ischemia in histidine decarboxylase knockout mice by reducing glutamate excitotoxicity. Free Radicals Biol. Med. 48, 727–735. [DOI] [PubMed] [Google Scholar]
- Dai H.; Fu Q.; Shen Y.; Hu W.; Zhang Z.; Timmerman H.; Leurs R.; Chen Z. (2007) The histamine H3 receptor antagonist clobenpropit enhances GABA release to protect against NMDA-induced excitotoxicity through the cAMP/protein kinase A pathway in cultured cortical neurons. Eur. J. Pharmacol. 563, 117–123. [DOI] [PubMed] [Google Scholar]
- Karnushina I. L.; Palacios J. M.; Barbin G.; Dux E.; Joo F.; Schwartz J. C. (1980) Studies on a capillary-rich fraction isolated from brain: histaminic components and characterization of the histamine receptors linked to adenylate cyclase. J. Neurochem. 34, 1201–1208. [DOI] [PubMed] [Google Scholar]
- Stanimirovic D. B.; Bertrand N.; Merkel N.; Bembry J.; Spatz M. (1994) Interaction between histamine and adenosine in human cerebromicrovascular endothelial cells: modulation of second messengers. Metab. Brain Dis. 9, 275–289. [DOI] [PubMed] [Google Scholar]
- Takagi H.; Morishima Y.; Matsuyama T.; Hayashi H.; Watanabe T.; Wada H. (1986) Histaminergic axons in the neostriatum and cerebral cortex of the rat: a correlated light and electron microscopic immunocytochemical study using histidine decarboxylase as a marker. Brain Res. 364, 114–123. [DOI] [PubMed] [Google Scholar]
- Karlstedt K.; Senkas A.; Ahman M.; Panula P. (2001) Regional expression of the histamine H(2) receptor in adult and developing rat brain. Neuroscience 102, 201–208. [DOI] [PubMed] [Google Scholar]
- Suzuki G.; Chen Z.; Sugimoto Y.; Fujii Y.; Kamei C. (1999) Effects of histamine and related compounds on regional cerebral blood flow in rats. Methods Find. Exp. Clin. Pharmacol. 21, 613–617. [PubMed] [Google Scholar]
- Chen Z. (2001) Effect of clobenpropit on regional cerebral blood flow in rat hippocampus. Acta Pharmacol. Sin. 22, 355–360. [PubMed] [Google Scholar]
- Zhao J. J.; Liu Y.; Chen X. L.; Liu J. X.; Tian Y. F.; Zhang P. B.; Kang Q. Y.; Qiu F.; Yang P. B. (2006) Effect of histamine on intracortical blood vessels of rats. Nan Fang Yi Ke Da Xue Xue Bao 26, 1284–1287. [PubMed] [Google Scholar]
- Yang P. B.; Chen X. L.; Zhao J. J.; Zhang J. S.; Zhang J. F.; Tian Y. M.; Liu Y. (2010) Effect of histamine on regional cerebral blood flow of the parietal lobe in rats. Lasers Med. Sci. 25, 711–717. [DOI] [PubMed] [Google Scholar]
- Fan Y. Y.; Hu W. W.; Dai H. B.; Zhang J. X.; Zhang L. Y.; He P.; Shen Y.; Ohtsu H.; Wei E. Q.; Chen Z. (2011) Activation of the central histaminergic system is involved in hypoxia-induced stroke tolerance in adult mice. J. Cereb. Blood Flow Metab. 31, 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur C.; Ling E. A. (2008) Blood brain barrier in hypoxic-ischemic conditions. Curr. Neurovasc. Res. 5, 71–81. [DOI] [PubMed] [Google Scholar]
- Hossmann K. A. (2009) Pathophysiological basis of translational stroke research. Folia Neuropathol. 47, 213–227. [PubMed] [Google Scholar]
- Dux E.; Joo F. (1982) Effects of histamine on brain capillaries. Fine structural and immunohistochemical studies after intracarotid infusion. Exp. Brain Res. 47, 252–258. [DOI] [PubMed] [Google Scholar]
- Gross P. M.; Teasdale G. M.; Graham D. I.; Angerson W. J.; Harper A. M. (1982) Intra-arterial histamine increases blood-brain transport in rats. Am. J. Physiol. 243, H307–317. [DOI] [PubMed] [Google Scholar]
- Sztriha L.; Joo F.; Szerdahelyi P. (1987) Histamine H2-receptors participate in the formation of brain edema induced by kainic acid in rat thalamus. Neurosci. Lett. 75, 334–338. [DOI] [PubMed] [Google Scholar]
- Irisawa Y.; Adachi N.; Liu K.; Arai T.; Nagaro T. (2008) Alleviation of ischemia-induced brain edema by activation of the central histaminergic system in rats. J. Pharmacol. Sci. 108, 112–123. [DOI] [PubMed] [Google Scholar]
- Tosaki A.; Szerdahelyi P.; Joo F. (1994) Treatment with ranitidine of ischemic brain edema. Eur. J. Pharmacol. 264, 455–458. [DOI] [PubMed] [Google Scholar]
- Nemeth L.; Deli M. A.; Falus A.; Szabo C. A.; Abraham C. S. (1998) Cerebral ischemia reperfusion-induced vasogenic brain edema formation in rats: effect of an intracellular histamine receptor antagonist. Eur. J. Pediatr. Surg. 8, 216–219. [DOI] [PubMed] [Google Scholar]
- Danton G. H.; Dietrich W. D. (2003) Inflammatory mechanisms after ischemia and stroke. J. Neuropathol. Exp. Neurol. 62, 127–136. [DOI] [PubMed] [Google Scholar]
- White B. C.; Sullivan J. M.; DeGracia D. J.; O’Neil B. J.; Neumar R. W.; Grossman L. I.; Rafols J. A.; Krause G. S. (2000) Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J. Neurol. Sci. 179, 1–33. [DOI] [PubMed] [Google Scholar]
- Iadecola C.; Salkowski C. A.; Zhang F.; Aber T.; Nagayama M.; Vogel S. N.; Ross M. E. (1999) The transcription factor interferon regulatory factor 1 is expressed after cerebral ischemia and contributes to ischemic brain injury. J. Exp. Med. 189, 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly E. S. Jr.; Winfree C. J.; Springer T. A.; Naka Y.; Liao H.; Yan S. D.; Stern D. M.; Solomon R. A.; Gutierrez-Ramos J. C.; Pinsky D. J. (1996) Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J. Clin. Invest. 97, 209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loddick S. A.; Rothwell N. J. (1996) Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J. Cereb. Blood Flow Metab. 932–940. [DOI] [PubMed] [Google Scholar]
- Takahashi H. K.; Yoshida A.; Iwagaki H.; Yoshino T.; Itoh H.; Morichika T.; Yokoyama M.; Akagi T.; Tanaka N.; Mori S.; Nishibori M. (2002) Histamine regulation of interleukin-18-initiating cytokine cascade is associated with down-regulation of intercellular adhesion molecule-1 expression in human peripheral blood mononuclear cells. J. Pharmacol. Exp. Ther. 300, 227–235. [DOI] [PubMed] [Google Scholar]
- Akdis C. A.; Simons F. E. (2006) Histamine receptors are hot in immunopharmacology. Eur. J. Pharmacol. 533, 69–76. [DOI] [PubMed] [Google Scholar]
- Azuma Y.; Shinohara M.; Wang P. L.; Hidaka A.; Ohura K. (2001) Histamine inhibits chemotaxis, phagocytosis, superoxide anion production, and the production of TNFalpha and IL-12 by macrophages via H2-receptors. Int. Immunopharmacol. 1, 1867–1875. [DOI] [PubMed] [Google Scholar]
- Hiraga N.; Adachi N.; Liu K.; Nagaro T.; Arai T. (2007) Suppression of inflammatory cell recruitment by histamine receptor stimulation in ischemic rat brains. Eur. J. Pharmacol. 557, 236–244. [DOI] [PubMed] [Google Scholar]
- Packard K. A.; Khan M. M. (2003) Effects of histamine on Th1/Th2 cytokine balance. Int. Immunopharmacol. 3, 909–920. [DOI] [PubMed] [Google Scholar]
- Igaz P.; Novak I.; Lazaar E.; Horvath B.; Heninger E.; Falus A. (2001) Bidirectional communication between histamine and cytokines. Inflammation Res. 50, 123–128. [DOI] [PubMed] [Google Scholar]
- Eng L. F.; Ghirnikar R. S.; Lee Y. L. (2000) Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem. Res. 25, 1439–1451. [DOI] [PubMed] [Google Scholar]
- Swanson R. A.; Ying W.; Kauppinen T. M. (2004) Astrocyte influences on ischemic neuronal death. Curr. Mol. Med. 4, 193–205. [DOI] [PubMed] [Google Scholar]
- Dong Y.; Benveniste E. N. (2001) Immune function of astrocytes. Glia 36, 180–190. [DOI] [PubMed] [Google Scholar]
- Feuerstein G. Z.; Wang X.; Barone F. C. (1998) The role of cytokines in the neuropathology of stroke and neurotrauma. Neuroimmunomodulation 5, 143–159. [DOI] [PubMed] [Google Scholar]
- Silver J.; Miller J. H. (2004) Regeneration beyond the glial scar. Nat. Rev. Neurosci. 5, 146–156. [DOI] [PubMed] [Google Scholar]
- Zhang R. L.; Zhang Z. G.; Chopp M. (2005) Neurogenesis in the adult ischemic brain: generation, migration, survival, and restorative therapy. Neuroscientist 11, 408–416. [DOI] [PubMed] [Google Scholar]
- Palmer T. D.; Takahashi J.; Gage F. H. (1997) The adult rat hippocampus contains primordial neural stem cells. Mol. Cell Neurosci. 8, 389–404. [DOI] [PubMed] [Google Scholar]
- Reynolds B. A.; Weiss S. (1992) Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255, 1707–1710. [DOI] [PubMed] [Google Scholar]
- Felling R. J.; Levison S. W. (2003) Enhanced neurogenesis following stroke. J. Neurosci. Res. 73, 277–283. [DOI] [PubMed] [Google Scholar]
- Chen J.; Zhang Z. G.; Li Y.; Wang Y.; Wang L.; Jiang H.; Zhang C.; Lu M.; Katakowski M.; Feldkamp C. S.; Chopp M. (2003) Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann. Neurol. 53, 743–751. [DOI] [PubMed] [Google Scholar]
- Jin K.; Sun Y.; Xie L.; Childs J.; Mao X. O.; Greenberg D. A. (2004) Post-ischemic administration of heparin-binding epidermal growth factor-like growth factor (HB-EGF) reduces infarct size and modifies neurogenesis after focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 399–408. [DOI] [PubMed] [Google Scholar]
- Lu D.; Goussev A.; Chen J.; Pannu P.; Li Y.; Mahmood A.; Chopp M. (2004) Atorvastatin reduces neurological deficit and increases synaptogenesis, angiogenesis, and neuronal survival in rats subjected to traumatic brain injury. J. Neurotrauma 21, 21–32. [DOI] [PubMed] [Google Scholar]
- Molina-Hernandez A.; Velasco I. (2008) Histamine induces neural stem cell proliferation and neuronal differentiation by activation of distinct histamine receptors. J. Neurochem. 106, 706–717. [DOI] [PubMed] [Google Scholar]
- Escobedoa I.; Molina-Hernándeza A.; Velasco I. J. (2009) Histamine promotes neuronal differentiation of midbrain neural stem cells. Dev. Biol. 331, 502. [Google Scholar]
- Steinmetz M. P.; Horn K. P.; Tom V. J.; Miller J. H.; Busch S. A.; Nair D.; Silver D. J.; Silver J. (2005) Chronic enhancement of the intrinsic growth capacity of sensory neurons combined with the degradation of inhibitory proteoglycans allows functional regeneration of sensory axons through the dorsal root entry zone in the mammalian spinal cord. J. Neurosci. 25, 8066–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry C. E.; Jennings R. B.; Reimer K. A. (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74, 1124–1136. [DOI] [PubMed] [Google Scholar]
- Dirnagl U.; Simon R. P.; Hallenbeck J. M. (2003) Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 26, 248–254. [DOI] [PubMed] [Google Scholar]
- Kitagawa K.; Matsumoto M.; Tagaya M.; Hata R.; Ueda H.; Niinobe M.; Handa N.; Fukunaga R.; Kimura K.; Mikoshiba K.; et al. (1990) ’Ischemic tolerance’ phenomenon found in the brain. Brain Res. 528, 21–24. [DOI] [PubMed] [Google Scholar]
- Pera J.; Zawadzka M.; Kaminska B.; Szczudlik A. (2004) Influence of chemical and ischemic preconditioning on cytokine expression after focal brain ischemia. J. Neurosci. Res. 78, 132–140. [DOI] [PubMed] [Google Scholar]
- Atochin D. N.; Clark J.; Demchenko I. T.; Moskowitz M. A.; Huang P. L. (2003) Rapid cerebral ischemic preconditioning in mice deficient in endothelial and neuronal nitric oxide synthases. Stroke 34, 1299–1303. [DOI] [PubMed] [Google Scholar]
- Moncayo J.; de Freitas G. R.; Bogousslavsky J.; Altieri M.; van Melle G. (2000) Do transient ischemic attacks have a neuroprotective effect?. Neurology 54, 2089–2094. [DOI] [PubMed] [Google Scholar]
- Gidday J. M. (2006) Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci. 7, 437–448. [DOI] [PubMed] [Google Scholar]
- He P.; Fan Y. Y.; Zhang L. Y.; Hu W. W.; Chen Z. (2009) Effect of endogenous histamine on ischemic preconditioning induced cerebral ischemic tolerance. Zhejiang Da Xue Xue Bao Yi Xue Ban 38, 579–583. [DOI] [PubMed] [Google Scholar]
- Malinowska B.; Godlewski G.; Schlicker E. (1998) Histamine H3 receptors--general characterization and their function in the cardiovascular system. J. Physiol. Pharmacol. 49, 191–211. [PubMed] [Google Scholar]
- Pollard H.; Moreau J.; Arrang J. M.; Schwartz J. C. (1993) A detailed autoradiographic mapping of histamine H3 receptors in rat brain areas. Neuroscience 52, 169–189. [DOI] [PubMed] [Google Scholar]
- Chazot P. L.; Hann V.; Wilson C.; Lees G.; Thompson C. L. (2001) Immunological identification of the mammalian H3 histamine receptor in the mouse brain. NeuroReport 12, 259–262. [DOI] [PubMed] [Google Scholar]
- Tiligada E.; Zampeli E.; Sander K.; Stark H. (2009) Histamine H3 and H4 receptors as novel drug targets. Expert Opin. Invest. Drugs 18, 1519–1531. [DOI] [PubMed] [Google Scholar]
- Crush K. G. (1970) Carnosine and related substances in animal tissues. Comp. Biochem. Physiol. 34, 3–30. [DOI] [PubMed] [Google Scholar]
- Gariballa S. E.; Sinclair A. J. (2000) Carnosine: physiological properties and therapeutic potential. Age Ageing 29, 207–210. [DOI] [PubMed] [Google Scholar]
- Hipkiss A. R. (2005) Glycation, ageing and carnosine: are carnivorous diets beneficial?. Mech. Ageing Dev. 126, 1034–1039. [DOI] [PubMed] [Google Scholar]
- Hipkiss A. R.; Preston J. E.; Himsworth D. T.; Worthington V. C.; Keown M.; Michaelis J.; Lawrence J.; Mateen A.; Allende L.; Eagles P. A.; Abbott N. J. (1998) Pluripotent protective effects of carnosine, a naturally occurring dipeptide. Ann. N.Y. Acad. Sci. 854, 37–53. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick J. C.; Fisher H.; Flancbaum L. (1991) Mobilization of renal carnosine and histidine to histamine during compound-48/80-induced shock. Nephron 59, 299–303. [DOI] [PubMed] [Google Scholar]
- Flancbaum L.; Fitzpatrick J. C.; Brotman D. N.; Marcoux A. M.; Kasziba E.; Fisher H. (1990) The presence and significance of carnosine in histamine-containing tissues of several mammalian species. Agents Actions 31, 190–196. [DOI] [PubMed] [Google Scholar]
- Jin C. L.; Yang L. X.; Wu X. H.; Li Q.; Ding M. P.; Fan Y. Y.; Zhang W. P.; Luo J. H.; Chen Z. (2005) Effects of carnosine on amygdaloid-kindled seizures in Sprague-Dawley rats. Neuroscience 135, 939–947. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Song L.; Cheng X.; Yang Y.; Luan B.; Jia L.; Xu F.; Zhang Z. (2011) Carnosine pretreatment protects against hypoxia-ischemia brain damage in the neonatal rat model. Eur. J. Pharmacol. 667, 202–207. [DOI] [PubMed] [Google Scholar]
- Stvolinsky S.; Kukley M.; Dobrota D.; Mezesova V.; Boldyrev A. (2000) Carnosine protects rats under global ischemia. Brain Res. Bull. 53, 445–448. [DOI] [PubMed] [Google Scholar]
- Min J.; Senut M. C.; Rajanikant K.; Greenberg E.; Bandagi R.; Zemke D.; Mousa A.; Kassab M.; Farooq M. U.; Gupta R.; Majid A. (2008) Differential neuroprotective effects of carnosine, anserine, and N-acetyl carnosine against permanent focal ischemia. J. Neurosci. Res. 86, 2984–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekcetin C.; Kiray M.; Ergur B. U.; Tugyan K.; Bagriyanik H. A.; Erbil G.; Baykara B.; Camsari U. M. (2009) Carnosine attenuates oxidative stress and apoptosis in transient cerebral ischemia in rats. Acta Biol. Hung. 60, 137–148. [DOI] [PubMed] [Google Scholar]