

Polyketides are an important class of natural products that often possess potent biological activity and intriguing chemical structures. Among the methods for constructing these ensembles, the stereoselective addition of allylmetal reagents[1] - particularly allyl boron reagents[2] - to prochiral carbonyls holds particular prominence. With Type I allylation reagents[3], this reaction not only delivers functionality that is strategically positioned for establishing appropriate oxygenation patterns, but its stereochemical predictability allows ready access to acetate, propionate, and isobutyrate synthetic equivalents. A limitation of many allylation reactions, however, is that they deliver products bearing a terminal alkene; if one desires additional substitution or functionality on the olefin, additional synthetic manipulations are often required.[4] In this regard, the vinylogous aldol reaction has proven particularly important as it delivers enoate-derived homoallylic alcohols. Unfortunately, even with the tremendous emphasis placed on the development of catalytic enantioselective vinylogous aldol reactions, an efficienct syn-selective asymmetric propionate version is still unavailable, as is a version that delivers quaternary centers.[5] In this report, we document the first examples of the enantioselective catalytic 1,2-diboration of 1,3-dienes (eq. 1, Scheme 1). As depicted in Scheme 1, the 1,2-diboration of 1,3-dienes delivers allylboron reagents (1) that are perfectly configured to participate in highly selective allylation reactions.[6,7,8] Importantly, with appropriate oxidative work-up, these reactions deliver vinylogous aldol equivalents that directly address the above-described synthesis limitations. Also of significant consequence, is that the allyl boron in the allylation product 2 may be subject to other useful bond forming reactions[9] that allow for chain-extending polyketide synthesis.
Scheme 1.

Strategy for chain-extending polyketide synthesis using 1,2-diboration of 1,3-dienes.
To develop the catalytic synthesis strategy in Scheme 1, efforts were first extended toward the development of an enantioselective 1,2-diboration of terminal dienes.[10] A recent study in our laboratory found that enantioselective 1,4-diboration of trans-1,3-dienes could be accomplished with Pt(dba)3 and a chiral phosphonite ligand.[11,12] While evaluation of alternate phosphorous donors revealed ligands that furnished the desired 1,2 diboration, selectivity was suboptimal. A more reliable strategy for obtaining the 1,2-diboration product was to replace the trans diene substrate with cis 1,3-dienes. This approach furnished 1,2-diboration products (3:1 to >20:1 1,2:1,4 selectivity) across a range of substrates and generally occured with excellent enantioselection. After optimization, the ligand structures and reaction conditions depicted in Table 1 were found to be optimal. With respect to polypropionate synthesis, the diboration of cis-pentadiene is paramount and this was found to occur in excellent enantioselectivity (95:5 er) and good yield with ligand L2 (1, Table 1).[13] Aside from the phenyl substituted diene, all other cis dienes examined reacted with outstanding enantiocontrol when employing ligand L1. The diboration of 1,1-disubstituted dienes employing ligand L3 occured with uniformly high levels of stereocontrol (compounds 9–14). Notably, allylic silyl ethers do not engage in allylic borylation under the reaction conditions and tethered alkenes do not appear to perturb the reaction in a detrimental way.
Table 1.
Catalytic Enantioselective 1,2-Diboration of 1,3-Dienes[a]
 |
Reaction carried out at 60 °C for 12 h, followed by oxidation with 30% H2O2 and 3 M NaOH for 4 h. For products 1 to 8, 3 mol % Pt(dba)3, 6 mol % ligand, and THF solvent employed; for products 9 to 14 3 mol % Pt(dba)3, 3.6 mol % ligand employed. Percent yield of purified 1,2-diol, average of two experiments. Enantiomeric purity determined by GC analysis of a derivative employing a chiral stationary phase (see SI for details).
Significant features of the 1,2-bis(boronate) resulting from the diboration of cis-1,3-dienes are an embedded α-chiral allylboronate and a cis alkene. According to the seminal studies of Hoffmann, it was anticipated that these features would render allylation reactions highly selective.[14] In an initial experiment, commercially available cis-piperylene was subjected to catalytic diboration with Pt(dba)3 and ligand L2 in THF. The solvent was then removed in vacuo, CH2Cl2 added, and benzaldehyde introduced to the reaction mixture. Upon oxidative work-up, the allylation product was obtained in moderate isolated yield (48%), however, the stereoisomeric purity was excellent (>20:1 syn:anti, 94:6 er). Examination of the unpurified reaction mixture revealed significant amounts of bis(allylation) product presumably arising from addition of the initial adduct (15, Table 2) to a second equivalent of aldehyde. To minimize the amount of bis(allylation), the diboration was executed on a scale that delivered a two-fold excess of 1,2-bis(boronate) relative to aldehyde. This strategy provided good yields of the monoallylation product for a range of aldehyde substrates (Table 2). As might be expected, the allylation products were found to possess syn relative stereochemistry (>20:1 in all cases) and the product alkene was found to be in the trans configuration. With respect to synthetic utility, it is significant that aromatic, aliphatic, and α,β-unsaturated aldehydes all participate and the level of chirality transfer from the allylboronate is excellent. Other notable features are that α-branched aldehydes react (entry 6) as do those that bear α-oxygenation (entries 7–9). Importantly, the regioisomeric 1,4-diboration product is not only less reactive in allylation reactions, but any derived allylation product is easily removed by silica gel chromatography. A stereochemical model that correlates reactant configuration with product configuration is depicted in ts-1 (graphic, Table 2). Most likely to avoid an A(1,3) interaction with the cis substituent, the CH2B(pin) occupies an equatorial position in the chair-like transition structure. Bond reorganization then delivers the observed enantiomer of product with syn stereoselection and with a trans alkene.
Table 2.
Asymmetric Allylboration of Carbonyls[a]
 | ||||
|---|---|---|---|---|
| Entry | Product | % Yield[b] | er[c] | es[d] |
| 1 |
 16 |
71 | 94:6 | 98 |
| 2 |
 17 |
66 | 94:6 | 98 |
| 3 |
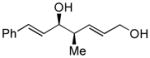 18 |
64 | 95:5 | >99 |
| 4 |
 19 |
66 | 96:4 | >99 |
| 5 |
 20 |
72 | 94:6 | 98 |
| 6 |
 21 |
62 | 93:7 | 96 |
| 7 |
 22 |
72 | 94:6 | 98 |
| 8 |
 23 |
90 | 97:3 | >99 |
| 9 |
 24 |
68 | 96:4 | >99 |
Diboration reaction carried out at 60 C for 12 h with ligand L2 for entries 1–7 and ligand L1 for entries 8–9; allylation at room temperature for 12 h with 2:1 ratio of diboron to aldehyde; oxidation with 30% H2O2 and 3 M NaOH for 12 h.
Percent yield of purified material, average of two experiments.
Enantiopurity determined by GC or SFC analysis employing a chiral stationary phase.
Enantiospecificity (es) calculated as follows: (%ee allylation product / %ee diboration product)*100; value ≥ 100% for entries 3,4 and 9, likely a result of error in the measurement of er.
As exemplified by the production of compounds 9–14 (Table 1), 1,2-diboration of 1,1-disubstituted dienes occurs with excellent selectivity. Similar to the case of cis dienes, it was anticipated that the intermediate bis(boronate) esters would participate in highly selective carbonyl allylation reactions. However, with 1,1-disubstitued dienes, the overall reaction sequence would furnish products bearing all-carbon quaternary centers.[15,16] Notably, the added steric encumbrance of these allylation products appeared to diminish the rate of secondary allylation such that high yields were obtained even when equimolar amounts of diene, B2(pin)2, and aldehyde were employed (Table 3). Also worth mention is that both the diboration and the allylation reactions proceeded cleanly in toluene solvent and this allowed the entire sequence to be accomplished in a single flask without solvent swapping operations. Of particular note, either diastereomer of product could be obtained in excellent yield and enantioselectivity simply by employing the appropriate diastereomer of diene substrates. For example, whereas diboration/allylation employing neral-derived diene 38 furnished product 26 in excellent selectivity, diboration/allylation employing geranial-derived diene 37 delivered diastereomer 27 with excellent levels of stereocontrol. Similar observations were made with propionaldehyde-derived products 28 and 29.
Table 3.
Asymmetric Allylboration of Carbonyls with γ,γ-Disubstituted Allylboronates.[a]
 |
Diboration reaction carried out at 60 °C for 12 h with ligand L3 and with [substrate] = 1.0 M in toluene. Allylations were conducted at 60 °C for 24 h with 1 equiv. of aldehyde relative to diene and B2(pin)2. Oxidation conducted with 30% H2O2 and 3 M NaOH for 4 h. Percentyield is of purified material and an average of two experiments. Enantiomer ratio determined by HPLC or SFC analysis employing a chiral stationary phase.
Experiment employed 3 equiv. i-PrCHO.
An attractive feature of the synthesis strategy described above is that the allylation product can be subjected to bond-forming reactions other than oxidation. As depicted in Scheme 2 (eq. 1), when diene 37 was subjected to diboration and then employed in allylation, oxidative work-up furnished allylic alcohol 27. Alternatively, subsequent to the allylation reaction, the intermediate allylboronate was subjected to homologation according to the Matteson protocol (eq. 2).[17] This delivered homoallylic alcohol 39, also in excellent yield and stereoselection. Lastly, it was found that the allylboronation product, when subjected to protodeboronation conditions similar to those described by Aggarwal[9d], was converted to the simple alkene product 40 (eq. 3). In this case, the protonation event occurred predominantly by an SE2′ pathway and delivered bishomoallylic alcohol 40 as the major product. Considering the range of transformations that are available to organoboronates, and the fact that the diboration/allylation sequence occurs cleanly in aprotic solvent, allylation intermediates may be directly transformed to a number of other useful building blocks.
Scheme 2.

Diboration-Allylation-Functionalization Sequence.
In conclusion, we have described the catalytic enantioselective 1,2-diboration of 1,3-dienes and have found that the 1,2-bis(boronate) products can be employed in versatile stereoselective allylation reactions and deliver a range of functionalized chiral building blocks. Further studies on the use of these reactions in complex molecule synthesis are in progress and will be reported in due course.
Acknowledgments
We are grateful to the NIH (NIGMS GM-59417) and the NSF (DBI-0619576, BC Mass. Spec. Center; CHE-0923264, BC X-ray Facility) for support and to AllyChem for a donation of B2(pin)2
References
- 1.Reviews on catalytic asymmetric allylation: Denmark SE. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h.Yanagisawa A. In: Comprehensive Asymmetric Catalysis, Supplement. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Vol. 2. Springer-Verlag; Berlin: 2004. p. 97.c) For selected recent examples: Jain P, Antilla JC. J Am Chem Soc. 2010;132:11884. doi: 10.1021/ja104956s.Rauniyar V, Hall DG. Angew Chem, Int Ed. 2006;45:2426. doi: 10.1002/anie.200504432.Wada R, Oisaki K, Kanai M, Shibasaki M. J Am Chem Soc. 2004;126:8910. doi: 10.1021/ja047200l.Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2006;128:12660. doi: 10.1021/ja0651308.
- 2.a) Hall DG. Pure Appl Chem. 2008;80:913. [Google Scholar]; b) Lachance H, Hall DG. Organic Reactions. 2008;73:1. [Google Scholar]
- 3.a) Denmark SE, Weber EJ. Helv Chim Acta. 1983;66:1655. [Google Scholar]; b) Roush WR. In: Comprehensive Organic Synthesis. Trost BM, editor. Vol. 2 Pergamon; Oxford: 1991. [Google Scholar]
- 4.For selected examples of asymmetric allylborations that deliver functionalized alkene products, see: Hoffmann RW, Dresely S. Angew Chem, Int Ed Engl. 1986;25:189.Corey EJ, Yu CM, Kim SS. J Am Chem Soc. 1989;111:5495.Lachance H, Lu X, Gravel M, Hall DG. J Am Chem Soc. 2003;125:10160. doi: 10.1021/ja036807j.Woodward AR, Burks HE, Chan LM, Morken JP. Org Lett. 2005;7:5505. doi: 10.1021/ol052312i.Rauniyar V, Hall DG. J Org Chem. 2009;74:4236. doi: 10.1021/jo900553f.Althaus M, Mahmood A, Suarez JR, Thomas SP, Aggarwal VK. J Am Chem Soc. 2010;132:4025. doi: 10.1021/ja910593w.Fernandez E, Pietruszka J, Frey W. J Org Chem. 2010;75:5580. doi: 10.1021/jo1008959.Chen M, Ess DH, Roush WR. J Am Chem Soc. 2010;132:7881. doi: 10.1021/ja103041u.
- 5.For recent reviews, see: Denmark SE, Heemstra JR, Jr, Beutner GL. Angew Chem, Int Ed. 2005;44:4682. doi: 10.1002/anie.200462338.Brodmann T, Lorenz M, Schackel R, Simsek S, Kalesse M. Synlett. 2009:174.
- 6.For recent catalytic enantioselective approaches to a-chiral allylboronates, see: Carosi L, Hall DG. Angew Chem, Int Ed. 2007;46:5913. doi: 10.1002/anie.200700975.Ito H, Ito S, Sasaki Y, Matsuura K, Sawamura M. J Am Chem Soc. 2007;129:14856. doi: 10.1021/ja076634o.Ito H, Ito S, Sasaki Y, Matsuura K, Sawamura M. Pure Appl Chem. 2008;80:1039.Guzman-Martinez A, Hoveyda AH. J Am Chem Soc. 2010;132:10634. doi: 10.1021/ja104254d.Ito H, Okura T, Matsuura K, Sawamura M. Angew Chem, Int Ed. 2010;49:560. doi: 10.1002/anie.200905993.Sasaki Y, Zhong C, Sawamura M, Ito H. J Am Chem Soc. 2010;132:1226. doi: 10.1021/ja909640b.
- 7.For relevant enantioselective carbonyl allylations that do not employ allyl metal reagents, see: Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340. doi: 10.1021/ja802001b.Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891. doi: 10.1021/ja805722e.Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514. doi: 10.1021/ja808857w.Skucas E, Zbieg JR, Krische MJ. J Am Chem Soc. 2009;131:5054. doi: 10.1021/ja900827p.Han SB, Han H, Krische MJ. J Am Chem Soc. 2010;132:1760. doi: 10.1021/ja9097675.Han SB, Gao X, Krische MJ. J Am Chem Soc. 2010;132:9153. doi: 10.1021/ja103299f.
- 8.For a review of relevant Ni-catalyzed carbonyl-diene coupling reactions, see: Miura M, Tamaru Y. Top Curr Chem. 2007;279:173.For selected recent references, see: Takimoto M, Kajima Y, Sato Y, Mori M. J Org Chem. 2005;70:8605. doi: 10.1021/jo051283m.Kimura M, Ezoe A, Mori M, Iwata K, Tamaru Y. J Am Chem Soc. 2006;128:8559. doi: 10.1021/ja0608904.Cho HY, Morken JP. J Am Chem Soc. 2008;130:16140. doi: 10.1021/ja806113v.Cho HY, Morken JP. J Am Chem Soc. 2010;132:7576. doi: 10.1021/ja101513d.
- 9.Review of stereospecific transformations of boronate esters, see: Thomas SP, French RM, Jheengut V, Aggarwal VK. Chem Rec. 2009;9:24. doi: 10.1002/tcr.20168.For selected recent examples: Stymiest JL, Bagutski V, French RM, Aggarwal VK. Nature. 2008;456:778. doi: 10.1038/nature07592.Schmidt F, Keller F, Vedrenne E, Aggarwal VK. Amgew Chem Int Ed. 2009;48:1149. doi: 10.1002/anie.200805272.Nave S, Sonawane RP, Elford TG, Aggarwal VK. J Am Chem Soc. 2010;132:17096. doi: 10.1021/ja1084207.Binanzer M, Fang GY, Aggarwal VK. Angew Chem, Int Ed. 2010;49:4264. doi: 10.1002/anie.201001223.Dutheuil G, Webster MP, Worthington PA, Aggarwal VK. Angew Chem, Int Ed. 2009;48:6317. doi: 10.1002/anie.200901194.Bagutski V, Elford TG, Aggarwal VK. Angew Chem Int Ed. 2011;50:1080. doi: 10.1002/anie.201006037.
- 10.For 1,2-diboration of 1,3-dienes in the absence of phosphine ligands, see: Ishiyama T, Yamamoto M, Miyaura N. Chem Commun. 1997:689.For enantioselective 1,2-diboraiton of terminal alkenes, see: Kliman LT, Mlynarski SN, Morken JP. J Am Chem Soc. 2009;131:13210. doi: 10.1021/ja9047762.
- 11.Burks HE, Kliman LT, Morken JP. J Am Chem Soc. 2009;131:9134. doi: 10.1021/ja809610h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Non-enantioselective 1,4-diene diboration: Ishiyama T, Yamamoto M, Miyaura N. Chem Commun. 1996:2073.Clegg W, Thorsten J, Marder TB, Norman NC, Orpen AG, Peakman TM, Quayle MJ, Rice CR, Scott AJ. J Chem Soc, Dalton Trans. 1998:1431.Morgan JB, Morken JP. Org Lett. 2003;5:2573. doi: 10.1021/ol034936z.
- 13.a) Sakaki J, Schweizer WB, Seebach D. Helv Chem Acta. 1993;76:2654. [Google Scholar]; b) Seebach D, Hayakawa M, Sakaki J, Schweizer WB. Tetrahedron. 1993;49:1711. [Google Scholar]; c) Alexakis A, Burton J, Vastra J, Benhaim C, Fournioux X, van den Heuvel A, Leveque JM, Maze F, Rosset S. Eur J Org Chem. 2000:4011. [Google Scholar]; d) Bee C, Han SB, Hassan A, Iida H, Krische MJ. J Am Chem Soc. 2008;130:2746. doi: 10.1021/ja710862u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffmann RW. Pure Appl Chem. 1988;60:123.Hoffmann RW, Neil G, Schlapbach A. Pure Appl Chem. 1990;62:1993.Hoffmann RW, Zeiss HJ. J Org Chem. 1981;46:1309.Hoffmann RW, Zeiss HJ. Angew Chem, Int Ed Engl. 1979;18:306.Andersen MW, Hildebrndt B, Kosher G, Hoffmann RW. Chem Ber. 1989;122:1777.Hoffmann RW, Dirich K, Kosher G, Strumer RR. Chem Ber. 1989;122:1783.For recent applications of these strategies, see: ref. 2b.
- 15.For reviews of enantioselective construction of quaternary centers, see: Das JP, Marek I. Chem Commun. 2011;47:4593. doi: 10.1039/c0cc05222a.Cozzi PG, Hilgraf R, Zimmermann N. Eur J Org Chem. 2007:5969.Trost BM, Jiang C. Synthesis. 2006:369.Christoffers J, Baro A. Adv Synth Catal. 2005;447:1473.Douglas CJ, Overman LE. Proc Natl Acad Sci USA. 2004;101:5363. doi: 10.1073/pnas.0307113101.Corey EJ, Guzman-Perez A. Angew Chem, Int Ed. 1998;37:388. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V.
- 16.For catalytic enantioselective construction of quaternary centers by allylation, see: Denmark SE, Fu J. J Am Chem Soc. 2001;123:9488. doi: 10.1021/ja016552e.Denmark SE, Fu J. Org Lett. 2002;4:1951. doi: 10.1021/ol025971t.For related asymmetric allylations with a g, g-disubstitued reagents, see: Hoffmann RW, Schlapbach A. Liebigs Ann Chem. 1991:1203.Skulte G, Marek I. J Am Chem Soc. 2006;128:4642. doi: 10.1021/ja060498q.
- 17.a) Sadhu KM, Matteson DS. Organometallics. 1985;4:1687. [Google Scholar]; b) Chen AC, Ren L, Crudden CM. Chem Commun. 1999:611. [Google Scholar]; c) Chen AC, Ren L, Crudden CM. J Org Chem. 1999;64:9704. [Google Scholar]; d) Ren L, Crudden CM. Chem Commun. 2000:721. [Google Scholar]