

Abstract
Background
Cardiac overload, a major cause of heart failure, induces the expression of the heat shock protein H11 kinase/Hsp22 (Hsp22).
Methods and Results
To determine the specific function of Hsp22 in that context, a knockout (KO) mouse model of Hsp22 deletion was generated. Although comparable to wild type mice in basal conditions, KO mice exposed to pressure overload developed less hypertrophy, and showed ventricular dilation, impaired contractile function, increased myocyte length and accumulation of interstitial collagen, faster transition into heart failure and increased mortality. Microarrays revealed that hearts from KO mice failed to transactivate genes regulated by the transcription factor STAT3. Accordingly, nuclear STAT3 tyrosine phosphorylation was decreased in KO. Silencing and over-expression experiments in isolated neonatal rat cardiomyocytes showed that Hsp22 activates STAT3 via production of interleukin-6 by the transcription factor NF-κB. In addition to its transcriptional function, STAT3 also translocates to the mitochondria where it increases oxidative phosphorylation. Both mitochondrial STAT3 translocation and respiration were significantly decreased in KO mice as well.
Conclusions
Hsp22 represents a previously undescribed activator of both nuclear and mitochondrial functions of STAT3, and its deletion in a context of pressure overload in vivo accelerates the transition into heart failure and increases mortality.
Keywords: Animals; Cardiomegaly; enzymology; genetics; Cell Nucleus; enzymology; genetics; Cells, Cultured; Collagen; metabolism; Gene Deletion; Gene Expression Profiling; HSP20 Heat-Shock Proteins; genetics; Heart Failure; enzymology; genetics; mortality; Interleukin-6; biosynthesis; Male; Mice; Mice, Knockout; Mitochondria, Heart; enzymology; genetics; Muscle Proteins; genetics; Myocytes, Cardiac; enzymology; NF-kappa B; metabolism; Oxidative Phosphorylation; Rats; STAT3 Transcription Factor; genetics
Keywords: Heart failure, Heat shock proteins, Overload, STAT
Expression of the heat shock protein (Hsp) H11 kinase/Hsp22 (Hsp22) is restricted to a limited number of tissues, including the heart and skeletal muscle, both in rodents and in humans[1], and it rapidly increases in response to excessive contractile workload (overload)[2]. We previously showed in a transgenic (TG) mouse model that cardiac-specific over-expression of Hsp22 induces a pattern of stable cardiac hypertrophy with normal function[2,3]. However, because of some redundancy in the function of small molecular weight Hsps[4], the precise function of Hsp22 in the response to overload remains unclear, and it is unknown whether Hsp22 expression is necessary for the physiological and molecular response of the heart exposed to increased stress by overload.
In the present study, we tested the hypothesis that Hsp22 expression is required for the activation of stress-responsive signaling pathways. We addressed that hypothesis using two complementary approaches, in vivo and in vitro. First, we generated a knockout (KO) mouse model to investigate the consequences of Hsp22 deletion in a context of pressure-induced overload, and found that a lack of Hsp22 accelerates ventricular dysfunction and remodeling, as well as the transition into heart failure. This phenotype corresponds to a lack of activation of genes trans-activated by STAT3, a stress-inducible transcription factor promoting cardiac cell survival and which is essential for adaptive growth upon overload[5]. Intriguingly, it was shown recently that mitochondrial translocation of STAT3 also promotes oxidative phosphorylation[6], and that function was also impaired in our KO model. Second, we manipulated Hsp22 expression in isolated cardiac myocytes to determine the signaling mechanisms involved.
These results illustrate a previously undescribed mechanism of cardiac response to overload, and in particular a novel mode of regulation of both nuclear and mitochondrial STAT pathways. Activation of this novel signaling may be of clinical relevance for patients exposed to chronic excessive workload and at risk of subsequent cardiac damage.
METHODS
Methods are further detailed in a supplementary file. All the animal investigations conform with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Generation of the construct for Hsp22 deletion
We generated a 12 Kb construct in which the first exon of the Hsp22 gene is targeted for deletion upon recombination. The left and right arms of the construct are 5 and 7 Kb long, respectively. The left arm is integrated by Xho I sites and the right arm by Xba I sites. The targeted exon is flanked by LoxP sites. Probes were generated outside of the construct on both sides for detection of positive clones by Southern blot, using Spe I and Eco RI digests, respectively.
Surgical procedures
Surgery was performed on 3 to 4 months-old male wild type (WT) and KO mice. To generate pressure overload, mice were submitted to transverse aortic constriction (TAC) with a 28-gauge needle as before[7]. All mice surviving the surgery were included in the study. Age-matched sham-operated control animals were included.
Tissue extraction and western blot
Heart tissues were homogenized at 4 °C in a lysis buffer[2,3,7,8]. Subcellular fractions were prepared by differential centrifugation[3]. Protein extracts were denatured, resolved on SDS-PAGE gels, and transferred to membranes. After incubation with the secondary antibody, detection was performed by chemiluminescence.
Histopathology
Samples were fixed in 10% formalin and cut in 7 μm-thick sections. Tissue sections were stained with Picric Acid Sirius Red to identify collagen[9]. Myocyte cross sectional area was determined on digitized images of TRITC-labeled wheat germ agglutinin stained sections[3,7]. TUNEL was performed as described[10]. Nuclear counterstaining was performed with DAPI.
Microarray data analysis
Messenger RNA was purified, reverse-transcribed[11,12], and subsequently transcribed into biotin-labeled synthetic antisense RNA[12]before hybridization (GeneChip 430.2.0, Affymetrix, Santa Clara, CA). Bioinformatics analysis of the microarray data was performed with the Welch’s t-test, using a P<0.05 and a fold change >1.2 to select significant genes[12]. For the gene density map, all genes were evenly divided into 20 groups based on fold change in each comparison, and then distributed in a 20x20 table. The number of genes in each cell was normalized to an expected number derived from randomized data. Transcription factor binding sites (TFBS) were obtained from the Molecular Signature Database[13]. Gene ontology (GO) and TFBS analyses were carried out with hypergeometric tests. Genes regulated by STAT3 were derived from a previous study[14]and from the Ingenuity Pathway analysis database. The STAT3 target genes were compared to other genes on the microarray using the cumulative distribution function (CDF) based on log2 ratio (fold change). Microarray data were deposited in the Gene Expression Omnibus (GEO) database (GSE24489).
Quantitative RT-PCR
Following reverse transcription of the mRNA of interest from 50 ng of total RNA, the cDNA was used for quantitative PCR (40 cycles of a 10 sec-step at 95°C and a 1 min-step at 60°C) using the SybrGreen method on a 7300 ABI-Prizm Sequence Detector (Applied Biosystems, Foster City, CA)[15]. Values are reported per cyclophilin transcript to correct for sample-to-sample RNA loading variations.
Mitochondrial oxygen consumption
After extraction, mitochondria (0.4 mg/ml) were incubated in a respiration buffer containing 100 mmol/L KCl, 50 mmol/L sucrose, 10 mmol/L HEPES, 5 mmol/L KH2PO4, pH 7.4 at 30°C. The following parameters of mitochondrial respiration were evaluated with a Clark-type electrode: 1. Substrate-dependent respiration rate (state 2 and state 4) in the absence of exogenous ADP; 2. ADP-stimulated respiration rate (state 3); 3. Respiration uncoupling in presence of 0.2 μmol/L carbonyl cyanide 4-trifluoromethoxyphenylhydrazone (FCCP).
Culture of cardiac myocytes
Neonatal rat cardiac myocytes were prepared as before[2,16,17]. The adenovirus harboring the Hsp22 sequence was described before[2,17]. An adenovirus harboring LacZ was used as a negative control. Hsp22 knockdown was performed as described previously[18], using luciferase silencing as a negative control[18]. IL-6 release was measured with the Quantikine rat IL-6 ELISA kit (R&D Biosystems, Minneapolis, MN). For DNA binding activities, myocytes were transfected with 20 moi Cignal lentivirus particles designed for STAT3 and NF-κB reporter assays (SABiosciences, Frederick, MD), and firefly luciferase activity was measured with the Luciferase Reporter Gene Assay (Roche, Indianapolis, IN).
Adult cardiac myocytes were prepared by retrograde perfusion of the hearts in presence of collagenase (type II; Worthington) and 0.1 mg/ml protease (type XIV, Sigma)[19]. Cell length, defined as the longest length parallel to the longitudinal axis of the myocytes, was measured by imaging system.
Statistical analysis
Results are presented as the mean ± standard error of the mean (SEM) for the number per group indicated in each Figure legend. The Student’s t test was used for two-group comparison. For more than two-group comparisons, statistical analysis was performed by two-by-two factorial designs (two groups exposed to two conditions), as well as one-way (multiple groups exposed to one condition) and two-way (multiple groups exposed to two conditions) analysis of variance (ANOVA) with Bonferroni post-hoc correction. Survival curves were compared with the Kaplan-Meier estimators, and their significant difference determined using the log rank test.
RESULTS
Hsp22 deletion does not affect basal cardiac structure and function
A construct targeting exon 1 of the Hsp22 gene (Figure 1a) was electroporated in 129 ES cells and subsequently injected in blastocysts of C57 mice. Mice with germline transmission of the construct were crossed with a CMV-Cre (C57) mouse to trigger the recombination of the construct, followed by crossing for five generations in a C57 background to obtain a homozygous mouse in which both Hsp22 alleles are deleted. KO mice were born at Mendelian distribution. The heterozygous mice express an amount of Hsp22 protein equivalent to that found in wild type (WT) mice (Figure 1a).
Figure 1. Characteristics of the KO mouse submitted to pressure overload.
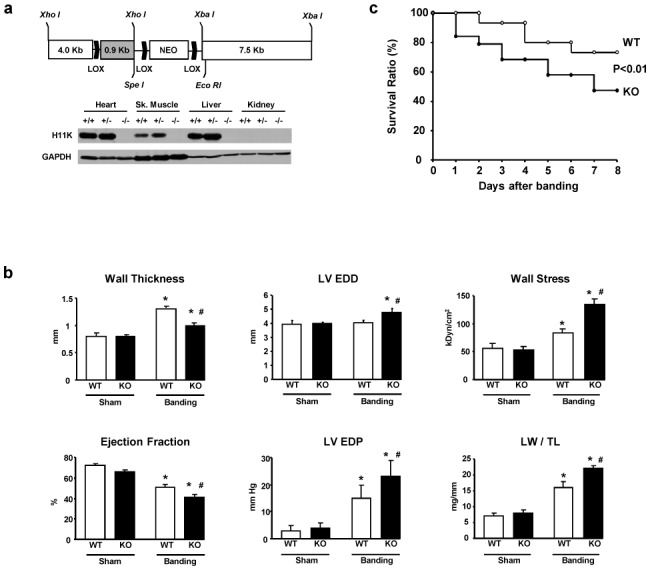
A. Generation of the Hsp22 KO mouse. Construct design (the gray box is exon 1) and Hsp22 expression in WT, heterozygous and KO mice, in various tissues. B. Functional characteristics of WT and KO mice (n=6 per group) in basal conditions (sham) and after two weeks TAC. The graphs show the LV septal wall thickness, end-diastolic diameter (EDD), wall stress, ejection fraction, end-diastolic pressure (EDP) and lung weight/tibial length (LW/TL). C. Survival curve of WT and KO mice after TAC and followed up to two weeks (n≥20 per group; there was no additional mortality in either group after day 7). *, P<0.05 versus corresponding sham; #, P<0.05 versus corresponding WT (2-by-2 analysis).
The physiological and morphological characteristics of the KO mouse in basal conditions are shown in the Supplemental Table 1. The KO mouse was comparable to WT in terms of body weight, left ventricular (LV) weight and lung weight. There was no difference between KO and WT mice in terms of cardiac dimensions measured by echocardiography, and of cardiac contractility, as assessed by ejected volume and developed pressures. At histology, there was no difference between KO and WT in terms of cell size, apoptosis, and collagen accumulation.
Hsp22 deletion precipitates heart failure upon pressure overload
Both WT and KO mice were submitted to transverse aortic constriction (TAC) for 2 weeks to determine the consequences of Hsp22 deletion on cardiac function and histology under cardiac stress. Upon TAC, Hsp22 was still detected specifically in myocytes as opposed to non-myocytic cells (Supplementary Figure S1). The pressure gradient induced by TAC was comparable between groups (80±10 versus 67±8 mmHg in WT and KO, respectively). Functional measurements by echocardiography and hemodynamics showed that WT mice responded to TAC by an increase in wall thickness, LV end-diastolic pressure, and lung weight/tibial length, as well as a decrease in ejection fraction (Figure 1b). All these parameters were also significantly affected in KO mice submitted to TAC. However, when compared to WT, KO mice showed significantly less wall thickening and increased LV diameter, resulting in higher wall stress, whereas the decrease in ejection fraction was significantly more important (Figure 1b). The deterioration of contractile function in hearts from banded KO mice was accompanied by a higher increase in LV end-diastolic pressure and lung weight/tibial length compared to WT (Figure 1b). Also, the mortality rate after TAC was doubled in KO compared to WT (Figure 1c). Considering the loss in cardiac function observed in KO mice after TAC, we assume that mice died of contractile dysfunction.
At histology, the myocyte cross-sectional area after TAC increased significantly more in WT than in KO mice (Figure 2a and b). Reciprocally, cell length increased significantly more in KO than in WT (Figure 2a and b), reflecting a pattern of eccentric hypertrophy in the KO, in agreement with the functional measurements shown in Figure 1. Measurement of cell volume confirmed that the KO mouse develops hypertrophy upon TAC, although to a significantly lesser extent than the WT (Figure 2b). The surface of myocardium covered by collagen after TAC was significantly higher in KO (Figure 2a and b), and this was accompanied by a higher expression of the matrix metalloproteinase (MMP) MMP2 in KO compared to WT (Supplementary Figure S2). However, TAC-induced increase in apoptosis was comparable between both groups at that time point (Figure 2b).
Figure 2.
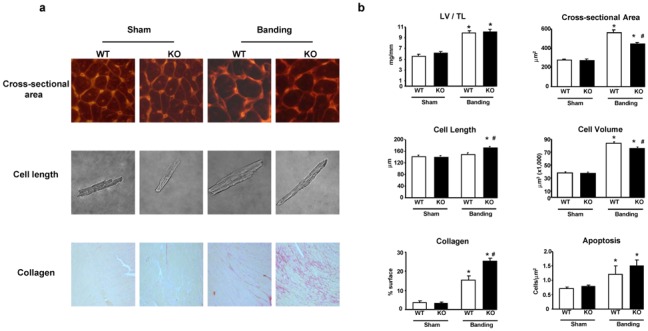
Morphological characteristics of WT and KO mice (n=6–8 per group) in basal conditions (sham) and after two weeks TAC. A. Representative examples of cross-sectional area, myocyte length and interstitial collagen accumulation (PSR staining). B. Quantitation of LV/TL, myocytes cross-sectional area, cell length and volume measured after myocytes isolation, and collagen accumulation and apoptosis (measured in all cell types) reported per myocardial surface. *, P<0.05 versus corresponding sham; #, P<0.05 versus corresponding WT (2-by-2 analysis).
Therefore, Hsp22 deletion does not affect cardiac structure and function in basal conditions, but it precipitates heart failure, increases mortality and accelerates ventricular remodeling in a context of pressure overload.
Hsp22 deletion impairs the activation of stress-activated survival pathways
Because KO mice show extensive phenotypic differences compared to WT at two weeks after TAC, we sought to determine which early events could be the cause, rather than the consequence, of altered function. For that purpose, mice were submitted to TAC for three days, a time point at which cardiac function was comparable between WT and KO mice (not shown). We showed before that Hsp22 activates the phosphatidylinositol-3 kinase (PI3K)/Akt growth pathway via the bone morphogenetic protein (BMP) receptor[18]. Phosphorylation of Akt (S473) was comparable between WT and KO in sham-operated animals (Figure 3). After TAC however, KO mice showed significantly less phosphorylation of Akt (60% of WT). Activation of the BMP receptor, as determined by Smad 1/5/8 phosphorylation[18], was significantly impaired in KO after TAC (Figure 3). Another pathway mediating cardiac cell growth and survival is represented by ERK1/2. The phosphorylation of both ERK1 and ERK2 was comparable between WT and KO mice in sham conditions (Figure 3). After TAC, phosphorylation of ERK1/2 increased in WT but dramatically decreased in KO (ERK1: 12% of WT; ERK2: 10% of WT; both, P<0.05). Other survival mechanisms activated by Hsp22 include the inducible isoform of nitric oxide synthase (iNOS)[8]and Hsp70[8]. In sham-operated KO mice, the abundance of iNOS was only 35% of that found in corresponding WT mice (Figure 3). After banding, iNOS expression was not significantly affected in WT but it was almost abolished in KO mice (Figure 3). Expression of Hsp70 after TAC was doubled in KO mice compared to WT (Figure 3), most likely as a compensatory mechanism.
Figure 3. Hsp22 deletion impairs the activation of stress-responsive pathways.
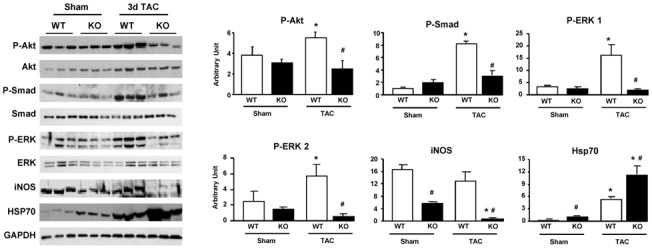
Immunoblotting for P-Akt, Akt, P-Smad 1/5/8, Smad 1, P-ERK 1, P-ERK 2, ERK 1/2, iNOS and Hsp70 in WT versus KO, both in sham conditions and after three days TAC. Bar graphs for n=5 per group. GAPDH is used as a loading control. *, P<0.05 versus corresponding sham; #, P<0.05 versus corresponding WT (2-by-2 analysis).
Hsp22 deletion impairs stress-activated gene expression
To explore the mechanism causing the deteriorating phenotype of the KO mouse upon TAC, both WT and KO mice were compared by microarray analysis in basal conditions, and three days and two weeks after TAC. Among over 14,000 genes analyzed, only 291 and 153 genes were up-regulated or down-regulated, respectively, in sham KO mice compared to WT (Figure 4a). After three days TAC, the number of genes up-regulated in the KO compared to the WT was similar to that found in sham-operated animals (248), whereas the number of genes down-regulated in KO mice increased by more than three-fold (Figure 4a). Two weeks after TAC, the number of differently regulated genes between WT and KO was again roughly similar to that found in sham (Figure 4a). Comparison of gene profiles by Venn diagram between sham and three days TAC (Figure 4b) shows that the identity of the regulated genes totally differs between both conditions. This difference is confirmed by the lack of similarity for significant ontology terms associated with the regulated genes (Supplemental Table 2). The large number of genes with lower expression in KO compared to WT 3 days after TAC together with the comparison of the Venn diagrams suggests a lack of activation of stress-induced genes at the acute phase of TAC in the KO mouse.
Figure 4. Microarray analysis of WT and KO mice.
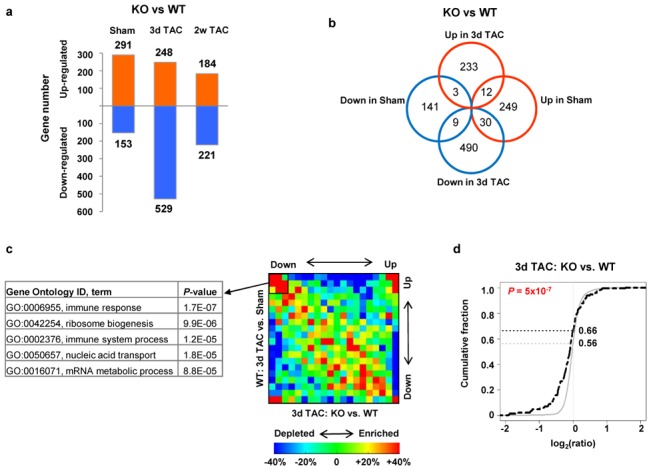
A. Number of genes differentially regulated between KO versus WT in sham, and after 3 days and 2 weeks TAC. B. Venn diagram showing differences of regulated genes between KO and WT in sham and 3 days TAC. C. Gene ontology categories, corresponding to genes with lower expression in KO versus WT 3 days after TAC, are indicated. The gene density map reflects distribution of genes, with color corresponding to the scale shown in the Figure. For example, “+40%” and “−40%”, shown as red and blue indicate 40% enrichment and 40% depletion, respectively. D. Expression changes of STAT3-regulated genes in Hsp22 KO 3 days after TAC. STAT3-annotated genes were compared to other genes on the microarray using the cumulative distribution function (CDF) based on log2 ratio (fold change). The black dotted curve is for STAT3-regulated genes, and the grey curve for other genes. For each curve, the fraction of genes at which log2(ratio)=0 is indicated. The P value indicating the difference between both curves is calculated from the Kolmogorov-Smirnov test.
Further analysis was performed by gene density map and by transcription factor binding sites (TFBS) analysis. The gene density map presented in Figure 4c shows, on the Y axis, the genomic differences between 3 days TAC and sham in WT mice, which represents the normal gene response to overload, and, on the X axis, the differences between KO and WT mice 3 days after TAC. A cluster of genes was found to be up-regulated in the normal response to overload but down-regulated in KO versus WT (Figure 4c). When grouped by ontology terms, these genes correspond mainly to the inflammatory response. TFBS analysis was used to determine whether this down-regulated inflammatory response in KO corresponds to specific transcription factors. Among the few TFBS that were found to be significantly associated with genes with lower expression in KO mice (Supplemental Table 3), we indeed found the cytokine-responsive interferon-stimulated response element (ISRE), which activates genes related to the inflammatory response.
A main activator of IRSE/inflammatory response in the heart is the transcription factor STAT, which is essential for the cardiac stress response to overload. To examine more specifically whether STAT plays a role in gene regulation in Hsp22 KO, we compared genes that were previously annotated as STAT3-regulated genes[14]with the other genes on the microarray, with respect to expression differences in WT versus KO 3 days after TAC (Figure 4d). We found that, compared to other genes, a higher fraction of STAT3-regulated genes have significantly lower expression in KO than in WT (Figure 4d), suggesting that these genes have an attenuated response to pressure overload in KO. These results were further validated by quantitative PCR, showing that representative STAT3-annotated genes were down-regulated in KO mice compared to WT, not only after TAC but also in sham conditions (Figure 5).
Figure 5. Validation of the microarray experiments by quantitative PCR.
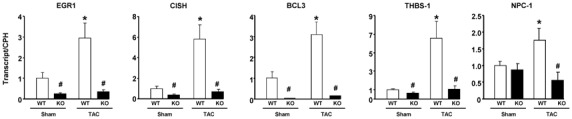
RNA expression of STAT3-regulated genes in hearts from WT versus KO mouse, in sham conditions or three days after TAC. Results are normalized per cyclophilin (CPH) transcript. *, P<0.05 versus corresponding sham; #, P<0.05 versus corresponding WT (2-by-2 analysis). Abbreviations: EGR-1, early growth response-1; CISH, cytokine-inducible SH2-containing protein; Bcl3, B cell leukemia/lymphoma-3; THBS-1, thrombospondin-1; NPC-1, Niemann Pick type C1.
Hsp22 modulates both nuclear and mitochondrial function of STAT3
Based on this information, the regulatory function Hsp22 on the STAT pathway was further explored in vivo. In the heart, the main subtypes of STAT transcription factors are STAT1 and STAT3, with STAT1 being mainly pro-apoptotic and STAT3 promoting survival[20]. STAT transcriptional activity is stimulated by tyrosine phosphorylation[5]. Beside its role as a transcription factor, STAT3 was recently shown to translocate to the mitochondria where it activates the electron transport chain upon serine phosphorylation[6]. Therefore, we tested in subcellular fractions (Figure 6a) whether Hsp22 expression affects the nuclear and/or mitochondrial regulation of STAT in vivo. For these experiments, we compared the consequences of down- versus up-regulation of Hsp22 expression by using not only our Hsp22 KO mouse, but also an Hsp22 TG mouse with cardiac-specific over-expression of Hsp22, which was described before[2].
Figure 6. Regulation of STAT by Hsp22.
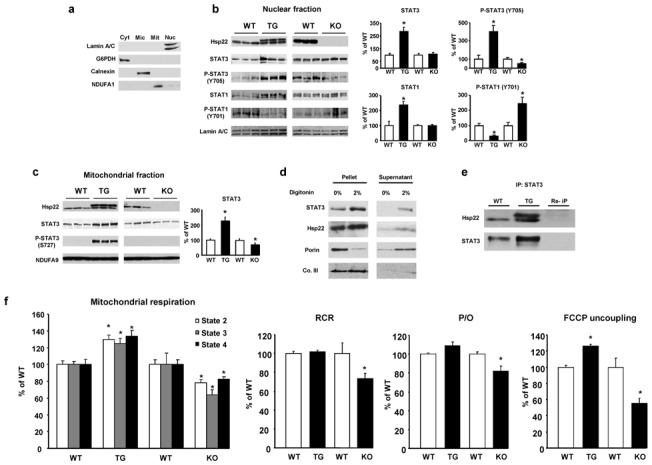
A. Subcellular fractionation, using markers for cytosol (cyt), microsomes (mic), mitochondria (mit) and nuclei (nuc). B. Expression of Hsp22, STAT3, Y705 P-STAT3, STAT1 and Y701 P-STAT1 in nuclear fractions from Hsp22 TG or KO mice and respective WT littermates. Lamin A/C is used as a loading control. Bar graphs for n=3 per group. *, P<0.05 versus corresponding WT (2-by-2 analysis). C. Expression of Hsp22, STAT3 and S727 P-STAT3 in mitochondrial fractions from Hsp22 TG or KO mice and respective WT littermates. The NADH dehydrogenase 1 alpha subunit 9 (NDUFA9) is used as a loading control. Bar graphs represent the mean±SEM for n=3 per group. *, P<0.05 versus corresponding WT (2-by-2 analysis). D. Treatment of mitochondrial fractions with 2% digitonin followed by centrifugation. Porin, used as a marker of outer mitochondrial membrane, is released into the supernatant upon addition of digitonin, whereas complex III (Co. III), a marker of the mitoplast, remains in the undigested pellet, together with both STAT3 and Hsp22. E. Immunoprecipitation of STAT3 in mitochondrial fractions followed by western blotting for both Hsp22 and STAT3 in a heart sample from both WT and TG mice. The upper band for Hsp22 in the TG sample corresponds to the hemagglutinin-tagged transgenic product. Repeated immunoprecipitation (Re-IP) on a TG protein sample refers to a second incubation of the protein sample with the same antibody. Re-IP should not provide any band upon immunoblotting if the protein of interest has been successfully depleted by the first immunoprecipitation. F. Measurement of mitochondrial respiration rate (states 2, 3 and 4), state 3/4 respiration coefficient ratio (RCR), ADP/O ratio and maximal respiration under uncoupling by FCCP in Hsp22 TG or KO mice and respective WT littermates (n=5 per group). *, P<0.05 versus corresponding WT (one-way ANOVA).
Immunoblotting in nuclear fractions from hearts of KO compared to WT mice showed no significant difference in the abundance of total STAT3, however tyrosine-phosphorylated (Y705) STAT3 was decreased by about 50% in the KO (Figure 6b). Reciprocally, both STAT3 and Y705 P-STAT3 were significantly more abundant in nuclear fractions from the TG mouse as compared to its corresponding WT (Figure 6b). Phosphorylation of nuclear STAT1 was also affected in both mouse models, however its phosphorylation pattern showed a mirror image compared to STAT3, i.e., it was increased in the KO and decreased in the TG mouse (Figure 6b).
Similar experiments were conducted in mitochondrial fractions. Expression of STAT3 was decreased by 40% in KO and increased by 2–3 fold in TG mice compared to WT (Figure 6c). Phosphorylation of mitochondrial STAT3 on S727 was also increased in the TG compared to WT (Figure 6c).
The intra-mitochondrial localization of STAT3 and Hsp22 was confirmed upon treatment of mitochondrial fractions with digitonin, which dissociates the outer mitochondrial membrane (OMM) but leaves intact the mitoplast, including the inner mitochondrial membrane (IMM) and the mitochondrial matrix. Treatment of mitochondria with 2% digitonin followed by centrifugation led to the release of porin, a marker of OMM, in the supernatant, whereas complex III, a marker of the mitoplast, was left in the pellet, together with most of both STAT3 and Hsp22 (Figure 6d). The small amount of STAT3 and Hsp22 released in the supernatant (Figure 6d) could be due to the presence of these proteins in the intermembrane space, or due to slight mitochondrial damage. Immunoprecipitation of STAT3 from these fractions showed a co-precipitation of Hsp22, suggesting that both proteins interact in the mitochondria (Figure 6e).
We determined next the impact of these findings on mitochondrial respiration, both in TG and KO sham-operated mice (Figure 6f). Absolute values of mitochondrial respiration are presented in Supplemental Table 4. Both respiration state 3 (ADP-stimulated), and state 2 and 4 (substrate-dependent) were reciprocally regulated in TG versus KO mice (Figure 6f). In addition, the ADP/O ratio and the respiratory control ratio of mitochondria isolated from KO were significantly decreased as compared to WT, reflecting impaired respiratory coupling, but unaffected in TG (Figure 6f). Maximal stimulation of mitochondrial respiration by the uncoupling agent carbonylcyanide 4 trifluoromethoxyphenylhydrazone (FCCP) resulted in a 50% lower respiration rate in KO compared to WT, whereas it was increased in TG mice (Figure 6f). Taken together, these results demonstrate a true limitation of the activity and coupling of the electron transport chain in KO mice, but an increased activity in TG mice.
Therefore, Hsp22 expression regulates the translocation, phosphorylation pattern and physiological function of STAT3, both in nuclei and in mitochondria.
Hsp22 promotes the activation of STAT3 by NF-κB
We investigated more mechanistically in isolated neonatal rat cardiac myocytes how Hsp22 expression modulates STAT3 function, using both adeno-mediated over-expression and silencing. Over-expression of Hsp22 in myocytes was sufficient to increase Y705 P-STAT3 phosphorylation in a dose-dependent manner, and overall STAT DNA binding activity, and to reduce STAT1 phosphorylation (Figure 7a), in agreement with our observations in vivo. Immunofluorescence of cardiac myocytes infected with the Hsp22 adenovirus also showed a nuclear accumulation of Y705 P-STAT3 (Figure 7b). This observation does not result from an artifact due to Hsp22 over-expression, since it was also found upon incubation of cardiac myocytes with the growth factor IGF-1 (Supplementary Figure S3).
Figure 7. Hsp22-mediated activation of STAT3 by NF-κB.
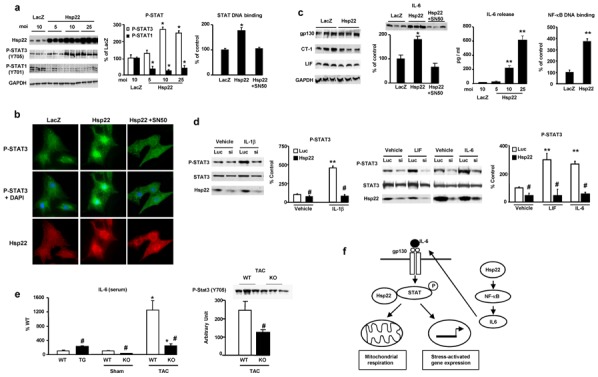
A. Impact of Hsp22 over-expression (5 to 25 moi) on tyrosine phosphorylation of STAT1 and STAT3 in isolated cardiac myocytes, and on STAT DNA binding activity in presence or in absence of SN50 (n=3 per group). *, P<0.05 versus LacZ control (two-way ANOVA with 2 factors). B. Immunofluorescence for Y705 P-STAT3 (with or without superimposition of nuclear DAPI) and for Hsp22 in cardiac myocytes infected with the Hsp22 adenovirus in presence or in absence of SN50, and compared to a LacZ control. C. Expression of gp130, CT-1, LIF and IL-6 in response to Hsp22 over-expression, response of IL-6 to Hsp22 over-expression in presence of SN50, IL-6 release (as measured by ELISA) in the extracellular medium of isolated neonatal rat cardiac myocytes (n=3 per group) in response to different moi of Hsp22 adenovirus, and DNA binding activity of NF-κB in response to Hsp22 over-expression (20 moi) as compared to LacZ control (n= 3 per group). **, P<0.01; *, P<0.05 versus LacZ control (one-way ANOVA with four factor levels). D. Impact of Hsp22 silencing on STAT3 phosphorylation in response to IL-1β, LIF and IL-6 stimulation in isolated cardiac myocytes infected with a hairpin harboring a sequence against luciferase (Luc) as a control or against Hsp22 (si). n=4 per group. **, P<0.01 versus corresponding vehicle; #, P<0.05 versus corresponding Luc control (two-way ANOVA). E. Measurement of plasma IL-6 concentration in TG and KO mice versus corresponding WT, as well as in KO versus WT three days after TAC, and STAT3 phosphorylation (Y705) in KO versus WT after TAC. *, P<0.01 versus corresponding sham; #, P<0.01 versus corresponding WT (two-way ANOVA with two factors). F. Summary of the findings.
Activation of STAT is promoted upon binding of the gp130 receptor by specific ligands, including interleukin-6 (IL-6), leukemia inhibitory factor (LIF) and cardiotrophin-1 (CT-1)[5]. Expression of gp130, LIF and CT-1 was not affected by Hsp22 but IL-6 abundance was significantly increased (Figure 7c). IL-6 is a cytokine, and its release in the culture medium, as measured by ELISA, dose-dependently increased in response to Hsp22 (Figure 7c). A known activator of IL-6 production in the heart is the transcription factor NF-κB[21]. Over-expression of Hsp22 significantly increased NF-κB DNA binding activity (Figure 7c). Addition of the NF-κB inhibitor SN50 to cardiac myocytes blocked Hsp22-mediated increase in STAT3 DNA binding (Figure 7a) and nuclear translocation (Figure 7b), and in IL-6 abundance (Figure 7c).
Using RNA silencing, we tested whether Hsp22 is required for NF-κB-mediated activation of STAT3. Hsp22 knockdown was performed using a short-hairpin siRNA, and a non-relevant short hairpin against luciferase was used for control experiments, as before[18]. In both groups, we measured STAT3 phosphorylation in basal conditions, upon stimulation of NF-κB, or upon activation of the gp130 receptor. In basal conditions, Hsp22 silencing already decreased STAT3 phosphorylation compared to the control (Figure 7d). Upon treatment of myocytes with interleukin-1β (IL-1β), a known activator of NF-κB, a significant increase in STAT3 phosphorylation was observed in the control group, and this was prevented by Hsp22 silencing (Figure 7d). Similarly, addition of the gp130 ligands IL-6 and LIF markedly increased STAT3 phosphorylation in control conditions, but not when Hsp22 was silenced (Figure 7d). This latter result implies that Hsp22-STAT3 interaction is necessary for the proper association of STAT3 with its receptor, and hence its phosphorylation.
These results were validated in vivo. Plasma concentration of IL-6, as measured by ELISA, was significantly increased in TG mice compared to WT, and decreased in KO compared to WT (Figure 7e). After TAC, IL-6 concentration increased markedly in WT mice but to a much lesser degree in KO (Figure 7e). Similarly, after TAC, Y705 STAT3 phosphorylation was reduced by about 50% in KO compared to WT (Figure 7e).
DISCUSSION
We demonstrate that Hsp22 deletion in a context of pressure overload in vivo promotes a pattern of ventricular eccentric hypertrophy and dilation, accelerates cardiac remodeling and the transition into heart failure, increases mortality, interferes with the activation of cell survival pathways, including Akt, ERK and iNOS, and prevents the activation STAT3-dependent stress-responsive genes. We show that Hsp22 deletion not only down-regulates the activity of STAT3 as a transcription factor, but it also impairs mitochondrial STAT3 abundance and oxidative phosphorylation. We further demonstrate in vitro that Hsp22 is both sufficient and necessary for proper activation of STAT3 by another stress-activated transcription factor, NF-κB (Figure 7f).
Hsp22 clearly represents a stress-responsive mechanism because the phenotype of the KO mouse is comparable to WT mice under basal conditions. We found that Hsp22 expression increases rapidly during ventricular hypertrophy due to overload[2]. Reproducing such increase in an Hsp22 TG mouse creates a pattern of cardiac hypertrophy with normal function[2,3]. We therefore hypothesized that Hsp22 deletion would impair the development of hypertrophy during overload. The results presented here show that, although cardiac hypertrophy is not prevented, it is significantly decreased. The cardiac phenotype of the KO mouse quickly evolves into a pattern of eccentric hypertrophy, which significantly increased cardiac wall stress, yet it did not result in greater ventricular mass.
The primary objective of the present study was to characterize the phenotype of the Hsp22 KO mouse under pressure overload. Based on the microarray data, we focused on STAT3 for the following reasons. First, the STAT pathway is typically a stress-responsive pathway, playing a modest role in normal conditions but becoming predominant under stress conditions[5], a characteristic that is shared with Hsp22 as shown here. Second, over-expression of STAT3 in the heart promotes the development of concentric, adaptive hypertrophy[5], which is comparable to that reported in the Hsp22 TG mouse[2,3]. Third, STAT3 is central to the cardioprotection by the second window, or delayed phase, of ischemic preconditioning (SWOP)[22]. Again, over-expression of Hsp22 provides cardioprotection that is quantitatively equivalent to that of SWOP[8]. Fourth, the major mediator of SWOP is iNOS[23], which is induced by NF-κB and STAT3 in response to cardiac stress[24]. We show here that Hsp22 is sufficient to activate both transcription factors and that iNOS expression is virtually abolished upon stress in the Hsp22 KO mouse. Fifth, the observed phenotype of eccentric hypertrophy is strikingly similar to that reported upon pressure overload in a gp130 KO model that prevents STAT activation[25]. Similarities between Hsp22 and STAT3 exist at the mitochondrial level as well. We show that Hsp22 localizes inside the mitochondria, where it co-precipitates with STAT3. A translocation of STAT3 to the mitochondria has been recently described, where it stimulates respiration by activation of complexes I and II[6]. Interestingly, a mitochondrial localization of Hsp22 has been reported in Drosophila, where it is associated with increased respiration, resistance against oxidative stress, and prolonged lifespan[26,27]. Our models of Hsp22 TG and KO might therefore offer a useful tool to further investigate in vivo the specific roles and mechanisms of regulation of mitochondrial STAT3.
In addition to changes in STAT3, the response of the Hsp22 KO mouse to pressure overload also includes an impaired activation of several signaling mechanisms involved in cardiac cell survival and growth, including Akt. We have shown that Hsp22 is an activator of Akt through stimulation of PI3K, which is itself activated by the BMP receptor[18]. Hsp22-mediated activation of Akt can be responsible for the subsequent stimulation of NF-κB[28]. However, Hsp22 also activates p38 MAP kinase, another well known activator of NF-κB[28]. Interestingly, p38 MAP kinase also mediates the S727 phosphorylation of STAT3 responsible for its mitochondrial translocation[6,29]. We also show here a deactivation of ERK1/2 at the acute phase of TAC in the KO, which could also be due to the absence of STAT3 activation because JAK, the upstream activator of STAT3, is an ERK activator[5]. Together with p38 MAP kinase, ERK can also phosphorylate S727 STAT3[30].
Although the role of STAT3 in the cardiac stress response is still emerging, its role in the mechanisms of growth and proliferation of cancer is well described. Deregulation of Hsp22 expression has been described in cancer as well[31]. Interestingly, a recent report showed that mitochondrial STAT3 is responsible for Ras-mediated cellular transformation[32]. As mentioned above, mitochondrial Hsp22 increases lifespan in Drosophila. It is therefore possible that a mitochondrial interaction between STAT3 and Hsp22 also participates in tumor growth.
In conclusion, Hsp22 represents a previously undescribed activator of both nuclear and mitochondrial functions of STAT3, and its deletion accelerates the transition into heart failure. Altogether, our observations provide a novel mechanism responsible for activation of stress-inducible signaling pathways in the cardiac response to overload. Pre-emptive activation of STAT3 by Hsp22 can be of therapeutic potential to activate critical survival pathways in patients with chronic cardiac overload who are at risk of subsequent myocardial damage and heart failure.
Supplementary Material
Acknowledgments
SOURCE OF FUNDING
Study supported by NIH grants HL 072863, HL 093415, HL033107, A6027211, HL069020 and AHA grant 0230017N.
Footnotes
DISCLOSURES
None
References
- 1.Danan IJ, Rashed ER, Depre C. Therapeutic potential of H11 Kinase for the ischemic heart. Cardiovasc Drug Revs. 2007;25:14–29. doi: 10.1111/j.1527-3466.2007.00002.x. [DOI] [PubMed] [Google Scholar]
- 2.Depre C, Hase M, Gaussin V, Zajac A, Wang L, Hittinger L, Ghaleh B, Yu X, Kudej RK, Wagner T, Sadoshima J, Vatner SF. H11 Kinase is a novel mediator of myocardial hypertrophy in vivo. Circ Res. 2002;91:1007–1014. doi: 10.1161/01.res.0000044380.54893.4b. [DOI] [PubMed] [Google Scholar]
- 3.Hedhli N, Wang L, Wang Q, Rashed E, Tian Y, Sui X, Madura K, Depre C. Proteasome activation during cardiac hypertrophy by the chaperone H11 Kinase/Hsp22. Cardiovasc Res. 2008;77:497–505. doi: 10.1093/cvr/cvm054. [DOI] [PubMed] [Google Scholar]
- 4.Willis MS, Patterson C. Hold me tight: Role of the heat shock protein family of chaperones in cardiac disease. Circulation. 2010;122:1740–1751. doi: 10.1161/CIRCULATIONAHA.110.942250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol Ther. 2008;120:172–185. doi: 10.1016/j.pharmthera.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, Baker DP, Wolfman A, Stuehr D, Hassan MO, Fu XY, Avadhani N, Drake JI, Fawcett P, Lesnefsky EJ, Larner AC. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Depre C, Wang Q, Yan L, Hedhli N, Peter P, Chen L, Hong C, Hittinger L, Ghaleh B, Sadoshima J, Vatner DE, Vatner SF, Madura K. Activation of the cardiac proteasome during pressure overload promotes ventricular hypertrophy. Circulation. 2006;114:1821–1828. doi: 10.1161/CIRCULATIONAHA.106.637827. [DOI] [PubMed] [Google Scholar]
- 8.Depre C, Wang L, Sui X, Qiu H, Hong C, Hedhli N, Ginion A, Shah A, Pelat M, Bertrand L, Wagner T, Gaussin V, Vatner SF. H11 Kinase prevents myocardial infarction by pre-emptive preconditioning of the heart. Circ Res. 2006;98:280–288. doi: 10.1161/01.RES.0000201284.45482.e8. [DOI] [PubMed] [Google Scholar]
- 9.Hedhli N, Lizano P, Hong C, Fritzky L, Dhar S, Liu H, Tian Y, Gao S, Madura K, Vatner SF, Depre C. Proteasome inhibition decreases cardiac remodeling after initiation of pressure overload. Am J Physiol Heart Circ Physiol. 2008;295:H1385–H1393. doi: 10.1152/ajpheart.00532.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Depre C, Kim SJ, John AS, Huang Y, Rimoldi OE, Pepper JR, Dreyfus GD, Gaussin V, Pennell DJ, Vatner DE, Camici PG, Vatner SF. Program of cell survival underlying human and experimental hibernating myocardium. Circ Res. 2004;95:433–440. doi: 10.1161/01.RES.0000138301.42713.18. [DOI] [PubMed] [Google Scholar]
- 11.Qiu H, Dai H, Jain K, Shah R, Hong C, Pain J, Tian B, Vatner D, Vatner S, Depre C. Characterization of a novel cardiac isoform of the cell cycle-related kinase that is regulated during heart failure. J Biol Chem. 2008;283:22157–22165. doi: 10.1074/jbc.M710459200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiu H, Tian B, Resuello R, Natividad F, Peppas A, Shen Y, Vatner D, Vatner S, Depre C. Sex-specific regulation of gene expression in the aging monkey aorta. Physiol Genomics. 2007;29:169–180. doi: 10.1152/physiolgenomics.00229.2006. [DOI] [PubMed] [Google Scholar]
- 13.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Nat Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dauer D, Ferraro B, Song B, Yu B, Mora L, Buettner R, Enkemann S, Jove R, Haura E. Stat3 regulates genes common to both wound healing and cancer. Oncogene. 2005;24:3397–3408. doi: 10.1038/sj.onc.1208469. [DOI] [PubMed] [Google Scholar]
- 15.Depre C, Shipley G, Chen W, Han Q, Doenst T, Moore M, Stepkowski S, Davies P, Taegtmeyer H. Unloaded heart in vivo replicates fetal gene expression of cardiac hypertrophy. Nature Medicine. 1998;4:1269–1275. doi: 10.1038/3253. [DOI] [PubMed] [Google Scholar]
- 16.Depre C, Wang L, Tomlinson J, Gaussin V, Abdellatif M, Topper J, Vatner S. Characterization of pDJA1, a cardiac-specific chaperone found by genomic profiling of the post-ischemic swine heart. Cardiovasc Res. 2003;58:126–135. doi: 10.1016/s0008-6363(02)00845-3. [DOI] [PubMed] [Google Scholar]
- 17.Hase M, Depre C, Vatner S, Sadoshima J. H11 has dose-dependent and dual hypertrophic and proapoptotic functions in cardiac myocytes. Biochem J. 2005;388:475–483. doi: 10.1042/BJ20041314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sui X, Li D, Qiu H, Gaussin V, Depre C. Activation of the bone morphogenetic protein receptor by H11Kinase/Hsp22 promotes cardiac cell growth and survival. Circ Res. 2009;104:887–895. doi: 10.1161/CIRCRESAHA.108.192328. [DOI] [PubMed] [Google Scholar]
- 19.Xie L-H, Chen F, Karagueuzian HS, Weiss JN. Oxidative stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ Res. 2009;104:79–86. doi: 10.1161/CIRCRESAHA.108.183475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stephanou A, Brar BK, Knight RA, Latchman DS. Opposing actions of STAT-1 and STAT-3 on the Bcl-2 and Bcl-x promoters. Cell Death Differ. 2000;7:329–330. doi: 10.1038/sj.cdd.4400656. [DOI] [PubMed] [Google Scholar]
- 21.Craig R, Larkin A, Mingo AM, Thuerauf DJ, Andrews C, McDonough PM, Glembotski CC. p38 MAPK and NF-kappa B collaborate to induce interleukin-6 gene expression and release. Evidence for a cytoprotective autocrine signaling pathway in a cardiac myocyte model system. J Biol Chem. 2000;275:23814–23824. doi: 10.1074/jbc.M909695199. [DOI] [PubMed] [Google Scholar]
- 22.Xuan YT, Guo Y, Han H, Zhu Y, Bolli R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci U S A. 2001;98:9050–9055. doi: 10.1073/pnas.161283798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo Y, Jones WK, Xuan Y-T, Tang X-L, Bao W, Wu W-J, Han H, Laubach VE, Ping P, Yang Z, Qiu Y, Bolli R. The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene. Proc Nat Acad Sci USA. 1999;96:11507–11512. doi: 10.1073/pnas.96.20.11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dawn B, Xuan YT, Guo Y, Rezazadeh A, Stein AB, Hunt G, Wu WJ, Tan W, Bolli R. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc Res. 2004;64:61–71. doi: 10.1016/j.cardiores.2004.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirota H, Chen J, Betz U, Rajewski K, Gu Y, Ross J, Muller W, Chien K. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:89–198. doi: 10.1016/s0092-8674(00)80729-1. [DOI] [PubMed] [Google Scholar]
- 26.Kim HJ, Morrow G, Westwood JT, Michaud S, Tanguay RM. Gene expression profiling implicates OXPHOS complexes in lifespan extension of flies over-expressing a small mitochondrial chaperone, Hsp22. Exp Gerontol. 2010;45:611–620. doi: 10.1016/j.exger.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 27.Morrow G, Battistini S, Zhang P, Tanguay RM. Decreased lifespan in the absence of expression of the mitochondrial small heat shock protein Hsp22 in Drosophila. J Biol Chem. 2004;279:43382–43385. doi: 10.1074/jbc.C400357200. [DOI] [PubMed] [Google Scholar]
- 28.Hall G, Hasday J, Rogers T. Regulating the regulator: NF-kappaB signaling in heart. J Mol Cell Cardiol. 2006;41:580–591. doi: 10.1016/j.yjmcc.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 29.Gollob JA, Schnipper CP, Murphy EA, Ritz J, Frank DA. The functional synergy between IL-12 and IL-2 involves p38 mitogen-activated protein kinase and is associated with the augmentation of STAT serine phosphorylation. J Immunol. 1999;162:4472–4481. [PubMed] [Google Scholar]
- 30.Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–2637. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- 31.Smith C, Yu Y, Kulka M, Aurelian L. A novel human gene similar to the protein kinase (PK) coding domain of the large subunit of Herpes Simplex virus type 2 ribonucleotide reductase (ICP10) codes for a serine-threonine PK and is expressed in melanoma cells. J Biol Chem. 2000;275:25690–25699. doi: 10.1074/jbc.M002140200. [DOI] [PubMed] [Google Scholar]
- 32.Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.