

Abstract
Owing to their unique functions in regulating glucose, lipid and cholesterol metabolism, PPARs (peroxisome proliferator-activated receptors) have drawn special attention for developing drugs to treat type-2 diabetes. By combining the lipid benefit of PPAR-alpha agonists (such as fibrates) with the glycemic advantages of the PPAR-gamma agonists (such as thiazolidinediones), the dual PPAR agonists approach can both improve the metabolic effects and minimize the side effects caused by either agent alone, and hence has become a promising strategy for designing effective drugs against type-2 diabetes. In this study, by means of the powerful “core hopping” and “glide docking” techniques, a novel class of PPAR dual agonists was discovered based on the compound GW409544, a well-known dual agonist for both PPAR-alpha and PPAR-gamma modified from the farglitazar structure. It was observed by molecular dynamics simulations that these novel agonists not only possessed the same function as GW409544 did in activating PPAR-alpha and PPAR-gamma, but also had more favorable conformation for binding to the two receptors. It was further validated by the outcomes of their ADME (absorption, distribution, metabolism, and excretion) predictions that the new agonists hold high potential to become drug candidates. Or at the very least, the findings reported here may stimulate new strategy or provide useful insights for discovering more effective dual agonists for treating type-2 diabetes. Since the “core hopping” technique allows for rapidly screening novel cores to help overcome unwanted properties by generating new lead compounds with improved core properties, it has not escaped our notice that the current strategy along with the corresponding computational procedures can also be utilized to find novel and more effective drugs for treating other illnesses.
Introduction
Diabetes mellitus is a group of metabolic diseases that has been classified as a disease of glucose overproduction by tissues without enough insulin production, or a disease resulting from cells not responding to the insulin in human body [1]. Type-2 diabetes is the most common type among all the diabetes mellitus forms. The risk of developing type-2 diabetes (T2D) increases with age, obesity, cardiovascular disease (hypertension, dyslipidaemia), lack of physical activity, and family history of diabetes. Increasing dramatically in the US and worldwide, type-2 diabetes has reached epidemic scale. There are nearly 50 million individuals (US) and 314 million individuals (worldwide) with the metabolic syndrome [2]. People suffering from overweight or obesity are of huge risk for developing T2D.
Peroxisome Proliferator-Activated Receptor (PPAR) has drawn increased attention as a drug discovery target by regulating glucose and lipid metabolism [3]. PPAR, and its subtypes PPARα and PPARγ, belong to the superfamily of nuclear receptors that function as transcription factors activated by several ligands. PPARs played a vitally important role in treating obesity, atherogenic dyslipidemia, hypertension, and insulin resistance as main therapeutic targets [4]. The primary function of PPARα is to act as regulator responding to transport and degradation of free fatty acids as well as reverse cholesterol transport by peroxisomal and beta-oxidation pathways [5]. A class of lipid-lowering drugs, such as fenofibrate and gemfibrozil, specially activate PPARα [6], [7], [8]. PPARγ played a significant role in transcriptionally regulating lots of physiological pathways, including adipocyte differentiation and glucose homeostasis [9]. Thiazolidinediones (TZDs) are a class of the antidiabetic drugs, which act by activating the special PPARγ [10]. If used alone, although each of the antidiabetic drugs could enhance the insulin sensitivity and hence lower glucose or fatty acid levels in type-2 diabetic patients [9], some side effects would be caused, such as weight gain, fluid accumulation, and pulmonary edema [11].
Recently, new dual agonists have received considerable attention for developing powerful drugs against diabetes. The strategy of dual agonists was aimed to treat both insulin resistance and dyslipidemia [12]. A critical challenge for developing dual agonists is how to identify the receptor subtype selectivity ratio [13].
Many studies have indicated that computational approaches, such as structural bioinformatics [14], [15], molecular docking [16], [17], pharmacophore modelling [18], QSAR techniques [19], [20], [21], [22], [23], [24], as well as a series of user-friendly web-server predictors developed recently, such as GPCR-2L [25] for identifying G protein-coupled receptors and their types, EnzClassPred [26] for predicting enzyme class, iLoc-Euk [27] and iLoc-Hum [28] for predicting subcellular localization of eukaryotic and human proteins, NR-2L [29] and iNR-PhysChem [30] for identifying nuclear receptors and their subfamilies, and HIVcleave [31] for predicting HIV protease cleavage sites in proteins [32], [33], can timely provide very useful information and insights for drug development. The software of “Core Hopping” [34] is another very powerful and cutting-edge computational technique that is particularly useful for de novel drug design [35].
Encouraged by the aforementioned researches in successfully utilizing various computational approaches for drug development, the present study was initiated in an attempt to screen the fragment database for finding new PPAR dual agonists for treating type-2 diabetes. To realize this, the techniques of “core hopping” with glide docking [34], [36] as well as molecular dynamic simulation were utilized to analyze the binding interactions between the agonist and PPARs in hoping that the findings thus obtained may provide useful insights for developing new and powerful agonists against diabetes mellitus.
Materials and Methods
The L-tyrosine analogue GW409544 was obtained by modifying the structure of farglitazar, a dual agonist for both PPARα and PPARγ [37]. The main difference between GW409544 and farglitazar is that the former contains a vinylogous amide as the L-tyrosine N-substituent [37]. That is why we chose GW409544 as a starting template structure for designing the new PPAR dual agonists.
The representative complex crystal structures of PPARα (PDB ID 1k7l) and PPARγ (PDB ID 1k74) with the same ligand GW409544 [37] were download from the PDB Bank [38], and were to be used for the molecular modelling studies.
All the calculations were carried out on Dell Precision™ T5500 computer with Schrodinger software package [34], [36] and Desmond 2.4 [39].
1. Preparation of Receptor Structures and Databases
The proteins with PDB codes 1k71 and 1k74 were chosen for modeling. In addition to the available knowledge of their 3D (dimensional) structures, the reasons of selecting the two proteins as receptors are as follows. (1) The two proteins contain the same ligand GW409544 as PPARα and PPARγ do, and their binding affinities with the ligand are also quite similar; however, the former selectivity is about 10-fold weaker than the latter [37]. (2) The source organism of both PPARα and PPARγ was from human.
In the process of preparing receptors for modelling, the protein preparation facility [40] was used that included the operations of assigning bond orders, adding hydrogen, treating metals, treating disulfides, deleting waters and alleviating potential steric clashes, adjusting bond order, building missing heavy atom and formal charges, as well as minimizing energy with the OPLS2005 force field [41] and refining the protein by imposing the 0.3 Å RMSD limit as the constraint.
The protein binding-site was identified by the SiteMap tool embedded in Schrodinger Suite 2009 (www.schrodinger.com) as described in [42], [43], [44]. The binding-site encompassed the ligand GW409544, which was observed in the crystal structures of both PPARα (1k71) and PPARγ (1k74) as a ligand.
The information of the binding pocket of a receptor for its ligand is very important for drug design, particularly for conducting mutagenesis studies [14]. In the literatures, the binding pocket of a protein receptor to a ligand is usually defined by those residues that have at least one heavy atom (i.e., an atom other than hydrogen) with a distance ≤5 Å from a heavy atom of the ligand. Such a criterion was originally used to define the binding pocket of ATP in the Cdk5-Nck5a* complex [45] that has later proved quite useful in identifying functional domains and stimulating the relevant truncation experiments [46]. The similar approach has also been used to define the binding pockets of many other receptor-ligand interactions important for drug design [15], [17], [47], [48]. In this study, we also used the same criterion [45] to define the binding pockets of proteins 1k7l and 1k74 for the ligand GW409544. A close-up view for the protein-ligand interaction at the binding pocket thus defined is shown in Fig. 1 , where panel A is for the interaction between PPARα (1k71) and GW409544, while panel B for the interaction between PPARγ (1k74) and GW409544.
Figure 1. Illustration to show the conformation obtained by docking GW409544 and Comp#1, respectively, to (A) PPARα (1k7l) and (B) PPARγ (1k74).

The binding pocket is defined by those residues that have at least one heavy atom with a distance of 5 Å from the ligand [45]. The ligand GW409544 (in grey color) was extracted from the crystal structure while the ligand Comp#1 (rendered by three colors: grey for Core A; red for Core B; and blue for Core C) was generated by the “core-hopping” method. The hydrophobic surface of the receptor is colored in green. The blue dotted lines indicate the H-bond interactions of the receptor with its ligand. The red helix is a part of AF-2 function domain. See the text for further explanation.
Because the natural ligands of PPARs are fatty acids, the binding site of PPARs is almost hydrophobic. Several hydrophobic interactions with three arms of the Y-shaped ligand binding to the site are taken as the key point for designing the new PPARs agonist [49]. The PPAR binding site is composed of three arms, namely Arm I, Arm II, and Arm III, as explicitly marked in Fig. 1 . The first arm has mainly polar character including the AF2 (transcriptional activation function 2) helix indicated by red ribbon. The hydrophilic head group of the PPAR ligands forms a network of hydrogen bonds with AF2 of Arm-I; while the hydrophobic tail of PPAR agonist is either interacts with Arm II or Arm III. The network hydrogen bonds forms an important conformation for AF2-helix to generate a charge clamp, thus reducing the mobility of AF2 via binding a ligand and hence make it able to regulate the gene expression [4]. The drug-like database and the fragment database derived from ZINC [50] were used for virtual screening and core hopping searching, respectively.
2. Molecular Docking with Core Hopping Method
Many useful insights for drug design could be gained via molecular docking studies (see, e.g., [14], [17], [51], [52]). To acquire even more useful information for drug design, a new docking algorithm called “Core Hopping” [36] was adopted in this study that is featured by having the functions to perform both the fragment-based replacing and molecular docking.
Core Hopping [36] is a very powerful and cutting-edge technique for de novel drug design because it can significantly improve the binding affinity of the receptor with its ligands, e.g., GW409544 ( Fig. 2 ) in the current study. The binding conformation thus obtained will be taken as an initial structure for further optimization by searching the fragment database to find the optimal cores that are attached to other parts of the template.
Figure 2. Illustration to show how to generate the 25 derivative compounds from GW409544.

(A) Structure of GW409544 and its three Cores: Core A (black), Core B (red), and Core C (blue). (B) Five compounds derived from GW409544 by changing Core C to C1, C2, C3, C4 and C5 respectively but fixing Core A and Core B at Core B1. Panels (C), (D), (E), and (F) each show another five compounds generated by following the similar procedure but fixing Core B at B2, B3, B4, and B5, respectively. See the text for further explanation.
During the process of core hopping, the 1st step is to define the possible points at which the cores are attached. It is performed in the “Define Combinations” step from the Combinatorial Screening panel in Schrodinger2009 (www.schrodinger.com). The 2nd step is to define the “receptor grid file”, which was done in the “Receptor Preparation” panel. The 3rd step is the cores preparation that was operated with the “Protocore Preparation” module to find the cores attaching to the scaffold using the fragment database derived from ZINC [50]. The 4th step is to align and dock the entire molecular structure built up by the core and scaffold. The cores are sorted and filtered by goodness of alignment and then redocked into the receptor after attaching the scaffold, followed by using the docking scores to sort the final molecules.
As the products of the core hopping operation, a total of 500 chemical compounds were prepared with the LigPre module [53], which consists of the procedures of generating possible states by ionization at target pH 7.0±2.0, desalting, retaining chiralities from 3D structure and geometry minimization with the OPLS2005 force field [41]. When the above steps were accomplished, all investigated compounds were docked into the receptor pocket through the rigid protein docking model with the Stand-Precision (SP) scoring function [54], [55] to estimate the binding affinities.
3. Molecular Dynamics
Many marvelous biological functions in proteins and DNA as well as their profound dynamic mechanisms, such as switch between active and inactive states [56], [57], cooperative effects [58], allosteric transition [59], [60], intercalation of drugs into DNA [61], and assembly of microtubules [62], can be revealed by studying their internal motions [63]. Likewise, to really understand the interaction mechanism of a receptor with its ligand, investigations should be aimed not only at their static structures but also at the dynamic process obtained by simulating their internal motions.
Here, the “Desmond 2.4 Package” [39] was adopted to study the internal motions of the receptor-ligand system. According to the software, the OPLS 2005 force fields [64], [65] was used to build aqueous biological systems, and the TIP3P model [66] was used to simulate water molecules. The orthorhombic periodic boundary conditions were set up to specify the shape and size of the repeating unit. In order to get an electrically neutral system, the minimum number of sodium and chloride ions needed to balance the system charge was placed randomly in the solvated system, and 0.15 mol/L sodium and chloride were then added to mimic the osmotic effect of water. Molecular dynamics simulations were carried out with the periodic boundary conditions in the NPT ensemble. The temperature and pressure were kept at 300 K and 1 atmospheric pressure using Nose-Hoover temperature coupling and isotropic scaling [67]. After all restrains were removed via the 3ns (nanosecond or 10−9 of a second) system minimization and relaxation, the operation was followed by running the 10 ns NPT production simulation and saving the configurations thus obtained in 2ps (picosecond or 10−12 of a second) intervals. All the molecular dynamics simulations were performed with a time step of 2fs (femtosecond or 10−15 of a second).
4. ADME Prediction
The QikProp [68], [69] is a program for predicting the ADME (absorption, distribution, metabolism, and excretion) properties of the compounds. With the QikProp software, a total of 44 properties of compounds can be predicted, including the principal descriptors and physiochemical properties.
All the compounds investigated need not the treatment for neutralization before using QikProp because it will be automatically done in QikProp. The normal mode was applied in the program. The property analyses for the partition coefficient (QP logP o/w), van der Waals surface area of polar nitrogen and oxygen atoms (PSA), predicted aqueous solubility (QP logSb ), and apparent MDCK permeability (QPP MDCKc) [70], were considered in the QikPro to evaluate the acceptability of the compounds.
Results and Discussion
1. Design of PPAR Dual Agonist and Modeling of PPAR Agonist Complex
The process of core hopping and the final agonists’ structures thus selected are illustrated in Fig. 2 , from which we can see the following. The structure of GW409544, which is conceived as an agonist model to develop novel therapeutic agents for treating metabolic disorder, may be divided into three parts, Core A, Core B, and Core C, as marked by dash lines. Considering the great importance of the acidic head in Core A for activating PPARs receptors, let us retain the Core A part during the core hopping calculation as described below.
The 1st core hopping operation was aimed at the Core C part (see red part of Fig. 2 ), generating five cores, Core C1, Core C2, Core C3, Core C4, and Core C5, respectively, to replace Core C. The 2nd core hopping operation was aimed at the Core B part (see blue part of Fig. 2 ), also respectively generating five cores, Core B1, Core B2, Core B3, Core B4, and Core B5 to replace Core B.
Consequently, we have a total of 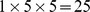 different combinations for the GW409544 derivatives thus generated. Subsequently, each of the 25 derivative compounds was docked into the two receptors PPARα (1k71) and PPARγ (1k74), respectively. Listed in
Table 1
are the 25 derivative compounds ranked roughly according to their docking scores to the receptors PPARα and PPARγ, respectively. The top ten compounds highlighted with boldface type in
Table 1
are those derivatives that are stronger than the original GW409544 in binding affinity with the two receptors. Of the top ten derivatives, the Comp#1, i.e., “Core A-Core B1-Core C1”, has the strongest binding affinity with both PPARα (1k71) and PPARγ (1k74), and hence it was singled out for further investigation.
different combinations for the GW409544 derivatives thus generated. Subsequently, each of the 25 derivative compounds was docked into the two receptors PPARα (1k71) and PPARγ (1k74), respectively. Listed in
Table 1
are the 25 derivative compounds ranked roughly according to their docking scores to the receptors PPARα and PPARγ, respectively. The top ten compounds highlighted with boldface type in
Table 1
are those derivatives that are stronger than the original GW409544 in binding affinity with the two receptors. Of the top ten derivatives, the Comp#1, i.e., “Core A-Core B1-Core C1”, has the strongest binding affinity with both PPARα (1k71) and PPARγ (1k74), and hence it was singled out for further investigation.
Table 1. The 25 compounds ranked roughly according to the strength of their docking scores.
| Rank | Compound | Combination of coresa | Docking scores (Kcal/mol) | ADME properties predicted | |||||
| PPARα (1k71) | PPARγ (1k74) | PSAb | logPo/wc | logSd | PMDCKe | ||||
| GW409544 | A-B-C | −14.41 | −15.16 | 102.39 | 5.78 | −6.80 | 28.08 | ||
| 1 | Comp#1 | A-B1-C1 | −16.10 | −16.19 | 159.92 | 5.85 | −6.53 | 109.76 | |
| 2 | Comp#24 | A-B5-C4 | −15.87 | −16.18 | 168.65 | 4.84 | −6.50 | 98.65 | |
| 3 | Comp#3 | A-B1-C3 | −14.89 | −16.08 | 160.40 | 4.75 | −6.59 | 108.32 | |
| 4 | Comp#14 | A-B3-C4 | −15.10 | −15.98 | 190.08 | 3.55 | −6.28 | 101.99 | |
| 5 | Comp#2 | A-B1-C2 | −14.97 | −15.92 | 195.19 | 4.10 | −6.81 | 101.82 | |
| 6 | Comp#13 | A-B3-C3 | −14.68 | −15.86 | 164.26 | 4.34 | −6.16 | 110.42 | |
| 7 | Comp#16 | A-B4-C1 | −14.26 | −15.71 | 166.47 | 5.03 | −6.92 | 106.95 | |
| 8 | Comp#17 | A-B4-C2 | −14.44 | −15.32 | 193.75 | 4.43 | −6.43 | 103.80 | |
| 9 | Comp#15 | A-B3-C5 | −14.42 | −15.29 | 190.02 | 3.52 | −6.57 | 102.48 | |
| 10 | Comp#12 | A-B3-C2 | −15.15 | −15.23 | 168.27 | 4.28 | −6.81 | 105.98 | |
| 11 | Comp#19 | A-B4-C4 | −14.21 | −15.25 | 180.89 | 4.64 | −6.70 | 109.39 | |
| 12 | Comp#4 | A-B1-C4 | −14.98 | −15.15 | 185.89 | 4.23 | −7.55 | 102.25 | |
| 13 | Comp#11 | A-B3-C1 | −16.15 | −15.11 | 168.04 | 5.12 | −7.74 | 108.03 | |
| 14 | Comp#25 | A-B5-C5 | −15.24 | −15.09 | 204.93 | 3.46 | −6.59 | 101.16 | |
| 15 | Comp#23 | A-B5-C3 | −15.58 | −15.06 | 176.72 | 4.89 | −7.70 | 104.35 | |
| 16 | Comp#5 | A-B1-C5 | −14.77 | −15.00 | 197.47 | 4.05 | −7.30 | 101.13 | |
| 17 | Comp#21 | A-B5-C1 | −14.78 | −14.98 | 152.24 | 6.45 | −9.10 | 106.45 | |
| 18 | Comp#18 | A-B4-C3 | −13.72 | −14.91 | 186.33 | 4.43 | −7.08 | 104.19 | |
| 19 | Comp#20 | A-B4-C5 | −14.42 | −14.75 | 197.70 | 4.26 | −7.20 | 101.65 | |
| 20 | Comp#9 | A-B2-C4 | −14.03 | −14.35 | 166.94 | 5.15 | −6.52 | 114.53 | |
| 21 | Comp#22 | A-B5-C2 | −15.10 | −14.26 | 150.45 | 5.55 | −8.30 | 107.48 | |
| 22 | Comp#7 | A-B2-C2 | −14.16 | −13.62 | 142.49 | 5.92 | −7.32 | 109.50 | |
| 23 | Comp#6 | A-B2-C1 | −12.00 | −9.14 | 142.06 | 6.94 | −9.40 | 120.29 | |
| 24 | Comp#10 | A-B2-C5 | −11.10 | −8.00 | 174.53 | 4.59 | −7.20 | 104.59 | |
| 25 | Comp#8 | A-B2-C3 | −13.07 | −7.64 | 156.10 | 5.18 | −6.25 | 124.85 | |
See Fig. 2 for the structure of cores.
The van der Waals surface area of polar nitrogen and oxygen atoms (7.0 to 200.0).
The predicted octanol/water partition coefficient (−2.0 to 6.5).
The predicted aqueous solubility, where S (mol dm–3) is the concentration of the solute in a saturated solution that is in equilibrium with the crystalline solid (−6.5 to 0.5).
The apparent MDCK permeability (nm/s); MDCK cells are considered to be a good mimic for the bloodbrain barrier. QikProp predictions are for non-active transport (<25 poor; >500 great).
Shown in Fig. 1 is the docked conformation of Comp#1 when aligned with GW409544 extracted from (A) the crystal complex in PPARα (1k7l) and (B) the crystal complex in PPARγ (1k74), respectively.
As described in [37], [71], the conversed H-bonding network formed by the polar acidic head of Core A in both GW409544 and Comp#1 to the four key residues of PPARα (or PPARγ), such as Ser280 (or Ser289), Tyr314 (or His323), Tyr464 (or Tyr473) and His440 (or His449), were observed in our docking study. This H-bonding network played the role in stabilizing the conformation of the AF2-helix in arm I (red helix in Fig. 1 ), which is vitally important for receptor-binding and activation [37], [72], [73], [74], [75], [76], [77].
The hydrophobic tail of both Core A and Core C of agonists are buried well in the hydrophobic arm I and arm II that are formed by hydrophobic residues as shown by the green surface in Fig. 1 . Compared with GW409544 (shown with grey color in Fig. 1 ), the compound of Comp#1 (purple color in Fig. 1 ) has more bulky molecular volume owing to the large hydrophobic Core C1, which is more fitted to the hydrophobic arm II, resulting in the much better binding affinity than GW409544 (cf. Table 1 ).
2. Molecular Dynamics Trajectory Analysis
Molecular dynamics can provide useful information for characterizing the internal motions of biomacromolecules with time. For a comparison study, the 10 ns molecular dynamics simulations were performed, respectively, for the crystal structures of PPARα (1k7l), PPARγ (1k74), as well as their complexes with GW409544 and Comp#1, i.e., PPARα-GW409544, PPARγ-GW409544, PPARα-Comp#1, and PPARγ-Comp#1. As we can see from Fig. 3 , all the characters concerned reached the simulation equilibrium within the 5ns (see panels A and C).
Figure 3. Illustration to show the outcomes of molecular dynamics simulations for Comp#1 ranked number 1 in Table 1 .

(A) The RMSD (root mean square deviation) of all backbone atoms for the receptor PPARα. (B) The RMSF (root mean square fluctuation) of the side-chain atoms for the receptor PPARα. (C) The RMSD (root mean square deviation) of all backbone atoms for the receptor PPARγ. (D) The RMSF (root mean square fluctuation) of the side-chain atoms for the receptor PPARγ. The green line indicates the outcome for the system of the receptor alone without any ligand, the red line for that of the receptor with the ligand GW409544, and the black line for that of the receptor with the ligand Comp#1. The curves involved with the AF2 helix region are framed with grey line. See the text for further explanation.
Meanwhile, the corresponding root mean square deviation (RMSD) value curves of the protein backbone for PPARα, PPARγ, PPARα-GW409544, PPARγ-GW409544, PPARα-Comp#1, and PPARγ-Comp#1 were also computed, respectively. It is interesting to see that the RMSD curves for PPARα-Comp#1 and PPARγ-Comp#1 are remarkably more stable than those of PPARα-GW409544 and PPARγ-GW409544, particularly for the case of PPARα (1k7l) system, where only a fluctuation of around 0.3 nm was observed when the complex system reached the plateau.
The detailed fluctuations of the aforementioned six different structures, as well as the root mean square fluctuations (RMSF) of their side-chain atoms, were also computed within 10 ns molecular dynamics simulations (see panels B and D of Fig. 3 ).
It is instructive to point out that the RMSF curve of PPARα-Comp#1 or PPARγ-Comp#1 is highly similar to that of PPARα-GW409544 or PPARγ-GW409544, respectively. This is especially remarkable in the binding site of AF2 helix region with the residues 459–465 for PPARα-Comp#1 and residues 469–477 for PPARγ-Comp#1 (see the grey frames in Fig. 3B and D ), indicating that the new designed compound, Comp#1, is very likely to have the same function for activating the AF2 helix as done by GW409544.
As a negative control, the similar molecular dynamics simulation was also performed for Comp#8 (A-B2-C3), which is ranked number 25 according to the strength of binding affinity with PPARα and PPARγ (cf. Table 1 ). The corresponding simulation results thus obtained are shown in the Fig. 4 , from which we can see that the fluctuating magnitudes of molecular dynamics for PPARα-Comp#8 and PPARγ-Comp#8, including the RMSD and RMSF, are much larger than those of PPARα-Comp#1 and PPARγ-Comp#1, especially for the binding site of AF2 helix region (see the gray frames in Fig. 4B and D ). These phenomena indicate that Comp#8 is not as good as Comp#1 in stably binding to PPARα and PPARγ, and hence Comp#8 might not have the same function for activating the AF2 helix as GW409544 had.
Figure 4. Illustration to show the outcomes of molecular dynamics simulations for Comp#8 ranked number 25 in Table 1 .

(A) The RMSD (root mean square deviation) of all backbone atoms for the receptor PPARα. (B) The RMSF (root mean square fluctuation) of the side-chain atoms for the receptor PPARα. (C) The RMSD (root mean square deviation) of all backbone atoms for the receptor PPARγ. (D) The RMSF (root mean square fluctuation) of the side-chain atoms for the receptor PPARγ. The green line indicates the outcome for the system of the receptor alone without any ligand, the red line for that of the receptor with the ligand GW409544, and the black line for that of the receptor with the ligand Comp#8. The curves involved with the AF2 helix region are framed with grey line.
3. ADME Prediction
Some pharmaceutically relevant properties of the new designed agonist derivatives as well as the original GW409544 compound, such as the “partition coefficient” (logPo/w), “van der Waals surface area of polar nitrogen and oxygen atoms” (PSA), “aqueous solubility” (logS), and “apparent MDCK permeability” (PMDCK), were predicted by means of the QP program embedded in the “Schrodinger2009 Software Package”. The results thus obtained are also listed in the Table 1 , respectively. Since PPARα and PPARγ have a more spacious pocket (∼1400 Å3) than any other nuclear hormone receptors [37], [72], it is quite natural that the agonist derivatives designed based on the two receptors by combining their three cores would have relatively large molecular weight (MW>500) and bulky volume, a trend quite similar to case in designing the inhibitors against the protein tyrosine phosphates (PTPase) [78].
As shown in Table 1 , the values calculated by the QP program, such as PAS, logPo/w, logS, and PMDCK for the newly designed agonists are all within the reasonable ranges. Although the higher logPo/w value of a compound, the stronger its affinity to PPARs is, it is not a good idea to excessively enhance logPo/w because this would induce bad distribution of the compound on fat and body fluid [70]. It should be pointed out that, rather than the experiential values within the range between −6.5 and 0.50, most of the log S values for the new agonists are quite close to that of GW409544. Such a phenomenon might result from the core A part which was kept unchanged during the process of designing the newly compounds as mentioned above. If the core A part was modified as well, the log S value would be further improved accordingly. Also, as mentioned above, the values for the four ADME properties listed in Table 1 are all within the acceptable range for human beings, indicating that most of the 25 compounds, particularly the top 10 derivatives found in this study as highlighted in Table 1 , can be utilized as candidates for the purpose of developing new drugs.
4. Conclusions
The goal of this study was to find new and more powerful dual agonists for PPARα and PPARγ. The new technique of “core hopping” adopted in this study allows for the rapid screening of novel cores to help overcome unwanted properties by generating new lead compounds with improved core properties. A set of 10 novel compounds were found in this regard. Compared with the existing dual agonist, the new agonists not only had the similar function in activating PPARα and PPARγ, but also assumed the conformation more favorable in binding to PPARα and PPARγ. It is anticipated that the new agonists may become potential drug candidates. Or at the very least, they may stimulate new strategy for developing novel dual agonists against type-2 diabetes.
Acknowledgments
The authors wish to thank the three anonymous Reviewers for their valuable suggestions, which were very helpful for strengthening the presentation of this study.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This study was supported by grants (20972112 and 21102103) from the National Natural Science Foundation of China; a grant (20091202110010) from the Doctoral Program of Higher Education of China; a grant (2011M500532) from the China Postdoctoral Science Foundation; a grant (09JCZDJC21600) from the Key Program of Tianjin Municipal Natural Science Foundation; a grant (2011KY43) from the Tianjin medical university, as well as Tianjin Institute of Pharmaceutical Research (TIPR). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Lewis SN, Bassaganya-Riera J, Bevan DR. Virtual Screening as a Technique for PPAR Modulator Discovery. PPAR Res. 2010;2010:861238. doi: 10.1155/2010/861238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carpino PA, Goodwin B. Diabetes area participation analysis: a review of companies and targets described in the 2008–2010 patent literature. Expert Opin Ther Pat. 2010;20:1627–1651. doi: 10.1517/13543776.2010.533171. [DOI] [PubMed] [Google Scholar]
- 3.Balakumar P, Rose M, Ganti SS, Krishan P, Singh M. PPAR dual agonists: are they opening Pandora's Box? Pharmacol Res. 2007;56:91–98. doi: 10.1016/j.phrs.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Markt P, Schuster D, Kirchmair J, Laggner C, Langer T. Pharmacophore modeling and parallel screening for PPAR ligands. J Comput Aided Mol Des. 2007;21:575–590. doi: 10.1007/s10822-007-9140-0. [DOI] [PubMed] [Google Scholar]
- 5.Shearer BG, Billin AN. The next generation of PPAR drugs: do we have the tools to find them? Biochim Biophys Acta. 2007;1771:1082–1093. doi: 10.1016/j.bbalip.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Issemann I, Prince RA, Tugwood JD, Green S. The peroxisome proliferator-activated receptor:retinoid X receptor heterodimer is activated by fatty acids and fibrate hypolipidaemic drugs. J Mol Endocrinol. 1993;11:37–47. doi: 10.1677/jme.0.0110037. [DOI] [PubMed] [Google Scholar]
- 7.Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999;341:410–418. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- 8.Gangloff M, Ruff M, Eiler S, Duclaud S, Wurtz JM, et al. Crystal structure of a mutant hERalpha ligand-binding domain reveals key structural features for the mechanism of partial agonism. J Biol Chem. 2001;276:15059–15065. doi: 10.1074/jbc.M009870200. [DOI] [PubMed] [Google Scholar]
- 9.Rosen ED, Spiegelman BM. PPARgamma : a nuclear regulator of metabolism, differentiation, and cell growth. J Biol Chem. 2001;276:37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 10.Blaschke F, Caglayan E, Hsueh WA. Peroxisome proliferator-activated receptor gamma agonists: their role as vasoprotective agents in diabetes. Endocrinol Metab Clin North Am 35: 561–574, ix. 2006. [DOI] [PubMed]
- 11.Rubenstrunk A, Hanf R, Hum DW, Fruchart JC, Staels B. Safety issues and prospects for future generations of PPAR modulators. Biochim Biophys Acta. 2007;1771:1065–1081. doi: 10.1016/j.bbalip.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Cronet P, Petersen JF, Folmer R, Blomberg N, Sjoblom K, et al. Structure of the PPARalpha and -gamma ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Structure. 2001;9:699–706. doi: 10.1016/s0969-2126(01)00634-7. [DOI] [PubMed] [Google Scholar]
- 13.Mochizuki K, Suruga K, Yagi E, Takase S, Goda T. The expression of PPAR-associated genes is modulated through postnatal development of PPAR subtypes in the small intestine. Biochim Biophys Acta. 2001;1531:68–76. doi: 10.1016/s0167-4889(01)00071-4. [DOI] [PubMed] [Google Scholar]
- 14.Chou KC. Review: Structural bioinformatics and its impact to biomedical science. Current Medicinal Chemistry. 2004;11:2105–2134. doi: 10.2174/0929867043364667. [DOI] [PubMed] [Google Scholar]
- 15.Chou KC. Molecular therapeutic target for type-2 diabetes. Journal of Proteome Research. 2004;3:1284–1288. doi: 10.1021/pr049849v. [DOI] [PubMed] [Google Scholar]
- 16.Wang JF, Chou KC. Insights from modeling the 3D structure of New Delhi metallo-beta-lactamase and its binding interactions with antibiotic drugs. PLoS ONE. 2011;6:e18414. doi: 10.1371/journal.pone.0018414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou KC, Wei DQ, Zhong WZ. Binding mechanism of coronavirus main proteinase with ligands and its implication to drug design against SARS. (Erratum: ibid., 2003, Vol.310, 675). Biochem Biophys Res Comm. 2003;308:148–151. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sirois S, Wei DQ, Du QS, Chou KC. Virtual Screening for SARS-CoV Protease Based on KZ7088 Pharmacophore Points. J Chem Inf Comput Sci. 2004;44:1111–1122. doi: 10.1021/ci034270n. [DOI] [PubMed] [Google Scholar]
- 19.Dea-Ayuela MA, Perez-Castillo Y, Meneses-Marcel A, Ubeira FM, Bolas-Fernandez F, et al. HP-Lattice QSAR for dynein proteins: Experimental proteomics (2D-electrophoresis, mass spectrometry) and theoretic study of a Leishmania infantum sequence. Bioorg Med Chem. 2008;16:7770–7776. doi: 10.1016/j.bmc.2008.07.023. [DOI] [PubMed] [Google Scholar]
- 20.Prado-Prado FJ, de la Vega OM, Uriarte E, Ubeira FM, Chou KC, et al. Unified QSAR approach to antimicrobials. 4. Multi-target QSAR modeling and comparative multi-distance study of the giant components of antiviral drug-drug complex networks. Bioorg Med Chem: in press. 2009. [DOI] [PubMed]
- 21.Prado-Prado FJ, Gonzalez-Diaz H, de la Vega OM, Ubeira FM, Chou KC. Unified QSAR approach to antimicrobials. Part 3: First multi-tasking QSAR model for Input-Coded prediction, structural back-projection, and complex networks clustering of antiprotozoal compounds. Bioorganic & Medicinal Chemistry. 2008;16:5871–5880. doi: 10.1016/j.bmc.2008.04.068. [DOI] [PubMed] [Google Scholar]
- 22.Prado-Prado FJ, Martinez de la Vega O, Uriarte E, Ubeira FM, Chou KC, et al. Unified QSAR approach to antimicrobials. 4. Multi-target QSAR modeling and comparative multi-distance study of the giant components of antiviral drug-drug complex networks. Bioorg Med Chem. 2009;17:569–575. doi: 10.1016/j.bmc.2008.11.075. [DOI] [PubMed] [Google Scholar]
- 23.Du QS, Huang RB, Wei YT, Pang ZW, Du LQ, et al. Fragment-Based Quantitative Structure-Activity Relationship (FB-QSAR) for Fragment-Based Drug Design. Journal of Computational Chemistry. 2009;30:295–304. doi: 10.1002/jcc.21056. [DOI] [PubMed] [Google Scholar]
- 24.Hou X, Du J, Fang H, Li M. 3D-QSAR Study on a Series of Bcl-2 Protein Inhibitors using Comparative Molecular Field Analysis. Protein and Peptide Letters. 2011;18:440–449. doi: 10.2174/092986611794927992. [DOI] [PubMed] [Google Scholar]
- 25.Xiao X, Wang P, Chou KC. GPCR-2L: Predicting G protein-coupled receptors and their types by hybridizing two different modes of pseudo amino acid compositions. Molecular Biosystems. 2011;7:911–919. doi: 10.1039/c0mb00170h. [DOI] [PubMed] [Google Scholar]
- 26.Concu R, Dea-Ayuela MA, Perez-Montoto LG, Bolas-Fernandez F, Prado-Prado FJ, et al. Prediction of enzyme classes from 3D structure: a general model and examples of experimental-theoretic scoring of peptide mass fingerprints of Leishmania proteins. J Proteome Res. 2009;8:4372–4382. doi: 10.1021/pr9003163. [DOI] [PubMed] [Google Scholar]
- 27.Chou KC, Wu ZC, Xiao X. iLoc-Euk: A Multi-Label Classifier for Predicting the Subcellular Localization of Singleplex and Multiplex Eukaryotic Proteins. PLoS One. 2011;6:e18258. doi: 10.1371/journal.pone.0018258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chou KC, Wu ZC, Xiao X. iLoc-Hum: Using accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Molecular Biosystems. 2012;8:629–641. doi: 10.1039/c1mb05420a. [DOI] [PubMed] [Google Scholar]
- 29.Wang P, Xiao X, Chou KC. NR-2L: A Two-Level Predictor for Identifying Nuclear Receptor Subfamilies Based on Sequence-Derived Features. PLoS ONE. 2011;6:e23505. doi: 10.1371/journal.pone.0023505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao X, Wang P, Chou KC. iNR-PhysChem: A Sequence-Based Predictor for Identifying Nuclear Receptors and Their Subfamilies via Physical-Chemical Property Matrix. PLoS ONE. 2012;7:e30869. doi: 10.1371/journal.pone.0030869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen HB, Chou KC. HIVcleave: a web-server for predicting HIV protease cleavage sites in proteins. Analytical Biochemistry. 2008;375:388–390. doi: 10.1016/j.ab.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 32.Chou KC. A vectorized sequence-coupling model for predicting HIV protease cleavage sites in proteins. Journal of Biological Chemistry. 1993;268:16938–16948. [PubMed] [Google Scholar]
- 33.Chou KC. Review: Prediction of HIV protease cleavage sites in proteins. Analytical Biochemistry. 1996;233:1–14. doi: 10.1006/abio.1996.0001. [DOI] [PubMed] [Google Scholar]
- 34.Schrodinger. Core Hopping. Schrödinger, LLC, New York, NY, 2009. 2009.
- 35.Li XB, Wang SQ, Xu WR, Wang RL, Chou KC. Novel Inhibitor Design for Hemagglutinin against H1N1 Influenza Virus by Core Hopping Method. PLoS One. 2011;6:e28111. doi: 10.1371/journal.pone.0028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.CombiGlide2.5. Schrodinger LLC, New York, NY, 2009. 2009.
- 37.Xu HE, Lambert MH, Montana VG, Plunket KD, Moore LB, et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A. 2001;98:13919–13924. doi: 10.1073/pnas.241410198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, et al. The Protein Data Bank. Acta Crystallogr D Biol Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 39.Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters 2006.
- 40.Prime2.1, Glide5.5. Schrodinger LLC, New York, NY, 2009. 2009.
- 41.Song Z, Kovacs FA, Wang J, Denny JK, Shekar SC, et al. Transmembrane domain of M2 protein from influenza A virus studied by solid-state (15)N polarization inversion spin exchange at magic angle NMR. Biophys J. 2000;79:767–775. doi: 10.1016/S0006-3495(00)76334-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Husslein T, Moore PB, Zhong Q, Newns DM, Pattnaik PC, et al. Molecular dynamics simulation of a hydrated diphytanol phosphatidylcholine lipid bilayer containing an alpha-helical bundle of four transmembrane domains of the influenza A virus M2 protein. Faraday Discuss: 201–208; discussion 225–246. 1998. [DOI] [PubMed]
- 43.Kochendoerfer GG, Salom D, Lear JD, Wilk-Orescan R, Kent SB, et al. Total chemical synthesis of the integral membrane protein influenza A virus M2: role of its C-terminal domain in tetramer assembly. Biochemistry. 1999;38:11905–11913. doi: 10.1021/bi990720m. [DOI] [PubMed] [Google Scholar]
- 44.Gandhi CS, Shuck K, Lear JD, Dieckmann GR, DeGrado WF, et al. Cu(II) inhibition of the proton translocation machinery of the influenza A virus M2 protein. J Biol Chem. 1999;274:5474–5482. doi: 10.1074/jbc.274.9.5474. [DOI] [PubMed] [Google Scholar]
- 45.Chou KC, Watenpaugh KD, Heinrikson RL. A Model of the complex between cyclin-dependent kinase 5 (Cdk5) and the activation domain of neuronal Cdk5 activator. Biochemical & Biophysical Research Communications. 1999;259:420–428. doi: 10.1006/bbrc.1999.0792. [DOI] [PubMed] [Google Scholar]
- 46.Zhang J, Luan CH, Chou KC, Johnson GVW. Identification of the N-terminal functional domains of Cdk5 by molecular truncation and computer modeling. PROTEINS: Structure, Function, and Genetics. 2002;48:447–453. doi: 10.1002/prot.10173. [DOI] [PubMed] [Google Scholar]
- 47.Chou KC. Insights from modelling the 3D structure of the extracellular domain of alpha7 nicotinic acetylcholine receptor. Biochemical and Biophysical Research Communication. 2004;319:433–438. doi: 10.1016/j.bbrc.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 48.Housaindokht MR, Bozorgmehr MR, Bahrololoom M. Analysis of ligand binding to proteins using molecular dynamics simulations. J Theor Biol. 2008;254:294–300. doi: 10.1016/j.jtbi.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 49.Feige JN, Gelman L, Tudor C, Engelborghs Y, Wahli W, et al. Fluorescence imaging reveals the nuclear behavior of peroxisome proliferator-activated receptor/retinoid X receptor heterodimers in the absence and presence of ligand. J Biol Chem. 2005;280:17880–17890. doi: 10.1074/jbc.M500786200. [DOI] [PubMed] [Google Scholar]
- 50.Irwin JJ, Shoichet BK. ZINC–a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao QH, Gao QZ, Wei J, Chou KC. Docking and Molecular Dynamics Study on the Inhibitory Activity of Novel Inhibitors on Epidermal Growth Factor Receptor (EGFR). Medicinal Chemistry. 2011;7:24–31. doi: 10.2174/157340611794072698. [DOI] [PubMed] [Google Scholar]
- 52.Cai L, Wang Y, Wang JF, Chou KC. Identification of Proteins Interacting with Human SP110 During the Process of Viral Infections. Medicinal Chemistry. 2011;7:121–126. doi: 10.2174/157340611794859343. [DOI] [PubMed] [Google Scholar]
- 53.LigPre2.3. Schrodinger LLC, New York, NY, 2009. 2009.
- 54.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 55.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 56.Chou KC. The biological functions of low-frequency phonons: 3. Helical structures and microenvironment. Biophysical Journal. 1984;45:881–890. doi: 10.1016/S0006-3495(84)84234-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang JF, Chou KC. Insight into the molecular switch mechanism of human Rab5a from molecular dynamics simulations. Biochem Biophys Res Commun. 2009;390:608–612. doi: 10.1016/j.bbrc.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 58.Chou KC. Low-frequency resonance and cooperativity of hemoglobin. Trends in Biochemical Sciences. 1989;14:212. doi: 10.1016/0968-0004(89)90026-1. [DOI] [PubMed] [Google Scholar]
- 59.Chou KC. The biological functions of low-frequency phonons: 4. Resonance effects and allosteric transition. Biophysical Chemistry. 1984;20:61–71. doi: 10.1016/0301-4622(84)80005-8. [DOI] [PubMed] [Google Scholar]
- 60.Chou KC. The biological functions of low-frequency phonons: 6. A possible dynamic mechanism of allosteric transition in antibody molecules. Biopolymers. 1987;26:285–295. doi: 10.1002/bip.360260209. [DOI] [PubMed] [Google Scholar]
- 61.Chou KC, Mao B. Collective motion in DNA and its role in drug intercalation. Biopolymers. 1988;27:1795–1815. doi: 10.1002/bip.360271109. [DOI] [PubMed] [Google Scholar]
- 62.Chou KC, Zhang CT, Maggiora GM. Solitary wave dynamics as a mechanism for explaining the internal motion during microtubule growth. Biopolymers. 1994;34:143–153. doi: 10.1002/bip.360340114. [DOI] [PubMed] [Google Scholar]
- 63.Chou KC. Review: Low-frequency collective motion in biomacromolecules and its biological functions. Biophysical Chemistry. 1988;30:3–48. doi: 10.1016/0301-4622(88)85002-6. [DOI] [PubMed] [Google Scholar]
- 64.Javangula KC, Batchelor TJ, Jaber O, Watterson KG, Papagiannopoulos K. Combined severe pectus excavatum correction and aortic root replacement in Marfan's syndrome. Ann Thorac Surg. 2006;81:1913–1915. doi: 10.1016/j.athoracsur.2005.03.045. [DOI] [PubMed] [Google Scholar]
- 65.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. Journal of Physical Chemistry B. 2001;105:6474–6487. [Google Scholar]
- 66.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 1983;79:926–935. [Google Scholar]
- 67.Hoover WG. Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 68.Jorgensen WL, Duffy EM. Prediction of drug solubility from structure. Adv Drug Deliv Rev. 2002;54:355–366. doi: 10.1016/s0169-409x(02)00008-x. [DOI] [PubMed] [Google Scholar]
- 69.QikProp3.2. Schrodinger LLC, New York, NY, 2009. 2009.
- 70.Singh KD, Kirubakaran P, Nagarajan S, Sakkiah S, Muthusamy K, et al. Homology modeling, molecular dynamics, e-pharmacophore mapping and docking study of Chikungunya virus nsP2 protease. J Mol Model. 2011. [DOI] [PubMed]
- 71.Ebdrup S, Pettersson I, Rasmussen HB, Deussen HJ, Frost Jensen A, et al. Synthesis and biological and structural characterization of the dual-acting peroxisome proliferator-activated receptor alpha/gamma agonist ragaglitazar. J Med Chem. 2003;46:1306–1317. doi: 10.1021/jm021027r. [DOI] [PubMed] [Google Scholar]
- 72.Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell. 1999;3:397–403. doi: 10.1016/s1097-2765(00)80467-0. [DOI] [PubMed] [Google Scholar]
- 73.Michalik L, Zoete V, Krey G, Grosdidier A, Gelman L, et al. Combined simulation and mutagenesis analyses reveal the involvement of key residues for peroxisome proliferator-activated receptor alpha helix 12 dynamic behavior. J Biol Chem. 2007;282:9666–9677. doi: 10.1074/jbc.M610523200. [DOI] [PubMed] [Google Scholar]
- 74.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 75.Yue L, Ye F, Xu X, Shen J, Chen K, et al. The conserved residue Phe273(282) of PPARalpha(gamma), beyond the ligand-binding site, functions in binding affinity through solvation effect. Biochimie. 2005;87:539–550. doi: 10.1016/j.biochi.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 76.Ji CG, Zhang JZ. Protein polarization is critical to stabilizing AF-2 and helix-2′ domains in ligand binding to PPAR-gamma. J Am Chem Soc. 2008;130:17129–17133. doi: 10.1021/ja807374x. [DOI] [PubMed] [Google Scholar]
- 77.Liu XY, Wang SQ, Xu WR, Tang LD, Wang RL, et al. Docking and Molecular Dynamics Simulations of Peroxisome Proliferator Activated Receptors Interacting with Pan Agonist Sodelglitazar. Protein Pept Lett. 2011. [DOI] [PubMed]
- 78.Liu G, Xin Z, Pei Z, Hajduk PJ, Abad-Zapatero C, et al. Fragment screening and assembly: a highly efficient approach to a selective and cell active protein tyrosine phosphatase 1B inhibitor. J Med Chem. 2003;46:4232–4235. doi: 10.1021/jm034122o. [DOI] [PubMed] [Google Scholar]