

Abstract
F1F0 ATPases have been identified in most bacteria, including mycoplasmas which have very small genomes associated with a host-dependent lifestyle. In addition to the typical operon of eight genes encoding genuine F1F0 ATPase (Type 1), we identified related clusters of seven genes in many mycoplasma species. Four of the encoded proteins have predicted structures similar to the α, β, γ and ε subunits of F1 ATPases and could form an F1-like ATPase. The other three proteins display no similarity to any other known proteins. Two of these proteins are probably located in the membrane, as they have three and twelve predicted transmembrane helices. Phylogenomic studies identified two types of F1-like ATPase clusters, Type 2 and Type 3, characterized by a rapid evolution of sequences with the conservation of structural features. Clusters encoding Type 2 and Type 3 ATPases were assumed to originate from the Hominis group of mycoplasmas. We suggest that Type 3 ATPase clusters may spread to other phylogenetic groups by horizontal gene transfer between mycoplasmas in the same host, based on phylogeny and genomic context. Functional analyses in the ruminant pathogen Mycoplasma mycoides subsp. mycoides showed that the Type 3 cluster genes were organized into an operon. Proteomic analyses demonstrated that the seven encoded proteins were produced during growth in axenic media. Mutagenesis and complementation studies demonstrated an association of the Type 3 cluster with a major ATPase activity of membrane fractions. Thus, despite their tendency toward genome reduction, mycoplasmas have evolved and exchanged specific F1-like ATPases with no known equivalent in other bacteria. We propose a model, in which the F1-like structure is associated with a hypothetical X0 sector located in the membrane of mycoplasma cells.
Introduction
Mycoplasmas are small bacteria that infect humans and animals and evolved from low-GC content firmicutes in a process involving a drastic reduction of genome size, resulting in present-day species with typical 1 Mb-genomes [1]. Mycoplasmas have lost genes from most functional categories and display the complete disappearance of several metabolic pathways and the elimination of many redundant genes. Like other members of the class Mollicutes, the bacteria of the Mycoplasma genus lack genes involved in the synthesis of cell-wall components, amino-acids, lipids, co-factors and nucleic acid precursors.
The cellular apparatus involved in the basic maintenance and expression of genetic information is essentially similar in most mycoplasmas [2], but the enzymes involved in energy metabolism may differ considerably between, even in those with very reduced genomes [3]. The repertoires of genes encoding membrane proteins such as lipoproteins and transporters are also highly diverse in mycoplasmas, probably reflecting the ability of the different species to infect animal species as diverse as mammals, birds, fishes and arthropods. Thus, despite the massive genome reduction that has marked their evolution and their general absence in natural environments, mycoplasmas have conquered a wide range of complex animals and seem to be able to adapt rapidly to new hosts. Phylogenomic studies based on 16S rDNA and other genes have shown that mycoplasmas are frequently associated with particularly long branches [4]. Moreover, going against the widespread view that mycoplasmas evolve purely by gene loss, recent studies have shown that horizontal gene transfers (HGT) between species in the same host may have increased the genetic potential of mycoplasmas, potentially facilitating adaptation to the host. Three examples of HGT have been reported to date, in mycoplasmas pathogenic to humans [3], birds [5] and ruminants [6]. In the species infecting birds and ruminants, several genes thought to have been subject to HGT were parts of typical mobile elements including integrative conjugative elements (ICEs), insertion sequences (ISs) and restriction-modification systems (RMSs) but many others encoded transporters, lipoproteins and hypothetical proteins potentially involved in host-specificity and pathogenicity. The genes thought to have been subject to HGT in human urogenital species encoded ISs, RMSs, hypothetical proteins and two proteins related to F1F0 ATPase subunits α (atpA) and β (atpD). Strikingly, genes annotated atpA and atpD were also found in the lists of genes thought to have been exchanged between bird mycoplasma species and between ruminant mycoplasma species.
All the mycoplasma genomes examined to date contain a typical complete operon encoding the eight subunits of the F1F0 ATPase ( Figure 1 ). The F1F0 ATPase is thought to function primarily in ATP hydrolysis and maintenance of the electrochemical gradient in mycoplasmas, rather than in the generation of ATP [7]. Nevertheless, the genes encoding the subunits of this complex were considered to be essential in several species in which global transposon mutagenesis was carried out [8], [9], [10]. Surprisingly, in addition to the F1F0 ATPase operon, extra copies of atpA and atpD, organized in pairs, have been identified and annotated in many mycoplasma genomes although no specific study has ever investigated this remarkable trait. Indeed, although most bacteria have F1F0 ATPases, the presence of extra genes located outside of the traditional operon has been reported in very few cases including those encoding atypical forms of F1F0 ATPase (N-ATPases) in some marine and halotolerant bacteria, in the pathogens Burkolderia spp. and in the archaea Methanosarcina acetivorans and Methanosarcina barkeri [11]. Thus, in the general context of gene loss and redundancy elimination during the evolution of mycoplasmas, the presence of additional copies was entirely unexpected. Moreover, the prediction that these extra copies of atpA and atpD may have been exchanged during three unrelated HGT events between mycoplasmas was puzzling.
Figure 1. ATPase F1F0 in mycoplasmas.
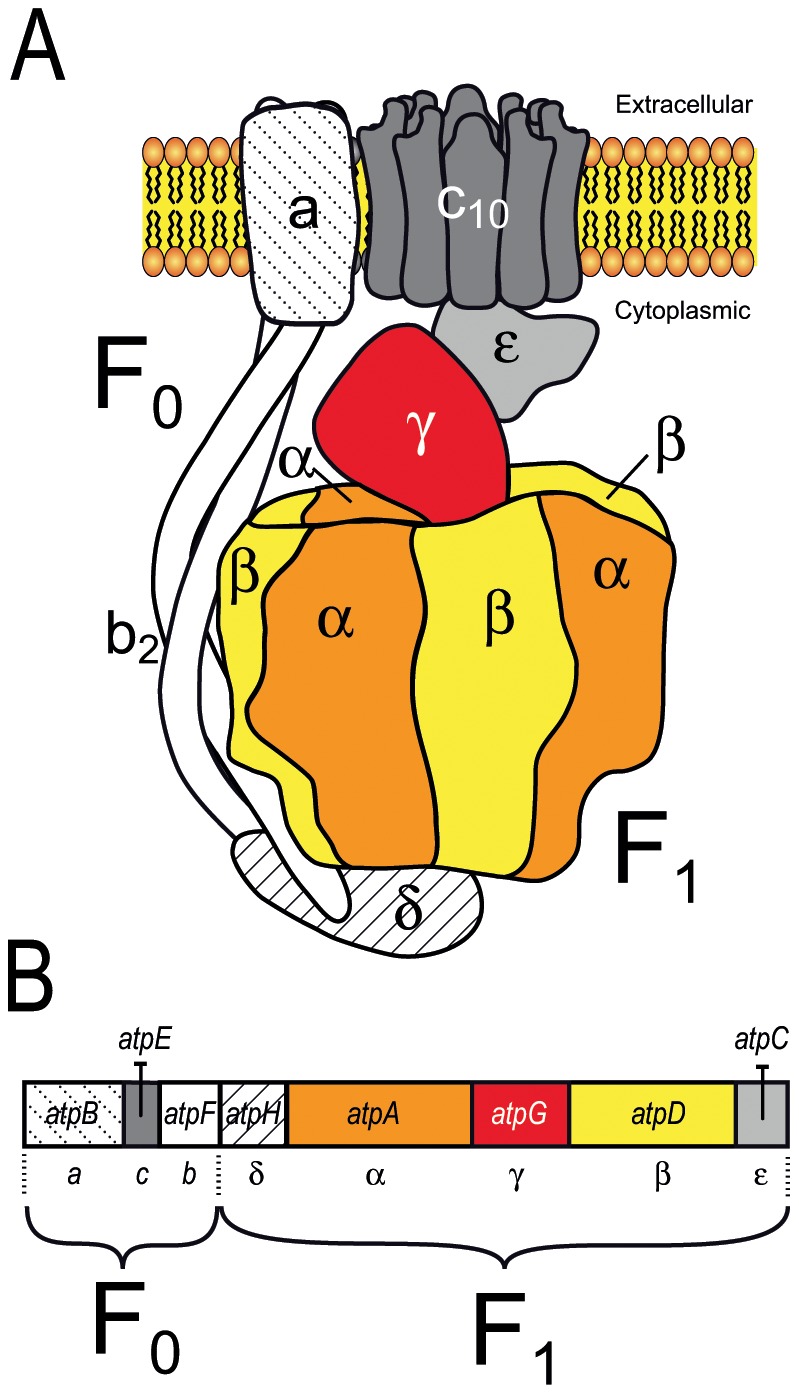
A. Bacterial ATPase F1F0. B. Organization of the operon encoding the ATPase F1F0 in mycoplasmas. In E. coli and mycoplasma species, the F1F0 ATPase operon and likely the 3D structure are similar.
We investigated the evolution of these genes and their relationship to the F1F0 ATPase, by carrying out a global phylogenomic and functional study with the ruminant pathogen M. mycoides subsp. mycoides (Mmm). We found that the extra copies of atpA and atpD belonged to a seven-gene cluster that had emerged during mycoplasma evolution and was spread by HGT. The proteins encoded by these genes were associated with a membrane ATPase activity. Based on our in silico analysis and experimental results, we propose a structural model of an F1-likeX0 ATPase specific to mycoplasmas.
Materials and Methods
Comparative genomics and phylogeny
Genome analysis and comparisons were carried out with MolliGen, a database for mollicute genomes (http://molligen.org) [12], and MBGD (http://mbgd.genome.ad.jp/) [13]. Protein sequences were named by their mnemonics (Table S2). GenBank IDs of the Type 3 ATPase proteins of Mmm are NP_975598.1 (MSC_0618), NP_975599.1 (MSC_0619), NP_975600.1 (MSC_0620), NP_975601.1 (MSC_0621), NP_975602.1 (MSC_0622), NP_975603.1 (MSC_0623) and NP_975604.1 (MSC_0624). Multiple alignments were generated with MAFFT [14] and MUSCLE [15] software. Unreliable sites were removed from the alignments with Gblocks [16] and Jalview [17]. Species and protein phylogenies were determined by the neighbour-joining (NJ), minimum evolution (ME) and maximum parsimony (MP) methods, in the MEGA4 package [18], and the maximum likelihood (ML) method in the Seaview [19] (integrating the phyML software [20]) or MEGA5 [21] packages. For trees constructed by the NJ, ME and MP methods, the “gap complete deletion” option was chosen and bootstrap analysis was carried out with 500 replicates. For phylogenetic reconstructions inferred from concatenated proteins α and β, the model best fitting the pattern of evolution, according to the Akaike information criterion (AIC), calculated with ProtTest [22], was LG+G+F. This model was used for ML phylogeny. The evolutionary divergence of sequences was estimated by the Poisson correction method in MEGA4 [18]. All positions containing gaps and missing data were eliminated from the dataset (complete deletion option). In silico predictions for operon structure were retrieved from the Database for prOkaryoticOperOns, (http://csbl1.bmb.uga.edu/OperonDB), [23]).
Prediction of protein secondary structure and membrane topology
PSIPRED v.3.0 and pGenTHREADER were used to predict secondary structure and fold recognition, respectively. These programs are available from the PSIPRED Protein Structure Prediction Server (http://bioinf.cs.ucl.ac.uk/psipred/). The TMpred (http://www.ch.embnet.org/software/TMPRED_form.html) [24] and TMHMM (http://www.cbs.dtu.dk/services/TMHMM) [25] methods were combined for the prediction of transmembrane segments. SignalP-4.0 [26] was used to query signal peptide cleavages. The F1 and F1-like complex models of Mmm were drawn by similarity with the crystal structure of the E. coli F1-ATPase (Pdb id: 3oaa) [27] with the help of the Pymol software (http://www.pymol.org) [28]. The F0 subunits were drawn according to the ac12 and b2 models [29], [30]. The X0 proteins were schematized on the basis of their predicted 2D structures.
Strains and culture conditions
The vaccine strain T1/44 (CIRAD-EMVT/PANVAC; Batch 002) of Mmm and the Δ619 mutant derived from it were grown at 37°C in modified Hayflick medium [31] supplemented with 20% horse serum, 0.4% pyruvate and 0.2% glucose. The growth of the T1/44 and mutant strains was monitored by measuring absorbance at 640 nm.
Mutant selection and complementation
The Mmm Δ619 mutant was isolated from a library of Mmm T1/44 transformed with the plasposon PMT85/2res [32], by PCR screening with the specific probes MSC620.3 and MT85.1 (Table S1). A pure culture of the mutant was established after three cloning steps, by passage of the culture through filters with 0.2 µm pores. The purity of the mutant culture was checked by PCR, with the same primers used for the PCR screening assay (Table S1). A plasmid for the complementation of Δ619 was constructed as follows: the spiralin promoter was amplified by PCR with the PS1 and PS2 primers (Table S1), which include XbaI and XhoI sites. The amplified DNA was digested with XbaI and the resulting fragment was inserted into XbaI-linearized pMYSO1 [33] to generate pCC1. The genomic region corresponding to the MSC_0619-MSC_0618 genes was amplified by PCR from T1/44 DNA with the MSC619.10 and MSC618.5 primers (Table S1). The PCR product was digested with XhoI and inserted into the XhoI site downstream from the spiralin gene promoter of pCC1. The resulting plasmid was named pCC2. T1/44 and Δ619 were transformed with 20 µg of pCC2 or pMYSO1, in the presence of polyethylene glycol, as described elsewhere [34].
Protein extraction and preparation of membrane-enriched fractions
Total protein extracts were prepared from exponentially growing Mmm cells of the T1/44 and Δ619 strains. Cells were collected by centrifugation (12,000×g for 30 min, 4°C). The pellets were dispersed in phosphate-buffered saline (PBS, 139 mM NaCl, 1.9 mM KCl, 1.9 mM KH2PO4, 8 mM Na2HPO4,12 H2O pH 7.4) and washed three times in the same buffer. Cells were lysed by three one-minute cycles of sonication on ice with a microprobe (Vibra-Cell sonicator, Branson). For the isolation of membrane-enriched fractions, mycoplasma cells were washed three times in a washing buffer (50 mM Tris-HCl, 150 mM NaCl pH 7.4) and resuspended in lysis buffer (50 mM Tris HCl pH 7.4, 0.4 mM PMSF). The cells were lysed by three cycles of sonication for one minute on ice and the lysates were centrifuged for 10 min at 500×g. The supernatants were centrifuged for 1 h at 30,000×g (4°C). The pellets were dispersed in 50 mM Tris HCl pH 7.4 and centrifuged again for 1 h at 20,000×g (4°C). The final pellets were resuspended in 50 mM Tris HCl pH 7.4, frozen in liquid nitrogen and stored at −85°C until use. Protein concentration was determined with the DC reagent kit (BioRad), using bovine serum albumin as the standard. The total metabolic activity of the fractions was evaluated by measuring their capacity to generate H2O2 in response to the addition of glycerol. In T1/44, the transport of glycerol across the membrane and its phosphorylation during transport to generate glycerol-3-P leads to the production of H2O2, the formation of which is catalysed by the membrane-located L-α-glycerophosphate oxidase [35]. H2O2 was quantified as previously described [36].
Generation of monospecific polyclonal antibodies
Peptide sequences for the MSC_0618, MSC_0619, MSC_0620 and MSC_0624 genes were retrieved from the Mmm genome sequence available from EMBL/GenBank/DDBJ entry BX293980. Transmembrane fragments were predicted with Phobius [37]. Protein fragments of 27 to 77 amino acids in length were selected on the basis of two criteria: (i) regions lying outside transmembrane fragments and (ii) regions with no UGA-encoded tryptophan residues. The corresponding gene regions were amplified by PCR from a genomic DNA template from Mmm strain T1/44 and inserted into pAff8c for the production of recombinant proteins in Escherichia coli strain BL21(DE3), these proteins then being purified as previously described [38]. The purity of the recombinant proteins, as assessed by SDS-PAGE and Coomassie blue staining, exceeded 80%. Quality control was also carried out for the proteins, by electrospray ionization mass spectrometry, before the removal of an aliquot of 500 µg of the recombinant protein for rabbit immunization. Serum samples were obtained and subjected to affinity purification according to national guidelines (Swedish permit 84-02) and standard procedures [39]. The affinity purification process was used to remove non-specific antibodies and antibodies binding to the fusion tag. The specificities of the monospecific polyclonal antibodies were assessed by dot and western blotting before use. Three antibodies – MSC618002, MSC619001, and MSC620002 – were finally selected. The rabbit polyclonal serum against N-ter LppQ (anti-LppQ) was a gift from Prof. J. Frey [40].
Protein electrophoresis, immunoblotting and mass spectrometry
Proteins were separated by electrophoresis, as previously described [41], in 10% polyacrylamide gels. For protein immunodetection, ∼10 µg of protein from T1/44 or Δ619 was loaded onto the gel. Proteins were electroblotted onto a nitrocellulose membrane in a semidry transfer unit (Trans-Blot® SD semi-dry electrophoretic transfer cell, BioRad) and probed with the polyclonal antibodies MSC618002, MSC619001, MSC620002 and Anti-LppQ, followed by horseradish peroxidase-conjugated secondary antibodies (Sigma). The Supersignal® West Pico Chemiluminescent Substrate kit (WestPicoLuminol, Pierce) was used for detection. For the identification of proteins in membranes by LC-MS/MS, membrane-enriched fractions were separated by electrophoresis in 10% polyacrylamide gels. Coomassie blue G250 staining was used to visualize the protein bands. The gel was sliced into 16 sections, which were subjected to trypsin (Sigma) digestion. The resulting peptides were further analysed by online capillary liquid chromatography (LC Packings) coupled to an MS/MS orbitrap mass spectrometer (ThermoFinnigan). Peptides were separated on a 75 µm (internal diameter)×15 cm C18 PepMap column (LC Packings), with a flow rate of 200 nl/min. Peptides were eluted with a 5–40% linear gradient of solvent B over a period of 35 min (solvent A was 0.1% formic acid in 5% acetonitrile, and solvent B was 0.1% formic acid in 80% acetonitrile). The mass spectrometer was operated in positive-ion mode, at a needle voltage of 2.2 V and a capillary voltage of 49 V. Data were acquired in a data-dependent mode consisting of full-scan MS over the range m/z 300–1700 or five full-scan MS/MS for the five most intense ions in the preceding MS spectra. MS/MS data were acquired with a 2 m/z-unit ion isolation window and relative collision energy of 35%. Peptides were identified with SEQUEST, through the Bioworks 3.2 interface (Thermo-Finnigan), using a database containing both direct and antisense sequences for Mmm coding sequence (CDS) and intergenic sequence data.
ATPase activity assays
ATPase assays were performed with EnzChek (Invitrogen), in the presence of 80 µM ATP and membrane-enriched fractions (60 µg protein), at room temperature. Absorbance at 360 nm was measured every 90 s, for 40–50 min, following the addition of ATP. A control assay lacking the membrane-enriched fraction was performed for the deduction of background ATP-self-hydrolysis values. Absorbance was converted into the amount of free orthophosphate (Pi) released, by comparison with standard curves for Pi (0.01 mM to 0.12 mM KH2PO4). The statistical t-student test at 0.05 significance level was applied to compare the Pi released with the membrane-enriched fraction from the T1/44 and Δ619strains.
RT-PCR analysis
Total RNA was extracted with Trizol from exponentially growing T1/44 and Δ619 cells subjected to DNase I treatment. We then synthesized cDNA from 500 ng of total RNA, with the SuperScript II reverse transcriptase (Invitrogen) and gene-specific primers, according to the manufacturer's instructions. A control lacking the reverse transcriptase was also included for each sample and was processed in tandem. For the amplification of intergenic regions, we used the following cycling parameters: 95°C for 5 min, followed by 35 cycles of 95°C for 40 s, 54°C for 40 s and 72°C for 40 s, with a final extension at 72°C for 7 min. The primers used are described in Table S1. The quality of the RNA was checked with the MSC679.1 and MSC679.2 primers for amplification of the gap gene. The amplified products were run on a 1% agarose gel and visualized with ethidium bromide staining.
Limited proteolysis assays
Exponentially growing Mmm cells were collected by centrifugation, washed with PBS pH 7.5 and dispersed in 200 µl of PBS pH 8.4. Limited proteolysis was performed on intact cells or on cells disrupted by brief sonication. One volume of agarose beads coated with trypsin (Promega) was added to 2.5 volumes of suspended cells. Samples were incubated at 37°C for 6 hours. Trypsin was removed by centrifugation and protein profiles were analysed by western blotting with MSC618002, MSC620002 and Anti-LppQ polyclonal antibodies.
Results
Extra copies of genes related to F1F0 ATPase subunits alpha and beta are found in mycoplasma genomes
Clusters of eight genes encoding the typical F1F0 ATPase have been identified in all mollicutes with the exception of plant pathogenic phytoplasmas. Extra copies of atpA (subunit α, F1-sector) and atpD (subunit β, F1-sector) genes, organized in pairs, have also been annotated in several mycoplasma genomes. Blastp queries have identified such extra copies in many species ( Figure 2A ). In the Hominis phylogenetic group which includes mycoplasmas pathogenic to humans and various animals, all eight of the available genomes contain one to three extra copies of the atpA-like/atpD-like gene pair. In the Pneumoniae group which also contains human and animal pathogens, two of the five species had extra copies. One extra pair of genes was found in the genome of the human urogenital pathogen Ureaplasma parvum (serovar 3 strain ATCC 700970) and in the other two complete Ureaplasma genomes available (serovar 3 strain ATCC 27815 and serovar 10 strain ATCC 33699, not shown). In the genome of the bird pathogen Mycoplasma gallisepticum, one pair of atpA-like/atpD-like genes was found. An additional gene related to atpD was also detected at another locus on the chromosome. Within the Spiroplasma phylogenetic group, an extra pair of genes was found in the genomes of the ruminant pathogens Mmm, Mycoplasma mycoides subsp. capri and Mycoplasma capricolum subsp. capricolum. By contrast, no extra copies of genes related to the F1F0 ATPase were predicted in the genomes of Mesoplasma florum (isolated from the surface of a lemon tree flower) and Spiroplasma citri (plant pathogen transmitted by insects). All the proteins encoded by the additional atpA-like and atpD-like genes were predicted to contain the corresponding Prosite motif (PS00152 ATPASE ALPHA BETA) and most were annotated accordingly.
Figure 2. Distribution and evolution of extra copies of atpA-like and atpD-like genes in mollicutes.

A. The number of typical F1F0 ATPase operons and of extra copies of atpA-like/atpD-like pairs of genes are indicated for each species. * In M. gallisepticum, one of the two extra copies only contains a truncated atpD-like gene. The 16S rDNA phylogenetic tree was generated by the ML method; bootstrap values of more than 50% are indicated. Bacillus subtilis was chosen as an outgroup species. Phylogenetic groups are indicated: S, Spiroplasma; H, Hominis; P, Pneumoniae; AP, Acholeplasma/Phytoplasma. Mnemonic codes are indicated in brackets besides species names when useful. B. The amino acid sequences of the proteins encoded by the atpA-like and atpD-like genes were concatenated and a multiple alignment was generated. Protein sequences of Type 1 atpA and atpD genes from M. pulmonis and B. subtilis (GenBank ID: atpA, NP_391564.1; atpD, NP_391562.1) were used as outgroup. The multiple sequence alignment was curated with GBLOCK to remove unreliable sites and a final round of manual editing was performed with Jalview. Phylogenetic trees were generated by ML, NJ and MP methods. The tree represented was obtained by the ML method. The aLRT/Bootstrap values corresponding to these three methods are indicated on the branches, in the following order: ML/NJ/MP. Sequences are labelled by their mnemonics, see also Table S2 for details.
We characterized the evolutionary relationship between the typical F1F0 ATPase and these extra copies of the atpA and atpD genes, by generating multiple alignments of the corresponding α-like and β-like protein sequences and inferring phylogenetic trees. A selection of F1F0 ATPase homologs from non mollicute bacteria and some related N-ATPases [11] were included in this analysis. The general shape of the tree was identical for both proteins (the tree obtained for α and α-like proteins is shown in Figure 3 ), indicating a clear divergence of the mycoplasma-specific extra copies from both genuine bacterial F1F0 ATPase homologs and N-ATPase homologs. Indeed, the typical F1F0 ATPase subunits from mollicutes clustered with the orthologous proteins found in most bacteria, in a sub-tree with a topology resembling that for 16S rDNA. These typical F1F0 ATPase copies and atypical N-ATPases are referred to hereafter as Type 1 and Type 1′, respectively.
Figure 3. Evolution of atpA and atpA-like genes in bacteria.

The phylogenetic tree was inferred from the amino acid sequences of ATPase alpha subunits encoded by atpA and atpA-like genes. Multiple alignment was generated with MUSCLE. The phylogenetic tree was generated by the ML method. Branches corresponding to Type 1, Type 1′, Type 2 and Type 3 proteins were supported by 96–100% bootstrap values. The ML, NJ, MP and ME methods generated trees with similar topologies, except alternative branching of N-ATPases using NJ or ME (indicated by a star). Main bacterial groups are indicated. Proteins from mollicutes are named by their mnemonics, others by the species name. See Table S2 for details.
The additional copies of α-like and β-like subunits from mollicutes clustered into two phylogenetically remote branches, identified here as Type 2 and Type 3. Type 2 copies were found only in two distantly related species from the Hominis group: the rodent pathogen Mycoplasma pulmonis [42] and Mycoplasma mobile [43], which was isolated from a fish. Both these species also had Type 3 genes clustered with all the other extra copies, in a monophyletic group. The separation of the Types 1, 1′, 2 and 3 copies was strongly supported by an approximate likelihood-ratio test.
Thus, many mycoplasma genomes contain one or several extra copies of genes related to atpA and atpD and α-like and β-like subunits they encode diverge markedly from the typical F1F0 ATPase subunits found in mycoplasmas and most bacteria.
Type 3 clusters have been exchanged in the three documented HGT between mycoplasmas
A phylogenomic study of Type 2 and Type 3 gene pairs was conducted with concatenated α-like and β-like proteins ( Figure 2B ), to clarify the evolutionary relationships. As expected, Type 2 and Type 3 pairs of proteins clustered into two well-supported branches, indicative of their distant relationship. The topology of the Type 3 subtree was only partially consistent with the species tree inferred from 16S rDNA (see Figure 2A ). First, in some species (Mycoplasma arthritidis, M. pulmonis) pairs of homologous proteins were not found close together, as would be expected for recently duplicated genes. Second, the proteins were not distributed along the well-defined main phylogenetic branches for mollicutes. Proteins from Mycoplasma agalactiae (Hominis phylogenetic group) and species of the so-called mycoides cluster (Mmm, M. mycoides subsp. capri and M. capricolum subsp. capricolum, Spiroplasma phylogenetic group) were found to be closely related. This finding is consistent with published reports of HGT between ruminant pathogens [6]. Similarly, proteins from the urogenital species Mycoplasma hominis (Hominis group) and U. parvum (Pneumoniae group) were positioned on sister branches, consistent with hypothetical gene exchange between these species [3]. The complete pair of genes found in the bird pathogen M. gallisepticum (MGA_0488/MGA_0491) was not clearly related to any other cluster, but was located at some distance from U. parvum proteins in the tree, despite the common ancestry of these species. Moreover, trees including the protein encoded by the isolated atpD-like gene of M. gallisepticum (MGA_1321d) showed that this protein was strongly related to the homologous protein from Mycoplasma synoviae, consistent with HGT (data not shown).
We estimated the degree of relatedness between Type 3 α-like/β-like pairs of proteins, by calculating pairwise distances between homologous pairs, using M. agalactiae, M. hominis and M. gallisepticum as references ( Figure 4 , panels A, C and E). For the purpose of comparison, pairwise distances were also calculated between concatenated α/β pairs from the Type 1 F1F0 ATPase ( Figure 4 , panels B, D and F). The F1F0 ATPase pairwise distances were roughly related to the evolutionary distance estimated on the basis of 16S rDNA pairwise distances, but no such correlation was observed for Type 3 proteins. In all cases, the minimal pairwise distances were recorded for homologs for which HGT was predicted. The most remarkable illustration of this finding concerns the isolated M. gallisepticum β-like copy (MGA_1321d), for which calculated pairwise distance from M. synoviae homolog MS53_0464 was only 0.239 whereas the other β-like copy (MGA_0491) present in M. gallisepticum was much more distant (pairwise distance: 0.613) ( Figure 4E ). Two additional features emerged from these graphs. First, both Type 1 and Type 2 homologs presented high pairwise distances from the Type 3 references, with the highest values obtained for Type 2 homologs. This suggests that Type 2 and Type 3 gene pairs have diverged considerably since their evolutionary separation. Second, the range of pairwise distances between homologs from the Hominis group was higher for Type 3 pairs than for F1F0 ATPase pairs. For instance, when M. agalactiae proteins were used as references, pairwise distances ranged from 0.252 to 0.625 (Δ = 0.373) for Type 3 ( Figure 4A ) but only from 0.234 to 0.363 (Δ = 0.129) for Type 1 ( Figure 4B ). Thus, even within the Hominis phylogenetic group, the level of divergence between Type 3 pairs was high, suggesting a complex evolutionary history marked by HGT and/or bursts of evolution.
Figure 4. Evolutionary distance between clusters.

Amino acid sequences of genes encoding α-like and β-like proteins of the Type 2 and Type 3 clusters (panels A, C and E) and α- and β-subunits of F1F0 Type 1 clusters (panels B, D and F) were concatenated and multiple alignments were generated. Multiple sequence alignments were curated with GBLOCK to remove unreliable sites and a final round of manual editing was performed with Jalview. Evolutionary distances were calculated with Type 3 pairs thought to have been exchanged through HGT as references. These distances are shown as a function of the evolutionary distance between species inferred from 16S rDNA data. The Type 3 pairs concerned were MAG2930/2940 (M. agalactiae), MHO_3130/3120 (M. hominis) and MGA_1321d (M. gallisepticum). In the last case, the analysis was based exclusively on the truncated atpD-like gene. Homologs from a phylogenetic group are circled: H, Hominis; P, Pneumoniae; S, Spiroplasma.
Five genes colocalize with extra atpA-like and atpD-like copies
We analysed the genomic context of Type 2 and Type 3 atpA-like and atpD-like genes. A genomic region surrounding Type 2 atpA-like/atpD-like genes and including nine syntenic genes was common to M. mobile and M. pulmonis ( Figure 5A ). Two genes located downstream from the atpA-like/atpD-like genes encoded a phosphate acetyltransferase (eutD) and an acetate kinase (ack). These tandemly associated genes were found at various locations on the chromosomes of most mycoplasmas, whether or not atpA-like/atpD-like genes were present. By contrast, the five genes upstream from the atpA-like/atpD-like genes encoded conserved hypothetical proteins. None presented homologs that could be identified by blastp search in other mollicutes or any other organism. These genes are listed in Table S2. For convenience, the seven genes of the cluster were arbitrarily numbered 1 to 7. Gene 5 of the Type 2 cluster in strain UABCTIP of M. pulmonis was interrupted by a stop codon [42], as confirmed by PCR and sequencing. However, this mutation was not found in other three strains of M. pulmonis (not shown).
Figure 5. Genomic contexts of Type 2 and Type 3 clusters in mycoplasmas.

Homologous genes are indicated by boxes of the same colour, connected by dashed lines. The genomic regions containing the Type 2 (A) and Type 3 (B) clusters are framed in red. The schematic diagram was generated from screenshots obtained from the MBGD database. Mnemonics and gene names are indicated; genes from the clusters are numbered arbitrarily from 1 to 7. The genome structures of M. mycoides subsp. capri and M. capricolum subsp. capricolum were identical to that of Mmm.
Five genes encoding hypothetical proteins were also conserved upstream from most Type 3 atpA-like/atpD-like genes ( Figure 5B , Table S2). Blastp queries retrieved no putative homologs for the proteins encoded by these five genes. In several species, these clusters of genes were preceded by genes encoding lipoproteins, transmembrane proteins and hypothetical proteins. Phylogenetic reconstructions suggested that genes 1 to 5 and some of these upstream genes may have been exchanged, together with the atpA-like/atpD-like genes, during HGT. Complete Type 3 clusters including seven non disrupted genes were found in eleven of the twelve species in which Type 3 atpA-like/atpD-like genes were identified. The only exception was M. gallisepticum (strain R-low), the genome of which contained a seven-gene cluster in which gene 5 presented a non-sense mutation that was checked by PCR and sequencing. This mutation was also found in the recently sequenced genome of a derived, non-pathogenic strain after a large number of passages (strain R-High), but vaccine strain F had an intact cluster indicating the recent nature of the mutation [44]. All sequenced M. gallisepticum genomes also contained a degraded cluster with only partial sequences of genes 1 and 7. Remarkably, the genes located on either side of the degraded cluster were found to be closely related to genes flanking one of the M. synoviae clusters (Figure S1). This observation is consistent with the hypothesis of HGT between bird mycoplasmas. Additional Type 3 clusters presumably split by genome re-organization events were found in M. arthritidis and M. pulmonis ( Figure 5B ). In particular, the gene encoding Protein 2 in M. pulmonis was missing from the first part of the cluster but was found next to the atpA-like/atpD-like genes, suggesting that this cluster may have been reshuffled by several successive events. Only M. synoviae harboured two complete Type 3 clusters.
In summary, both Type 2 and Type 3 atpA-like/atpD-like pairs of genes were found clustered with five genes encoding proteins of unknown functions with no obvious similarities to any other proteins from databases. These clusters of seven genes were found in all the species from the Hominis group, consistent with a common ancestry. Horizontal gene transfer may account for the presence of Type 3 clusters in species of the Pneumoniae and Spiroplasma phylogenetic groups.
Type 2 and Type 3 clusters: indications for an F1-likeX0 structure
The Type 2 and Type 3 gene clusters did not display strong sequence similarity, but they were organized similarly and proteins encoded by genes 1, 2, 3 and 4 were of the similar lengths in the two types ( Figures 6B and S2). We investigated the function of Type 2 and Type 3 clusters further, by analysing and comparing the predicted proteins. This analysis is illustrated in Figure S3 for proteins 1–7 from Mmm.
Figure 6. Model of an F1-likeX0 ATPase encoded by the seven-gene clusters of Types 2 and 3 specific to mycoplasmas.
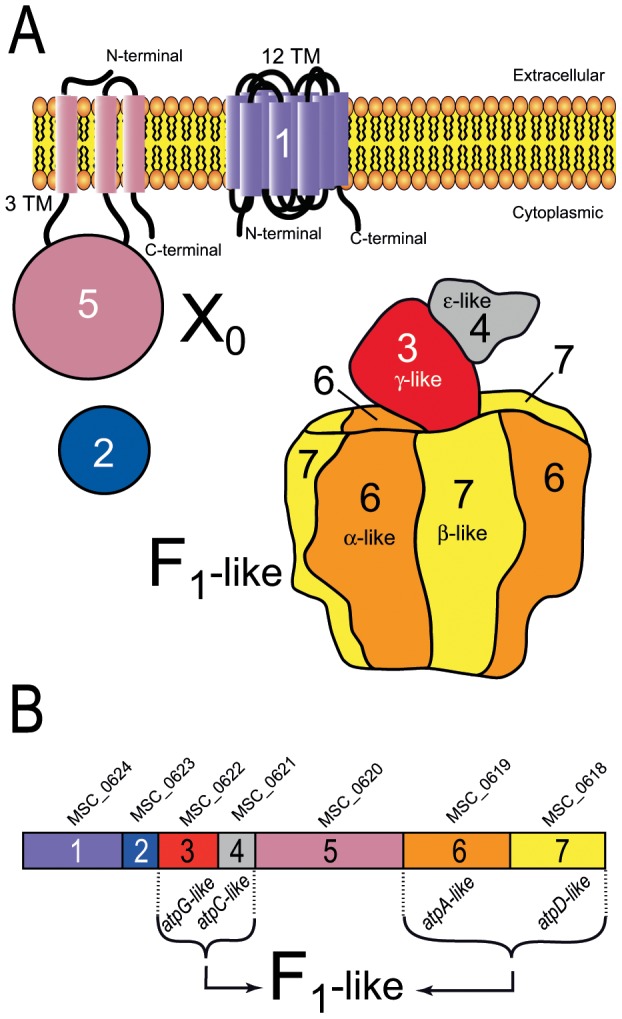
A. The F1-like complex model of Mmm was drawn by similarity with the crystal structure of the E. coli F1-ATPase (Pdb id: 3oaa) [27] with the help of the Pymol software (http://www.pymol.org) [28]. The X0 complex proteins of Mmm were schematized on the basis of 2D structure predictions. Proteins 1 and 5 are depicted associated with the membrane, in accordance with the predicted transmembrane segments. Based on in silico and experimental results, the F1-like complex, Protein 2 and the main part of Protein 5 were predicted to be cytoplasmic. Within this model, the F1-like and the X0 sectors are represented, but the way they could interact remains largely unclear. B. The genes of the clusters were arbitrarily numbered from 1 to 7. Gene names are indicated above the boxes representing the genes. TM, transmembrane segments. The proteins encoded by genes 3, 4, 6 and 7 were found to be related to the subunits γ, ε, α and β of the F1F0 ATPase, respectively.
In accordance with the atpA-like and atpD-like annotation found in most mycoplasma genomes, the primary and secondary structures of Proteins 6 and 7 displayed significant similarity to those of the α- and β-subunits of bacteria F1F0 ATPases, respectively. The presence of Walker consensus sequences A and B suggested that these proteins were able to bind nucleotides. The Walker A motif, also named P loop, interacts with the ATP phosphate groups while the Walker B motif is involved in ATP hydrolysis [45]. Protein 6 possessed the DELSEED-loop signature of the β-subunit, which is essential for the ε-subunit to play a role as an inhibitor [27], [46]. Protein 3 (∼36 kDa) displayed less than 22% and 14% amino-acid identity to γ-subunits of mycoplasma and other bacteria F1F0 ATPases, respectively. However, the predicted secondary structure included long helices at the N- and C-terminal ends, surrounding a mixed α-helix/β-strand domain (Figure S3C). Fold recognition software predicted a γ-subunit F1 ATPase with medium confidence (score = 41). Protein 4 was predicted to have eight strands at the N-terminal end and two helices at the C-terminal end (Figure S3D). Despite a very low level of sequence similarity to the ε-subunit of bacteria F1 ATPases (less than 11% similarity), the predicted secondary structure and the topology of Proteins 4 resembled the known structure of the ε-subunit of bacteria F1 ATPases. An ε-subunit fold was therefore predicted with a low level of confidence (score = 31).
Protein 7 (β-like) in cluster 2 (M. mobile and M. pulmonis) had an extension of about ∼30 kDa at the N-terminus. The resulting additional mass of ∼90 kDa located on top of the potential hetero-hexamer (αβ)3 might affect its connection with the peripheral stalk subunit. Moreover, no ORFs in clusters 2 and 3 were found to code the δ-subunit or b-subunit homologs that are essential for the formation of the peripheral stalk of the F1F0 ATPase. Without a peripheral stalk to anchor (αβ)3 to the membrane, the enzyme is likely to function as an ATPase rather than an ATP synthase. Together, Proteins 6, 7, 3 and 4 from both Type 2 and Type 3 could assemble into a functional complete F1-like ATPase ( Figure 6A ).
No sequence similarity was found between Proteins 2 and proteins of known function or structure. No transmembrane segment or signal peptide was predicted suggesting that Protein 2 was cytosolic and might therefore interact with the F1-like ATPase. Short (19 kDa) and long (35 kDa) versions of Protein 2 were found in Types 2 and 3, respectively. Based on molecular weight alone, the shorter version of the protein could be a homolog to the 19 kDa δ-subunit. However, its predicted secondary structure (Figure S3B) was clearly different from that of the δ-subunit which is an α-helix rich- protein.
According to TMHMM and TMPred predictions, Protein 1 (∼55 kDa) contained twelve transmembrane helices connected by short loops (Figure S3A) and both the N- and C-terminal ends were located on the cytoplasmic side of the membrane. Protein 5 was predicted to be anchored in the membrane through at least two transmembrane helices linked by a short loop near the C-terminus (amino acids 707–768 for MSC_0620 from Mmm, Figure S3E). The predicted secondary structure included N-terminal (amino acids 1–190) and C-terminal (aa 460–770) regions rich in helices and a central region (aa 190–460) containing both helices and strands. A cleavable (between positions 24 and 25) signal peptide mediating membrane targeting was predicted by SignalP-4.0 software. Finally, two models for the topology for Protein 5 were predicted depending on the program used. According to TMHMM predictions, Protein 5 is probably mostly exposed at the cell surface (aa 31–709), whereas TMpred predicted it to be mainly cytoplasmic using (Figure S3F). Fold recognition with pGenTHREADER software predicted no meaningful topology for Proteins 1 and 5. All the membrane-spanning segments of Proteins 1 and 5 were highly hydrophobic, with no charged residues as expected for rotor and stator F0 subunits. However, Proteins 1, 2 and 5 were clearly not homologous to any known F0 subunits of ATPases.
Experimental study of the Type 3 cluster in Mmm: evidence for membrane ATPase activity
We investigated whether the seven genes of the clusters related to F1 ATPase were actively expressed and whether they displayed any associated ATPase activity, by carrying out an experimental study on the Type 3 cluster of Mmm. This particular mycoplasma species was chosen because: (i) it is predicted to contain a single, apparently complete Type 3 cluster, (ii) Mmm is a major pathogen of ruminants and its biology and pathogenicity remain poorly understood, (iii) Mmm grows more rapidly than other mycoplasma species and several genetic tools have been developed for this species, including replicative oriC plasmids and transposon-based mutagenesis methods [47]. Proteins 1–7 of Mmm were named by their mnemonics MSC_0624-MSC_0618 (Table S2), with MSC_0618 and MSC_0619 corresponding to the β-like and α-like subunits, respectively.
The Type 3 cluster forms an operon in Mmm
In prokaryotic genomes, functionally related genes are frequently organized into operons. In silico predictions for operon structure were retrieved from the DOOR database (Database for prokaryotic operons, http://csbl1.bmb.uga.edu/OperonDB, [23]). The seven genes of Mmm Type 3 cluster were predicted to be organized into a single operon on the basis of several features including synteny and intergenic regions which were less than 6 nt in length. An operon structure was also predicted for all eight mycoplasma Type 3 clusters for which information was available from the DOOR database at the time of analysis. Interestingly, in both Mmm and other mollicutes (M. pulmonis, M. synoviae, U. parvum, M. agalactiae, M. capricolum subsp. capricolum, M. gallisepticum), the operon was predicted to contain several additional genes. Almost all these additional genes encoded proteins annotated as (conserved) hypothetical proteins or lipoproteins, with the exception of a glycyl-tRNA synthetase-encoding gene in M. pulmonis. In Mmm, the predicted operon contained the putative lipoprotein-encoding gene MSC_0625 and MSC_0626, encoding a predicted transmembrane protein, in addition to the Type 3 genes MSC_0618-MSC_0624. The actual sensitivity and specificity of the DOOR predictor for mycoplasma genomes remain unknown because no global experimental operon data are available for Type 3- and Type 2-containing genomes. However, only 10 to 18 operons of seven or more genes were predicted in all the genomes considered. This suggests that the prediction of operons is highly specific in these mycoplasma genomes.
Standard RT-PCR assays were then undertaken to demonstrate the production of transcripts overlapping the various CDS regions within the cluster in Mmm. Based on the DOOR predictions, we carried out nine RT-PCR assays with the primers listed in Table S1. An overview of the binding sites of the primers is provided in Figure 7A . Products of the expected size were obtained for eight out of nine RT-PCRs, confirming linked transcription of the open reading frames in the Type 3 gene cluster in Mmm. No product was detected when the MSC_0617-MSC_0618 intergenic region was amplified. These results suggest that the genes of the Type 3 cluster and at least three upstream genes constitute an operon expressed in Mmm.
Figure 7. Operon structure and expression of the genes of the Type 3 cluster in Mmm.

A. RT-PCR experiments were carried out on intergenic regions to demonstrate the co-transcription of the MSC_0618 to MSC_0627 genes. The region of the genome region surrounding the Type 3 cluster in Mmm is shown. Gene mnemonics and numbers are shown for the Type 3 cluster. The site of transposon insertion in the MSC_0619 disrupted mutant (Δ619) is indicated by an arrow. Expected sizes of the putative transcripts are indicated. Amplification products of the expected sizes were obtained with primers binding within and upstream from the cluster (+) but not downstream from the cluster (−). B. Immunodetection of proteins from the Type 3 cluster of T1/44 and Δ619. A control for membrane protein detection was included, in the form of an antibody against a membrane protein, LppQ (anti-LppQ serum kindly supplied by Prof. J. Frey). C. Nano-LC-MS/MS detection of Type 3 ATPase proteins. Numbers of scans and distinct peptides detected are indicated. D. Evaluation of the sensitivity of Protein 5 and β-like subunit to trypsin degradation. Intact and lysed cells of Mmm T1/44 were incubated with (+) or without (−) trypsin enzyme coated on beads for six hours. Protection against hydrolysis was assessed by immunodetection with antibodies raised against Protein 5 (MSC_0620) and β-like subunit (MSC_0618).
Protein production and localization in Mmm
We assessed the production of membrane-associated proteins from axenic cultures of Mmm grown in pyruvate-supplemented Hayflick culture medium. Membrane-enriched extracts were subjected to SDS-PAGE and western-blotting with polyclonal antibodies raised against synthetic peptides designed from the protein sequences of MSC_0618, MSC_0619 and MSC_0620. Each of the three polyclonal antibodies detected a single band corresponding to a protein of the expected size in the membrane protein extracts of Mmm ( Figure 7B ). The same western-blotting pattern was observed whatever the initial optical density at 640 nm of the mycoplasma culture batch (data not shown). These data suggest that MSC_0618-MSC_0620 proteins were constitutively produced by the mycoplasma cells in the culture conditions tested. All nine MSC_0618-MSC_0626 proteins were unambiguously identified by a proteomic approach based on nano-LC-MS/MS ( Figure 7C ) from the set of extrinsic and integral proteins detected in membrane protein extracts. Thus, Mmm produced the nine proteins of the cluster and all nine proteins remained associated with the membrane under the experimental conditions used to isolate membrane-enriched fractions.
As indicated above (Figure S3F), a cytoplasmic location was unambiguously predicted for five proteins, including the F1-like subunits MSC_0618 and MSC_0619. The other two proteins, MSC_0620 and MSC_0624, were predicted to contain transmembrane segments. The TMHMM and TMPred programs predicted twelve transmembrane segments with the N-terminus located in the cytoplasm for MSC_0624. For MSC_0620, no unambiguous topology model could be obtained with the prediction methods used. Indeed, TMHMM data suggested that amino-acid residues 31 to 709 were exposed at the surface, whereas residues 31–707 were predicted to be cytoplasmic by TMpred. We obtained experimental data for the orientation and topology of the putative ATPase complex, the membrane topology of MSC_0620 and the localization of the F1-like component MSC_0618, by carrying out limited proteolysis of Mmm cells with trypsin immobilized on agarose beads. The accessibility of proteins to trypsin digestion was assessed in intact cells and in cells disrupted by mild sonication. The proteolysis of MSC_0618 and MSC_0620 following the addition of trypsin was evaluated by western-blotting with the monospecific polyclonal antibodies MSC618002 and MSC620002, respectively. Under identical experimental conditions, no hydrolysis of MSC_0618 was observed after six hours ( Figure 7D ). By contrast, disruption of the cells before the addition of trypsin resulted in the nearly complete digestion of MSC_0618, suggesting that an intact plasma membrane was required to protect MSC_0618 against digestion. Thus, MSC_0618 (β-like subunit) is located in the cytoplasm, providing strong evidence in favour of a cytoplasmic location for the whole F1-like structure.
After treatment with trypsin for six hours, MSC_0620 digestion was observed in disrupted cells, but not in intact cells. As the polyclonal antibodies used in proteolysis experiments were raised against a polypeptide corresponding to amino acids 182–231, the trypsin accessibility assay indicates that this fragment was on the cytoplasmic side of the membrane. In the simplest topology model consistent with the experimental data, MSC_0620 is a protein exposed mostly on the cytoplasmic side of the membrane and possessing a membrane-anchoring domain consisting of two transmembrane segments at its C-terminal end. In this model, the orientation of MSC_0620 may allow it to interact with both the membrane and the F1-like structure of the complex.
An Mmm Δ619 mutant displays low levels of ATPase activity: the cluster is associated with ATPase activity
A previously obtained library of mutants generated by random insertion of the gentamycin resistance transposon Tn4001 in Mmm T1/44 [32] was screened for insertion in genes belonging to the Type 3 cluster. A mutant carrying Tn4001 inserted at position 307 of the MSC_0619 gene ( Figure 7A ) was identified by PCR as described in the Materials and Methods. The insertion of the 3.7 kbp transposon leaded to a predicted truncated protein of 108 amino acids whereas the complete protein includes 505 amino acids. Comparative growth studies were performed for preliminary characterization of the mutant phenotype. The T1/44 and Δ619 strains were grown in pyruvate-supplemented Hayflick medium at 37°C and the optical density at 640 nm was recorded over a period of 60 h. The time courses for growth were very similar, except that the cell density observed in stationary growth phase was repeatedly slightly lower for the mutant strain ( Figure 8A ). Both strains were able to acidify the culture medium, the pH decreasing from an initial value of 7.4 to 5.4 for T1/44, and to 5.8 for the mutant strain at t = 30 h.
Figure 8. Growth and ATPase activity of Mmm T1/44 and the Δ619 mutant.
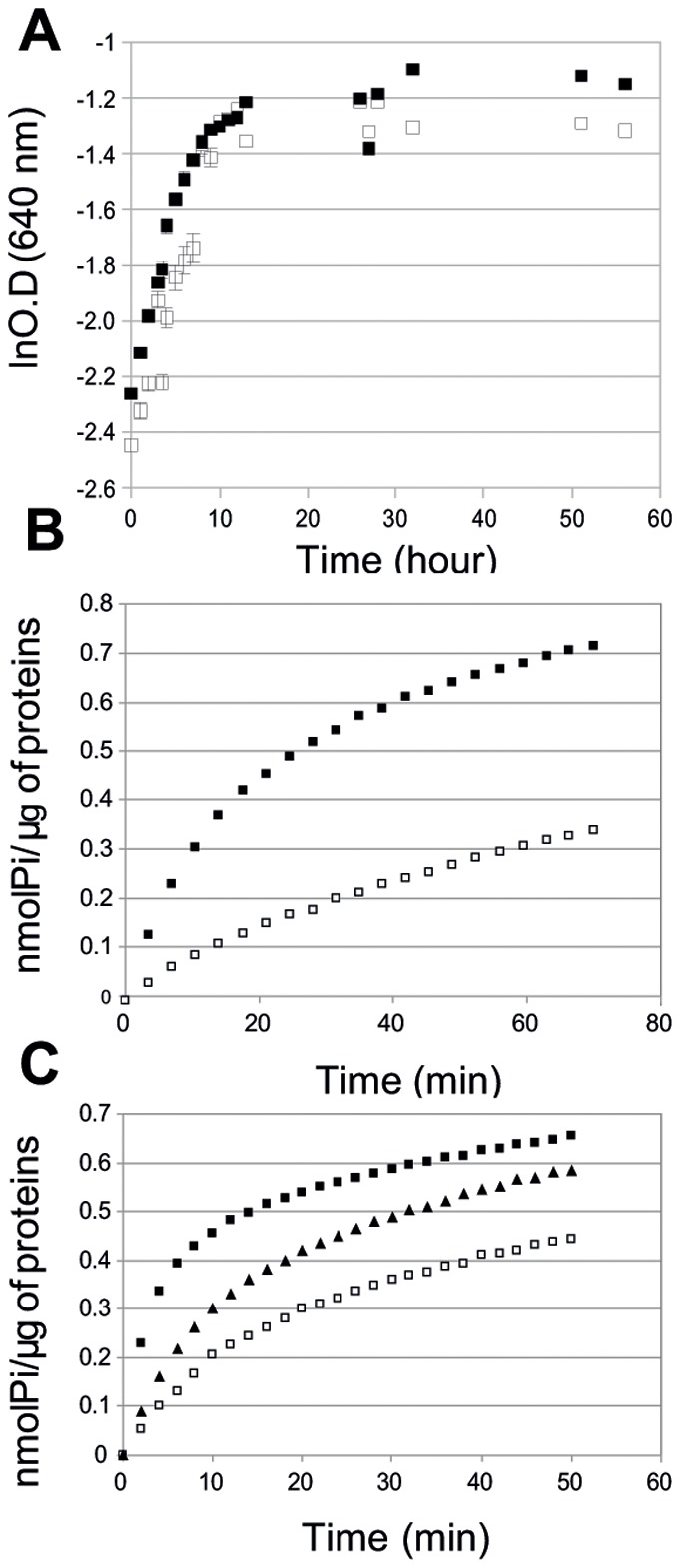
A. Growth of Mmm T1/44 (▪) and Δ619 (□) in Hayflick medium at 37°C. B. Rate of release of Pi from ATP in the presence of membrane preparations from T1/44 (▪) and Δ619 (□). The figure shows representative results of five independent experiments. C. Rate of release of Pi from ATP in the presence of membrane preparations from T1/44 (▪) and Δ619 (□) transformed with the control plasmid pMYSO1, and Δ619 complemented with the MSC_0619 (α-like) and MSC_0618 (β-like) proteins, generated from the plasmid pCC1 (▴).
We assessed expression of the MSC_0618-MSC_0626 genes in the Δ619 insertion mutant. As transcriptional analyses of the T1/44 provided strong evidence that the cluster containing genes MSC_0618 to MSC_0626 functioned as an operon, we expected the insertion of Tn4001 in MSC_0619 to affect expression only of MSC_0619 and MSC_0618, which is located immediately downstream from the gene carrying the inserted transposon. On a western-blot of membrane protein extracts from Δ619 cells, polyclonal antibodies directed against Protein 5 (MSC_0620) detected a band corresponding to a protein of the expected size for MSC_0620, indicating that this protein was produced in Δ619 ( Figure 7B ). However, MSC_0619 and MSC_0618 were not detected with the polyclonal antibodies. For confirmation of these results, we carried out LC-MS/MS analyses after separating the membrane-associated proteins by SDS-PAGE. This approach revealed the presence of the MSC_0620 to MSC_0626 proteins in the insertion mutant, but the absence of MSC_0619 or MSC_0618 ( Figure 7C ), consistent with the lack of expression of the two last genes of the cluster. This confirmed that the cluster MSC_0618-MSC_0626 functions as an operon.
In silico analyses strongly suggested a F1-likeX0 structure for the proteins encoded by the Type 3 cluster. We therefore investigated the roles of the proteins of the cluster in Mmm further, by evaluating the total ATPase activity of membrane extracts from axenic cultures of T1/44 and Δ619. The isolation of membrane-enriched fractions by sonication may lead to an artificial general loss of membrane protein activity. We assessed the ability of the membrane-enriched fractions to generate hydrogen peroxide in response to glycerol addition, as a means of evaluating the metabolic activity of these fractions. Indeed, in Mmm after import and phosphorylation of glycerol, the synthesis of H2O2 requires the activity of the membrane located enzyme L-α-glycerophosphate oxidase GlpO. In this control experiment, the membrane-enriched fractions obtained from the T1/44 and Δ619 strains produced H2O2 with similar kinetics, with specific rates of H2O2 production varying from 5 to 6 µmol. mg protein−1. h−1 for both strains. This indicated that the fractions prepared from both strains had similar overall levels of metabolic activity. ATPase activities as determined by an in vitro colorimetric assay are shown Figure 8B . In this experiment, we assessed the generation, by washed membrane vesicles, of Pi following the addition of ATP. The membrane-enriched fractions of Δ619 displayed significantly lower (p<0.05) levels of total ATPase activity than those of T1/44. Activity in Δ619 was only 20% that of T1/44, indicating strong correlation between MSC_0619 inactivation and the large loss of ATPase activity in membrane-enriched fractions. These data suggest that MSC_0618 and/or MSC_0619 proteins are involved in membrane-associated total ATPase activity. We tested this hypothesis, by carrying out a complementation assay. Mmm Δ619 was complemented with the pCC2 plasmid carrying MSC_0618 and MSC_0619. Both α-like and β-like proteins were produced in the complemented strain as shown by LC-MS/MS (not shown). Complementation partially restored the total ATPase activity of membrane-enriched fractions ( Figure 8C ), thus confirming the involvement of MSC_0618 and MSC_0619 in the maintenance of total ATPase activity in isolated membrane fractions.
Discussion
Evolution of Type 2 and Type 3 clusters
In addition to the typical operon encoding Type 1 F1F0 ATPase, an analysis of mycoplasma genomes detected clusters of seven genes, four of which were predicted to encode proteins related to the α-, β-, γ- and ε-subunits of the F1F0 ATPase.
The phylogenetic reconstructions inferred from the α- and β-subunits and comparisons of genomic contexts showed a distribution of these clusters into two well supported groups, identified here as Type 2 and Type 3. F1-like sectors potentially assembled from these clusters shared a common ancestry with genuine F1F0 ATPase, N-ATPases and, more distantly, with other ATPases such as V-type ATPase and type 3 secretion systems. The relative positions of the branches corresponding to Type 1 (F1F0 ATPases), Type 1′ (N-ATPases), Type 2 and Type 3 ATPases remain unclear, but the monophyletic distributions of the Type 2 and Type 3 ATPases indicated that they had not recently emerged by sporadic duplications in particular mycoplasma species.
For Type 3 ATPases, clustering was not entirely consistent with the three well established phylogenetic groups of mycoplasmas (Spiroplasma, Pneumoniae and Hominis). Moreover, Type 3 clusters in the same genome were not always clustered on sister branches, and therefore cannot be assumed to have arisen through recent duplication events. Sequence variability and the evolutionary distances calculated for β-subunits suggested that Type 2 and Type 3 ATPases evolved more rapidly than Type 1 F1F0 ATPases. This more rapid evolution of Types 2 and 3 ATPases was particularly marked for Proteins 1, 2, 3, 4 and 5, as blast queries did not even detect reciprocal relatedness. All of these data suggested a complex evolutionary scenario including HGT and a remarkably fast evolution of protein sequences.
The distribution of Type 3 ATPases among mycoplasmas suggested that the ancestor of the Hominis branch had this specific ATPase. Nearly all other Type 3 ATPases found in mycoplasmas from other phylogenetic groups were highly related with homologs from species of the Hominis group. This suggested a scenario with spreading from this group to the others by HGT. Only one single Type 3 ATPase of the bird pathogen M. gallisepticum failed to show any closely related homolog in the Hominis group. However, even in this particular case, the phylogenetic tree indicated a close relationship with Type 3 ATPases from the Hominis group, consistent with a hypothetical acquisition from this group. Interestingly, the other Type 3 cluster in M. gallisepticum genome is highly degraded but very closely related to a complete cluster in M. synoviae, another bird pathogen. This suggests that a large deletion occurred during or after gene transfer into M. gallisepticum. The phylogenetic reconstructions and analysis of genomic contexts also suggested HGT of Type 3 ATPases between ruminant mycoplasmas (M. agalactiae and species from the mycoides group) and between human mycoplasmas (M. hominis and U. parvum). In these cases, gene exchanges may have been extended to a pair of genes, located upstream of the Type 3 cluster and directly repeated up to four times (M. capricolum subsp. capricolum and M. mycoides subsp. capri) at this locus. These genes encoded a transmembrane protein with a predicted bacteriocin motif and a lipoprotein with a serine-protease domain. Such genes might be involved in pathogenicity, but they were not found systematically in the vicinity of Type 3 clusters. Thus, despite the fact that they may have been exchanged during the same HGT event, the functional relationship between these two groups of genes remains fairly hypothetical.
Several genomes are now available for some species harbouring Type 3 clusters (M. gallisepticum, M. hyopneumoniae, U. urealyticum/parvum, M. agalactiae, Mmm and M. mycoides subsp. capri). In all cases, the Type 3 clusters were found at the same locus in all strains. This indicated that HGT involving Type 3 clusters pre-dated intra-specific diversification. This observation also suggested that Type 3 ATPase spreading by HGT is probably much less frequent than movements of typical mobile elements (IS, ICE, RM) which occurrences and localizations often vary from one strain to another. No clear co-localization of the Type 3 clusters and mobile elements could be shown. However, we noticed that modifications of the chromosomal structure frequently occurred near the Type 3 locus for mycoplasmas from the mycoides group, including insertion of an ICE (M. mycoides subsp. capri GM12) or an IS (Mmm PG1) and a chromosomal inversion (M. capricolum subsp. capricolum). In M. agalactiae strain 5632, an ICE was also located upstream of the Type 3 cluster. These observations suggested that Type 3 clusters may tend to localize within highly dynamic chromosomal regions.
The N-type ATPases were found in the archaea Methanosarcina spp., in various marine bacteria and in pathogenic Burkholderia spp [11]. By contrast to Type 2 and Type 3 ATPases, N-type ATPases were encoded by operons containing genes typical of both F1 and F0 subunits. The phylogenetic trees ( Figure 2 ) confirmed their specific evolution from F-type ancestor. The N-type ATPase operons were always found in genomes in addition to the genuine F1F0 ATPase operon and an evolution including HGT was suspected. The presence of N-type ATPase operons on plasmids of marine bacteria is in accordance with this hypothesis. Thus, despite Type 2 and Type 3 ATPases are only distantly related to N-type ATPases, they share some features that might be significant of their biological role in the adaptation to the environment.
Functionality and expression of Type 2 and Type 3 clusters
In most species, the genes encoding Type 3 ATPases were found to be intact, suggesting biological significance. However, mutants with transposons integrated into the genes of Type 3 clusters were obtained by random mutagenesis in M. pulmonis [8] and M. arthritidis [10]. We also isolated an Mmm mutant lacking α-like and β-like subunits. These data demonstrate that Type 3 ATPases are not essential for life in axenic media. Moreover, the Mmm Δ619 did not grow significantly slower than T1/44, in axenic medium. This suggests that Type 3 ATPases are not involved in the central metabolism of mycoplasmas, instead possibly playing an adaptive role during interactions with the host.
Proteomic data for Mmm indicated that the seven genes of the Type 3 cluster were expressed in vitro. In all other mycoplasma species for which transcriptomic or proteomic data are available, the Type 2 and Type 3 clusters were found to be expressed. This was the case in M. mobile (Type 2 and Type 3) [43], [48], M. agalactiae strain 5632 [49] and Mycoplasma hyopneumoniae [50], [51].
In Mmm, RT-PCR and bioinformatics analyses indicated that the genes of the Type 3 cluster were organized into an operon that also included several upstream genes. The predicted horizontal transfer of these seven genes as a single block is also consistent with the idea they are functionally associated. Transcriptomic data obtained by Oneal et al. for M. hyopneumoniae showed that the expression of six of seven genes was increased by norepinephrine [52]. This stress hormone is produced by mammalian hosts during bacterial infections. Several bacterial species have been shown to respond to norepinephrine by an increase in growth rate and induction of virulence factors [53], [54]. This work showed not only that these genes may be coregulated, but also that two genes of the Type 3 cluster displayed more than 2-fold activation. The biological significance of this activation remains unclear, but these results suggest that Type 3 ATPase gene clusters maybe regulated during host-pathogen interactions.
The relationship between Type 2 and Type 3 ATPases and possible biological functions
The organizations of the cluster and the structural features of the predicted proteins strongly suggested a common ancestry for Type 2 and Type 3 predicted ATPases. Nevertheless, with the exception of highly conserved subunits, namely the α-like and β-like subunits, the predicted Type 2 and Type 3 cluster proteins displayed only very low level of sequence similarity. Moreover, specific 200–300 amino acid-long N-terminal extensions were observed on the β-like subunits of Type 2. These differences raised questions about the possible biological functions common to these ATPase types. Interestingly, six of the seven proteins of the M. mobile Type 2 ATPase were associated with an intracellular “jellyfish” structure [48] connected to the complex gliding machinery on the external side of M. mobile cells [55]. Type 2 α-like and β-like subunits were found to be located within the cell. Our data also suggested that Type 3 ATPases were present within the cells.
The Type 2 and Type 3 clusters each encode four proteins structurally related to subunits α, β, γ and ε that could assemble into a structure similar to the F1 sectors of F-type ATPases. This prediction is consistent with functional assays showing that the Type 3 cluster is associated with a membrane ATPase activity in Mmm. Thus, there seems to be a clear relationship between F1-like structures and ATP hydrolysis.
Two general functions have been associated with structures including proteins related to the α- and β-subunits of ATPases: (1) H+/Na+ pumps associated with ATPase activities (F-ATPases, N-ATPases and V-ATPases) and, (2) translocation systems (T3SS, T4SS and flagellum). In silico analysis of the proteins encoded by Type 2 and Type 3 clusters but not predicted to have an F1-like structure (i.e. Proteins 1, 2 and 5) identified no sequence or structural similarity to any protein involved in ion transport or translocation systems. Therefore, the biological significance of Type 2 and Type 3 ATPases remains largely unknown.
One first hypothesis regarding to the cellular function of the Type 2 and Type 3 ATPases is an ion transport activity, as documented for F1F0 ATPases and N-ATPases. However, this would imply that F1-like structures of Type 2 and Type 3 ATPases are associated with a membrane complex functionally equivalent to the F0 sector. The three remaining proteins encoded by both Type 2 and Type 3 clusters did not show any sequence or structural similarity with the subunits a, b and c that form the F0 sector of F-type ATPases. Protein 5 is a large, highly structured protein with three predicted transmembrane segments, suggesting it is anchored in the membrane with a main part of the protein being intracellular and potentially available for other interactions. Thus, Protein 5 could be the main part of a peripheral stalk analogous to that formed by a and b proteins of F0 sectors. In F1F0 ATPases, the peripheral stalk maintains the (αβ)3 complex, leading to the rotation of γ and the membrane-located c ring [56]. In V-Type ATPases, an analogous structure was found, despite any homology with F-type proteins was demonstrated. The existence of analogous structures in prokaryotes suggests that several structures have evolved to form a functional peripheral stalk. Whether this is the case for Protein 5 or not remains unclear. In many bacterial F1F0 ATPases, the juxtaposition of 12 proteins c form a ring inserted in the membrane, each c monomer containing two transmembrane segments. This ring is connected with subunits γ and ε. Twelve transmembrane helices were predicted in the Protein 1 of Type 2 and Type 3 ATPases. An evolutionary scenario involving duplications and fusions of gene encoding c protein is also tempting but we found no repeated motifs that could argue in favour of this hypothesis. The Protein 2 has the size of a delta subunit but, we found no sequence or structural similarity between those two. Therefore, the appealing hypothesis that Proteins 1, 2 and 5 could form a structure functionally analogous to F0 remains purely speculative.
A second hypothesis proposes that Type 2 and/or Type 3 F1-like structures would hydrolyse ATP to provide the energy required for a translocation system. This would bring these mycoplasmal ATPases closer to bacterial T3SS, T4SS and flagellum apparatus. The Type 2 and Type 3 ATPases are phylogenetically close to F1F0 ATPases, compared with these prokaryotic translocation systems. However, the possibility of an evolutionary convergence cannot be formally rejected. Interestingly, a translocation function was proposed by Nakane et al for the Type 2 α-like and β-like subunits of M. mobile [48]. In this scheme, Type 2 ATPase could be in charge of translocating gliding proteins. In favour of such proposal is the structure of Protein 1 which 12 transmembrane segments are reminiscent of transporter permeases.
In some evolutionary scenario, it has been proposed that ancestors of F- and V-type ATPases could have a translocation function, first of RNA then of proteins [57]. In such scenario, the γ subunit found in F1F0 ATPases would originate from a protein which translocation would have finally been blocked. Therefore, it might seem difficult to propose a translocation function for Type 2 and/or Type 3 ATPase, given that a γ-like protein was clearly identified. Nevertheless, a recent study of the FliJ protein that is essential for export of flagellum proteins in Salmonella enterica serovar Typhimurium showed a remarkable similarity with part of γ subunits of F-type ATPases [58]. Thus, a translocation function of molecules with high molecular weight (i.e. proteins, ADN or ARN) might not be ruled out for Type 2 and/or Type 3 ATPases.
Both in silico and experimental approaches indicated that Proteins 1 and 5 were anchored to the cytoplasmic membrane, suggesting a hypothetical X0 structure that might be physically connected to the F1-like structure. Furthermore, a possible interaction of F1-like structures with the F0 sector of genuine F1F0 ATPases cannot be ruled out. Indeed, in experimental conditions, the disruption of MSC_0619 led to the loss of about 80% of the total ATPase activities of membrane fractions from Mmm. This may not accurately reflect the contribution of Type 3 ATPase activity in vivo, but it does suggest that Type 3 ATPases are able to modify significantly the global ATP consumption associated with mycoplasmal membranes. A role of α–like and β–like subunits in the structural organization of the F1F0 ATPase complex might account for the much lower total ATPase activity recorded for membranes isolated from the mutant lacking the F1-like components.
Conclusion
In summary, we have shown that clusters of seven genes including four related to the F1 sectors of F1F0 ATPases are present in the genomes of many mycoplasma species, in addition to the typical F1F0 ATPase operon. Two types of potential ATPases were described, Type 2 and Type 3, both evolving rapidly, with a complex pattern of evolution for Type 3, involving probable HGT. Our studies confirmed that the Type 3 cluster present in Mmm encoded a functional membrane ATPase. This evolution of F1-like structures is specific of mycoplasmas, with no apparent equivalent in other bacteria. The biological functions of Type 2 and Type 3 ATPases remain unclear, but the remarkably rapid evolution of these F1-like structures and their probable association with membrane proteins unrelated to all known ion pump and translocation systems provides insights into the ability of mycoplasmas to recycle and modify universally conserved proteins for the development of novel functional associations.
Supporting Information
Primers used in this study.
(DOC)
Genes included in the Type 2 and Type 3 clusters.
(DOC)
Type 3 cluster region exchanged between bird mycoplasmas. The complete Type 3 cluster in M. synoviae and the deleted form present in M. gallisepticum are indicated under the bracket. The xenologous regions are connected by yellow bands. The schematic diagram was composed from screenshots obtained from the MBGD database. HP, Hypothetical Protein.
(TIF)
Conserved positions in Type 2 and Type 3 proteins. Concatenated amino acid sequences of the clusters were aligned by MAFFT. Overview of the conserved positions over the cluster was obtained by Jalview. Genes from the ATPase cluster were represented as coloured boxes on the top of the diagram. Conserved positions were coloured as blue bars. Identity threshold for colouring was 50%. Red bars indicate genes boundaries.
(TIF)
Secondary structure of Proteins 1 to 5 from Mmm . (A) Protein 1 (MSC_0624) contained twelve transmembrane helices. (B) Protein 2 (MSC_0623) is a α-helix rich protein. (C) Protein 3 (MSC_0622) presented long N-terminus (amino acids 2 to 84) and C-terminus (amino acids 222 to 290) helices surrounding a mixed α-helix/β-strand region (amino acids 85 to 221). (D) Protein 4 (MSC_0621) contained eight strands (amino acids 3 to 90) followed by two helices (amino acids 96 to 143). (E) Protein 5 (MSC_0620) displays N-terminus (amino acids 1 to 190) and C-terminus (amino acids 460 to 770) regions rich in helices and a central region (amino acids 190 to 460) with both helices and strands. It was predicted anchored into the membrane through at least two transmembrane helices near the C-terminus (amino acids 707 to 768). (F) Two topology models for MSC_0620 were predicted. TMHMM (left) suggested that Protein 5 was mainly (amino acids 31 to 709) surface-exposed while it was predicted to be mainly cytoplasmic using TMpred (right). The panels (B–E) were composed from PSIPRED drawings.
(TIF)
Acknowledgments
We thank Antony Cougnoux and Jean-Baptiste Guyon for technical support, Sandrine Eveillard for precious technical advices and Anthony Levasseur for fruitful discussions on the evolution of F1-like ATPases. We also thank Prof. Joachim Frey for providing the anti-LppQ serum used in this work.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by grants from the Institut National de Recherche Agronomique (http://www.inra.fr/), the Région Aquitaine (http://aquitaine.fr/), the University Bordeaux Segalen (http://www.univ-bordeauxsegalen.fr/fr/index.html) and the Agence Nationale pour la Recherche (grant EVOLMYCO ANR-07-GMGE-001, http://www.agence-nationale-recherche.fr/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Razin S, Yogev D, Naot Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol Mol Biol Rev. 1998;62:1156. doi: 10.1128/mmbr.62.4.1094-1156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sirand-Pugnet P, Citti C, Barre A, Blanchard A. Evolution of mollicutes: down a bumpy road with twists and turns. Res Microbiol. 2007;158:766. doi: 10.1016/j.resmic.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Pereyre S, Sirand-Pugnet P, Beven L, Charron A, Renaudin H. Life on arginine for Mycoplasma hominis: clues from its minimal genome and comparison with other human urogenital mycoplasmas. PLoS Genet. 2009;5:e1000677. doi: 10.1371/journal.pgen.1000677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciccarelli FD, Doerks T, von Mering C, Creevey CJ, Snel B. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311:1287. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- 5.Vasconcelos AT, Ferreira HB, Bizarro CV, Bonatto SL, Carvalho MO. Swine and poultry pathogens: the complete genome sequences of two strains of Mycoplasma hyopneumoniae and a strain of Mycoplasma synoviae. J Bacteriol. 2005;187:5577. doi: 10.1128/JB.187.16.5568-5577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sirand-Pugnet P, Lartigue C, Marenda M, Jacob D, Barre A. Being pathogenic, plastic, and sexual while living with a nearly minimal bacterial genome. PLoS Genet. 2007;3:e75. doi: 10.1371/journal.pgen.0030075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shirvan MH, Rottem S. Ion pumps and volume regulation in mycoplasma. Subcell Biochem. 1993;20:292. doi: 10.1007/978-1-4615-2924-8_10. [DOI] [PubMed] [Google Scholar]
- 8.Dybvig K, Lao P, Jordan DS, Simmons WL. Fewer essential genes in mycoplasmas than previous studies suggest. FEMS Microbiol Lett. 2010;311:55. doi: 10.1111/j.1574-6968.2010.02078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hutchison CA, Peterson SN, Gill SR, Cline RT, White O. Global transposon mutagenesis and a minimal Mycoplasma genome. Science. 1999;286:2169. doi: 10.1126/science.286.5447.2165. [DOI] [PubMed] [Google Scholar]
- 10.Dybvig K, Zuhua C, Lao P, Jordan DS, French CT. Genome of Mycoplasma arthritidis. Infect Immun. 2008;76:4008. doi: 10.1128/IAI.00516-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dibrova DV, Galperin MY, Mulkidjanian AY. Characterization of the N-ATPase, a distinct, laterally transferred Na+-translocating form of the bacterial F-type membrane ATPase. Bioinformatics. 2010;26:1476. doi: 10.1093/bioinformatics/btq234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barré A, de Daruvar A, Blanchard A. MolliGen, a database dedicated to the comparative genomics of Mollicutes. Nucleic Acids Res 32 Database issue. 2004. pp. 310. [DOI] [PMC free article] [PubMed]
- 13.Uchiyama I, Higuchi T, Kawai M. MBGD update 2010: toward a comprehensive resource for exploring microbial genome diversity. Nucleic Acids Res. 2010;38:365. doi: 10.1093/nar/gkp948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9:298. doi: 10.1093/bib/bbn013. [DOI] [PubMed] [Google Scholar]
- 15.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 17.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 19.Gouy M, Guindon S, Gascuel O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 20.Guindon S, Delsuc F, Dufayard JF, Gascuel O. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol. 2009;537:137. doi: 10.1007/978-1-59745-251-9_6. [DOI] [PubMed] [Google Scholar]
- 21.Tamura K, Peterson D, Peterson N, Stecher G, Nei M. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abascal F, Zardoya R, Posada D. ProtTest: selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 23.Mao F, Dam P, Chou J, Olman V, Xu Y. DOOR: a database for prokaryotic operons. Nucleic Acids Res. 2009;37:463. doi: 10.1093/nar/gkn757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hofmann K, Stoffel W. TMbase – A database of membrane spanning proteins segments. Biol Chem Hoppe-Seyler. 1993;347:166. [Google Scholar]
- 25.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 26.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 27.Cingolani G, Duncan TM. Structure of the ATP synthase catalytic complex (F(1)) from Escherichia coli in an autoinhibited conformation. Nat Struct Mol Biol. 2011;18:707. doi: 10.1038/nsmb.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeLano WL editor. The PyMOL Molecular Graphics System San Carlos, CA, USA: DeLano Scientific. 2002.
- 29.Rastogi VK, Girvin ME. Structural changes linked to proton translocation by subunit c of the ATP synthase. Nature. 1999;402:268. doi: 10.1038/46224. [DOI] [PubMed] [Google Scholar]
- 30.Del Rizzo PA, Bi Y, Dunn SD, Shilton BH. The “second stalk” of Escherichia coli ATP synthase: structure of the isolated dimerization domain. Biochemistry. 2002;41:6884. doi: 10.1021/bi025736i. [DOI] [PubMed] [Google Scholar]
- 31.Freund EA. Razin S, Tully JG, editors. Culture media for classic mycoplasmas. 1983. Methods in Mycoplasmology. San Diego: Academic Press.127.
- 32.Janis C, Bischof D, Gourgues G, Frey J, Blanchard A. Unmarked insertional mutagenesis in the bovine pathogen Mycoplasma mycoides subsp. mycoides SC: characterization of a lppQ mutant. Microbiology. 2008;154:2436. doi: 10.1099/mic.0.2008/017640-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lartigue C, Blanchard A, Renaudin J, Thiaucourt F, Sirand-Pugnet P. Host specificity of mollicutes oriC plasmids: functional analysis of replication origin. Nucleic Acids Res. 2003;31:6618. doi: 10.1093/nar/gkg848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.King KW, Dybvig K. Transformation of Mycoplasma capricolum and examination of DNA restriction modification in M. capricolum and Mycoplasma mycoides subsp. mycoides. Plasmid. 1994;31:311. doi: 10.1006/plas.1994.1033. [DOI] [PubMed] [Google Scholar]
- 35.Bischof DF, Janis C, Vilei EM, Bertoni G, Frey J. Cytotoxicity of Mycoplasma mycoides subsp. mycoides small colony type to bovine epithelial cells. Infect Immun. 2008;76:269. doi: 10.1128/IAI.00938-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vilei EM, Frey J. Genetic and biochemical characterization of glycerol uptake in Mycoplasma mycoides subsp. mycoides SC: its impact on H(2)O(2) production and virulence. Clin Diagn Lab Immunol. 2001;8:92. doi: 10.1128/CDLI.8.1.85-92.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kall L, Krogh A, Sonnhammer EL. Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res. 2007;35:432. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hamsten C, Neiman M, Schwenk JM, Hamsten M, March JB. Recombinant surface proteomics as a tool to analyze humoral immune responses in bovines infected by Mycoplasma mycoides subsp. mycoides small colony type. Mol Cell Proteomics. 2009;8:2554. doi: 10.1074/mcp.M900009-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nilsson P, Paavilainen L, Larsson K, Odling J, Sundberg M. Towards a human proteome atlas: high-throughput generation of mono-specific antibodies for tissue profiling. Proteomics. 2005;5:4337. doi: 10.1002/pmic.200500072. [DOI] [PubMed] [Google Scholar]
- 40.Abdo EM, Nicolet J, Frey J. Antigenic and genetic characterization of lipoprotein LppQ from Mycoplasma mycoides subsp. mycoides SC. Clin Diagn Lab Immunol. 2000;7:595. doi: 10.1128/cdli.7.4.588-595.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 42.Chambaud I, Heilig R, Ferris S, Barbe V, Samson D. The complete genome sequence of the murine respiratory pathogen Mycoplasma pulmonis. Nucleic Acids Res. 2001;29:2153. doi: 10.1093/nar/29.10.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaffe JD, Stange-Thomann N, Smith C, DeCaprio D, Fisher S. The complete genome and proteome of Mycoplasma mobile. Genome Res. 2004;14:1461. doi: 10.1101/gr.2674004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szczepanek SM, Tulman ER, Gorton TS, Liao X, Lu Z. Comparative genomic analyses of attenuated strains of Mycoplasma gallisepticum. Infect Immun. 2010;78:1771. doi: 10.1128/IAI.01172-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. Embo J. 1982;1:951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hara KY, Kato-Yamada Y, Kikuchi Y, Hisabori T, Yoshida M. The role of the betaDELSEED motif of F1-ATPase: propagation of the inhibitory effect of the epsilon subunit. J Biol Chem. 2001;276:23973. doi: 10.1074/jbc.M009303200. [DOI] [PubMed] [Google Scholar]
- 47.Janis C, Lartigue C, Frey J, Wroblewski H, Thiaucourt F. Versatile use of oriC plasmids for functional genomics of Mycoplasma capricolum subsp. capricolum. Appl Environ Microbiol. 2005;71:2893. doi: 10.1128/AEM.71.6.2888-2893.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakane D, Miyata M. Cytoskeletal “jellyfish” structure of Mycoplasma mobile. Proc Natl Acad Sci U S A. 2007;104:19523. doi: 10.1073/pnas.0704280104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nouvel LX, Sirand-Pugnet P, Marenda MS, Sagne E, Barbe V. Comparative genomic and proteomic analyses of two Mycoplasma agalactiae strains: clues to the macro- and micro-events that are shaping mycoplasma diversity. BMC Genomics. 2010;11:86. doi: 10.1186/1471-2164-11-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pinto PM, Chemale G, de Castro LA, Costa AP, Kich JD. Proteomic survey of the pathogenic Mycoplasma hyopneumoniae strain 7448 and identification of novel post-translationally modified and antigenic proteins. Vet Microbiol. 2007;121:93. doi: 10.1016/j.vetmic.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 51.Pinto PM, Klein CS, Zaha A, Ferreira HB. Comparative proteomic analysis of pathogenic and non-pathogenic strains from the swine pathogen Mycoplasma hyopneumoniae. Proteome Sci. 2009;7:45. doi: 10.1186/1477-5956-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oneal MJ, Schafer ER, Madsen ML, Minion FC. Global transcriptional analysis of Mycoplasma hyopneumoniae following exposure to norepinephrine. Microbiology. 2008;154:2588. doi: 10.1099/mic.0.2008/020230-0. [DOI] [PubMed] [Google Scholar]
- 53.Voigt W, Fruth A, Tschape H, Reissbrodt R, Williams PH. Enterobacterial autoinducer of growth enhances shiga toxin production by enterohemorrhagic Escherichia coli. J Clin Microbiol. 2006;44:2249. doi: 10.1128/JCM.00330-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freestone PP, Haigh RD, Lyte M. Specificity of catecholamine-induced growth in Escherichia coli O157: H7, Salmonella enterica and Yersinia enterocolitica. FEMS Microbiol Lett. 2007;269:228. doi: 10.1111/j.1574-6968.2006.00619.x. [DOI] [PubMed] [Google Scholar]
- 55.Miyata M. Centipede and inchworm models to explain Mycoplasma gliding. Trends Microbiol. 2008;16:12. doi: 10.1016/j.tim.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 56.Rees DM, Leslie AG, Walker JE. The structure of the membrane extrinsic region of bovine ATP synthase. Proc Natl Acad Sci U S A. 2009;106:21601. doi: 10.1073/pnas.0910365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mulkidjanian AY, Makarova KS, Galperin MY, Koonin EV. Inventing the dynamo machine: the evolution of the F-type and V-type ATPases. Nat Rev Microbiol. 2007;5:899. doi: 10.1038/nrmicro1767. [DOI] [PubMed] [Google Scholar]
- 58.Ibuki T, Imada K, Minamino T, Kato T, Miyata T. Common architecture of the flagellar type III protein export apparatus and F- and V-type ATPases. Nat Struct Mol Biol. 2011;18:282. doi: 10.1038/nsmb.1977. [DOI] [PubMed] [Google Scholar]
- 59.Papazisi L, Gorton TS, Kutish G, Markham PF, Browning GF. The complete genome sequence of the avian pathogen Mycoplasma gallisepticum strain R(low). Microbiology. 2003;149:2316. doi: 10.1099/mic.0.26427-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used in this study.
(DOC)
Genes included in the Type 2 and Type 3 clusters.
(DOC)
Type 3 cluster region exchanged between bird mycoplasmas. The complete Type 3 cluster in M. synoviae and the deleted form present in M. gallisepticum are indicated under the bracket. The xenologous regions are connected by yellow bands. The schematic diagram was composed from screenshots obtained from the MBGD database. HP, Hypothetical Protein.
(TIF)
Conserved positions in Type 2 and Type 3 proteins. Concatenated amino acid sequences of the clusters were aligned by MAFFT. Overview of the conserved positions over the cluster was obtained by Jalview. Genes from the ATPase cluster were represented as coloured boxes on the top of the diagram. Conserved positions were coloured as blue bars. Identity threshold for colouring was 50%. Red bars indicate genes boundaries.
(TIF)
Secondary structure of Proteins 1 to 5 from Mmm . (A) Protein 1 (MSC_0624) contained twelve transmembrane helices. (B) Protein 2 (MSC_0623) is a α-helix rich protein. (C) Protein 3 (MSC_0622) presented long N-terminus (amino acids 2 to 84) and C-terminus (amino acids 222 to 290) helices surrounding a mixed α-helix/β-strand region (amino acids 85 to 221). (D) Protein 4 (MSC_0621) contained eight strands (amino acids 3 to 90) followed by two helices (amino acids 96 to 143). (E) Protein 5 (MSC_0620) displays N-terminus (amino acids 1 to 190) and C-terminus (amino acids 460 to 770) regions rich in helices and a central region (amino acids 190 to 460) with both helices and strands. It was predicted anchored into the membrane through at least two transmembrane helices near the C-terminus (amino acids 707 to 768). (F) Two topology models for MSC_0620 were predicted. TMHMM (left) suggested that Protein 5 was mainly (amino acids 31 to 709) surface-exposed while it was predicted to be mainly cytoplasmic using TMpred (right). The panels (B–E) were composed from PSIPRED drawings.
(TIF)