

Abstract
Using a fibrin-based angiogenesis model, we have established that there is no canonical mechanism used by ECs to degrade the surrounding extracellular matrix (ECM), but rather the set of proteases used is dependent on the mural cells providing the angiogenic cues. Mesenchymal stem cells (MSCs) originating from different tissues, which are thought to be phenotypically similar, promote angiogenesis through distinct mechanisms. Specifically, adipose-derived stem cells (ASCs) promote utilization of the plasminogen activator-plasmin axis by ECs as the primary means of vessel invasion and elongation in fibrin. Matrix metalloproteinases (MMPs) serve a purpose in regulating capillary diameter and possibly in stabilizing the nascent vessels. These proteolytic mechanisms are more akin to those involved in fibroblast-mediated angiogenesis than to those in bone marrow-derived stem cell (BMSC)-mediated angiogenesis. In addition, expression patterns of angiogenic factors such as urokinase plasminogen activator (uPA), hepatocyte growth factor (HGF), and tumor necrosis factor alpha (TNFα) were similar for ASC and fibroblast-mediated angiogenesis, and in direct contrast to BMSC-mediated angiogenesis. The present study illustrates that the nature of the heterotypic interactions between mural cells and endothelial cells depend on the identity of the mural cell used. Even MSCs which are shown to behave phenotypically similar do not stimulate angiogenesis via the same mechanisms.
Keywords: Angiogenesis, Adipose-derived stem cells, Mesenchymal stem cells, Matrix metalloproteinases, Plasmin, Urokinase plasminogen activator
Introduction
Angiogenesis, the sprouting of new blood vessels from pre-existing vasculature, is an essential feature in the growth and maintenance of tissues. The diffusion limit of oxygen within tissue has widely been stated to be an obstacle in the creation of functional, viable tissue implants [1–3]. When hypoxic conditions exist within the body, interstitial cells, primarily from a mesenchymal lineage, respond by secreting pro-angiogenic factors which lead to the recruitment of vasculature to ischemic tissues [4, 5]. Understanding the mechanisms which govern this process will assist greatly towards the engineering of tissue or the treatment of pathological conditions characterized by aberrant vascularization.
The complex process of angiogenesis can be categorized into four distinguishable steps: basement membrane degradation, proliferation and invasion into the extracellular matrix (ECM), vessel maturation, and pruning of nonessential vasculature [6]. Proteases are intimately involved in each step of angiogenesis and therefore are essential to the entire angiogenic process. Prior groups have shown that, within fibrin, angiogenesis may proceed through the use of different protease families. Some groups have proposed that the plasmin axis of serine proteases is necessary for angiogenesis [7–9], while others have stated that endothelial cells (ECs) invade the ECM utilizing plasmin-independent matrix metalloproteinases (MMPs) [10, 11]. The heterotypic interactions that occur between ECs and interstitial cells along with cell-ECM interactions are necessary in the production of pro angiogenic factors and may determine which set of proteases are used [12, 13]. We have previously shown that two different types of mesenchymal cells promote EC proteolysis using different sets of proteases in the same angiogenic model [13].
Recent studies have suggested that mesenchymal stem cells (MSCs) may exist as pericytes in situ in all tissues where they are present [14, 15]. It is theorized that MSCs remain quiescent in this perivascular niche until local tissue injury occurs, at which point they function to prevent excessive scarring, promote angiogenesis, and possibly differentiate towards the lineage of the damaged tissue [16]. Since stem cell-ECM interactions play a significant role in determining cell behavior, it is particularly striking that MSCs from physically diverse tissues nevertheless possess similar multilineage differentiation potential.
The present study explores whether one type of MSC, adipose-derived stem cells (ASCs), behaves similarly to another type of MSC, bone marrow-derived stem cells (BMSCs), in promoting angiogenesis. Using an established angiogenesis coculture model [17], we show that ASCs modulate EC proteolysis using a different repertoire of proteases than BMSCs. Specifically, ASC-mediated vessel morphogenesis is highly dependent on the plasmin system and minimally on MMPs, whereas we have previously seen that BMSCs promote EC proteolysis solely through the use of MMPs [13]. This mechanism is similar to that which we have seen promoted by the presence of fibroblasts. We also find that the angiogenic cytokine profile of ASC-EC cocultures is similar to that of fibroblast-EC cocultures, rather than BMSC-EC cocultures. However, although ASC- and fibroblast-mediated angiogenesis proceed via similar proteolytic mechanisms, ASCs retain their multipotency and do not simply become perivascular fibroblasts. These data demonstrate that MSCs from different tissues, despite similar multipotencies, promote angiogenesis via distinct mechanisms.
Materials & Methods
HUVEC Isolation and Cell Culture
Human umbilical vein endothelial cells (HUVECs) were isolated from freshly harvested umbilical cords as previously described [18]. Briefly, the vein was flushed with sterile phosphate buffered saline (PBS) and then incubated with 0.1% collagenase type I (Worthington Biochemical, Lakewood, NJ) for 20 minutes at 37 °C. The digestion product and subsequent PBS wash were collected and centrifuged. The cell pellet was resuspended in EGM-2 (Lonza, Walkersville, MD) plated onto T-25 flasks, and allowed to attach overnight. PBS was used to wash away any red blood cells the following day. ASCs (Invitrogen, Carlsbad, CA) and BMSCs (Lonza) were cultured in 4.5 g/L DMEM (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS, Mediatech, Manassas, VA), 1% antibiotic-antimycotic (Mediatech), and 0.5 mg/mL gentamicin (Invitrogen). Normal human lung fibroblasts (NHLFs, Lonza) were cultured in Medium 199 (Invitrogen) supplemented with 10% FBS, 1% antibiotic-antimycotic, and 0.5 mg/mL gentamicin. All cultures were incubated at 37 °C and 5% CO2. Media were changed every 2–3 days and cells were harvested with 0.05% trypsin-EDTA (Invitrogen). HUVECs were used prior to passage four, while ASCs, BMSCs, and NHLFs were all used prior to passage ten.
Cell Transduction
To facilitate visualization and quantification of vessel networks, HUVECs were stably transduced with a gene encoding a fluorescent protein using the Phoenix Ampho Retrovirus Expression System (Orbigen, San Diego, CA). Specifically, the mCherry gene was cloned into the pBMN-Z entry plasmid as previously described [19]. Phoenix Ampho cells were transfected with the pBMN-mCherry plasmid using Lipofectamine 2000 (Invitrogen). Retroviral supernatant was collected, passed through a 0.45 μm syringe filter, and supplemented with 5 μg/mL Polybrene (Millipore, Billerica, MA) before being incubated with HUVECs for a period of eight hours. This process was repeated the following day.
Fibrin Tissue Assembly
Fibrin tissues were constructed as previously described [18]. Briefly, 10,000 Cytodex 3 microcarrier beads (Sigma-Aldrich) were subjected to a series of washes in EGM-2, and then added to a cell suspension containing four million HUVECs in a T-25 flask stood up vertically. To promote cell coating onto the beads, the flask was agitated by gentle shaking every 30 minutes for four hours. Afterwards, the bead solution was collected and transferred into a new T-25 flask with an additional 5 mL of EGM-2. This flask was cultured overnight at 37 °C and 5% CO2 to allow any remaining suspended cells to attach to the culture surface. The following day, fibrinogen (Sigma-Aldrich) was dissolved in serum-free EGM-2 to a final concentration of 2.5 mg/mL. The solution was passed through a 0.22 μm syringe filter before being combined with the appropriate interstitial cell type (100,000 cells/mL) and HUVEC-coated beads (50 beads/mL). For each gel to be constructed, 10 μL of a 50 U/mL thrombin solution (Sigma-Aldrich) was added to the center of a well of a 24-well tissue culture plate. Immediately prior to polymerization, 5% FBS was added to the fibrinogen solution followed by addition of 500 μL to each of the thrombin-spotted wells. The solution was initially allowed to set for five minutes at room temperature followed by incubation at 37 °C and 5% CO2 for an additional 30 minutes. Following polymerization, fully-supplemented EGM-2 and any chemical inhibitors were added. Medium was changed at day 1, 3, and 5 post-assembly.
Fluorescense Imaging and Vessel Quantification
Vessel formation was assessed seven days post-assembly. HUVECs expressing mCherry were imaged via fluorescence microscopy utilizing an Olympus IX51 equipped with a 100 W high-pressure mercury burner (Olympus America, Center Valley, PA), a QImaging QICAM 12-bit Color Fast 1394 camera (QImaging, Surry, BC, Canada), and QCapture Pro software (QImaging). Imaged beads were chosen at random provided that they met the condition that vessels emanating from a given bead did not form anastomoses with vessels from adjacent beads. At least five beads per condition were imaged at low magnification (4×) for each independent experiment. Total vessel network length was measured utilizing the Angiogenesis Tube Formation module in Metamorph Premier (Molecular Devices, Sunnyvale, CA). Minimum and maximum widths were entered into the program to differentiate vessels from noise and the bead, respectively.
Confocal Reflectance Imaging
Nonfluorescent HUVECs were used to assemble fibrin tissues in a Lab-Tek Chambered Coverglass (Thermo Fisher Scientific, Rochester, NY) essentially as described above. An Olympus FluoView FV1000 equipped with a 488 nm argon laser and accompanying software (Olympus) were used to visualize remodeling of the ECM after four days of culture. A 20/80 beam splitter was used to illuminate the sample and collect the subsequent backscattered light. This process was used to capture high magnification (120×) 50 μm z-stacks.
Western Blotting and Angiogenic Antibody Array
Fibrin constructs were digested in Garner Buffer (50mM Tris, pH 7.5; 150mM NaCl; 1mM phenylmethylsulfonyl fluoride; 1% Triton X-100). The tissues were homogenized twice for 30 seconds followed by three cycles of sonication for 10 seconds. The lysate was clarified by centrifugation at 14,000 rpm and 4 °C for 10 minutes. The supernatant was collected and total protein concentration was determined using a bicinchoninic acid assay (Thermo Fisher Scientific). After boiling, equal amounts of total protein from tissue lysates were loaded into a 10% tris-glycine gel (Invitrogen) and subjected to SDS-PAGE. The separated proteins were then transferred onto a poly(vinylidine fluoride) membrane and probed with a goat polyclonal anti-human uPA antibody (100 ng/mL, ab40841, Abcam, Cambridge, MA) or anti-human β-actin-HRP (200 ng/mL, sc-47778 HRP, Santa Cruz Biotechnologies, Santa Cruz, CA). After washing, the membrane was incubated with a horseradish peroxidase-conjugated anti-goat secondary antibody (80 ng/mL, sc-2033, Santa Cruz Biotechnologies). Protein expression was visualized using an enhanced chemiluminescence detection system. An angiogenesis antibody array (R&D Systems, Minneapolis, MN) was used to test the protein-level expression of a panel of angiogenic cytokines in tissue supernatants. For these experiments, tissues were homogenized in the absence of any lysis buffer. The supernatant was clarified by centrifugation at 14,000 rpm and 4 °C for 10 minutes. The supernatant was incubated with the membranes provided in the kit and developed according to the manufacturer’s protocol.
Reverse Transcription and Polymerase Chain Reaction
Mural cell and HUVEC RNA lysates were differentially obtained from tissue constructs as previously described [13]. Briefly, 2.5% (Sigma-Aldrich) trypsin was used to detach the overlying interstitial cell layer. Medium 199 supplemented with 20% FBS was used to neutralize the trypsin before it digested the fibrin gel any further. The remaining fibrin gel, with the HUVEC-coated beads, was washed in PBS and digested using 2.5% trypsin with gentle agitation. After neutralizing the trypsin, HUVECs were dissociated from beads by vigorous pipetting and collected via centrifugation. All cells were lysed and total RNA was purified using the SV Total RNA Isolation System (Promega, Madison, WI) and quantified using a Nanodrop ND-1000 (Thermo Fisher Scientific). Equal amounts of total RNA from each sample were used to create first-strand cDNA using the ImProm-II Reverse Transcription System (Promega). The cDNA was amplified via polymerase chain reaction using the primers listed in Table 1. The PCR product was separated via electrophoresis through a 2% agarose gel containing 1 μg/mL ethidium bromide and then imaged using an ultraviolet transilluminator and digital camera (Fotodyne, Hartland, WI). The images were inverted to facilitate visualization of bands. Quantitative PCR (qPCR) was performed using a 7500 Fast Real-Time PCR System and TaqMan Universal PCR Master Mix (Applied Biosystems, Carlsbad, CA). Predesigned qPCR primers for human uPA, membrane type 1 (MT1)-MMP, and 18s rRNA were selected from the TaqMan Gene Expression Assays database (Applied Biosystems). The ΔΔCT method was used to assess the relative quantity of each target gene.
Table 1.
PCR primers and annealing temperatures used
| Gene | Primer Sequences (5′ → 3′) | Amplicon (bp) | Annealing Temperature (°C) |
|---|---|---|---|
| TNFα |
F: CTCCAGTGGCTGAACCGCCG R: AGCGCTGAGTCGGTCACCCT |
327 | 63 |
| uPA |
F: GGCAGCAATGAACTTCATCAAGTTCC R: TATTTCACAGTGCTGCCCTCCG |
135 | 56 |
| MT1-MMP |
F: GCTTGCAAGTAACAGGCAAA R: AAATTCTCCGTGTCCATCCA |
589 | 57 |
| GAPDH |
F: CCCTGTTGCTGTAGCCGTA R: CCGGTGCTGAGTATGTCG |
442 | 58 |
Differentiation Assays
The adipogenic differentiation potential of the interstitial cell types was assessed by culturing 95,000 of each interstitial cell type in adipogenic differentiation medium (DMEM, 10% FBS, 1% antibiotic-antimycotic, 0.5 mg/mL gentamicin, 0.5 mM isobutyl-methylxanthine, 1 mM dexamethasone, 10 mM insulin, 200 mM indomethacin) [20] for 14 days. Lipid expression was assessed using an Oil Red O staining kit (American MasterTech, Lodi, CA) and following the manufacturer’s protocol. Osteogenic differentiation potential was assessed by culturing 95,000 of each interstitial cell type in osteogenic differentiation medium (DMEM, 10% FBS, 1% antibiotic-antimycotic, 0.5 mg/mL gentamicin, 0.1 mM dexamethasone, 50 mM ascorbate-2-phosphate, 10 mM β-glycerophosphate) [20] for 14 days. Calcium deposition was assessed via Alizarin Red S staining. Briefly, cells were washed in PBS followed by deionized water to ensure removal of any exogenous calcium. Cells were then fixed in 100% ethanol for 15 minutes and then stained with 0.2% Alizarin Red S solution (pH 6.4) for 30 min. Cells were subsequently washed with deionized water extensively to remove any unbound Alizarin Red S.
Other Reagents Used
GM6001 (10–50 μM, EMD Chemicals, Darmstadt, Germany), BB2516 (3.3 μM, Tocris Bioscience, Ellisville, MO), rhTIMP-1 (5 μg/mL, R&D Systems), and rhTIMP-2 (5 μg/mL, R&D Systems) were used for broad-spectrum MMP inhibition. ε-aminocaproic acid (10mM, EACA, Sigma-Aldrich) was used to inhibit the PA/plasmin axis. A previously characterized MT1-MMP function-blocking antibody (clone LEM-2/15.8, 5 μg/mL) [21] was graciously provided by Dr. Alicia Garcia Arroyo. For tissue cultures involving function-blocking antibodies, the antibody was added to the gel precursor solution as well as at every medium change.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism software (GraphPad Software, La Jolla, CA). Data are presented as mean ± standard deviation. One-way analysis of variance with a Bonferroni post test was used to assess statistical significance between data sets. Statistical significance was assumed when p < 0.05.
Results
ASC-Mediated Vessel Sprouting and Phenotype are Mediated by MMPs and Plasmin
To test the proteolytic dependence of ASC-mediated vessel morphogenesis, tissue constructs were treated with either the broad-spectrum MMP inhibitor GM6001 [22], the plasmin inhibitor ε-aminocaproic acid (EACA) [23], or a combination of the two (Figure 1a). Broad scale inhibition of MMPs did not have any effect on the total vessel network length, whereas inhibition of plasmin saw a 43% reduction in network length relative to the vehicle control. Combinatorial inhibition of both protease families resulted in an 81% decrease in network length relative to the vehicle, which represented a further 67% reduction relative to the EACA only condition (Figure 1b). Interestingly, inhibition of MMPs resulted in the formation of larger diameter vessels (Figure 1c) presumably due to the proteolytic activity of plasmin, as this phenotype was not observed when the plasmin system was inhibited. Inhibition with a separate MMP inhibitor, BB2516, phenocopied the results observed using GM6001 (data not shown), further elucidating the role of MMPs in this system. To test whether or not MMP inhibition is dose dependent, cocultures were treated with increasing concentrations of GM6001 (Figure 2). Even at the highest concentration, MMP inhibition failed to mitigate vessel formation.
Fig. 1.
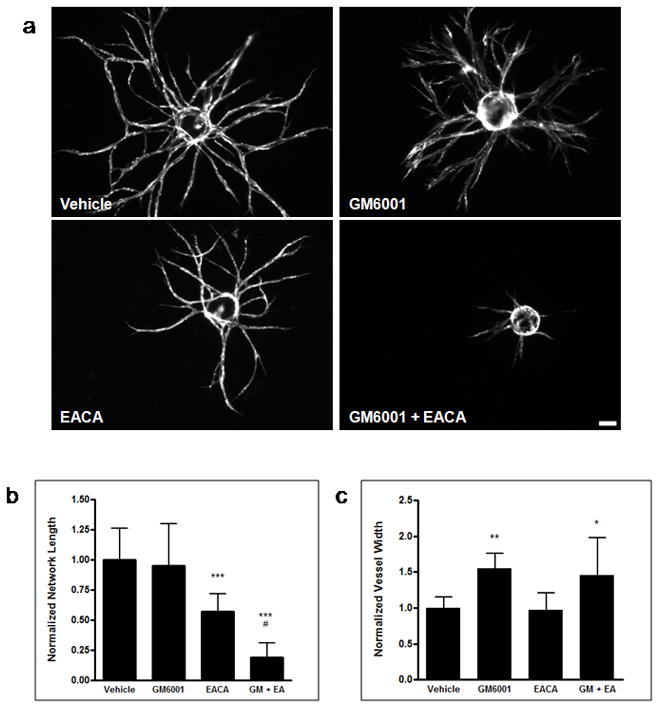
Broad spectrum protease inhibition reveals that ASC-mediated vessel invasion is plasmin dependent, while MMPs are required in regulating vessel diameter. HUVECs, transduced with mCherry, were coated on microcarrier beads and cultured within 2.5 mg/mL fibrin gels in the presence of ASCs. (a) Representative day 7 images of vessel formation in the presence of DMSO vehicle control, 10 μM GM6001, 10 mM EACA, or GM6001 + EACA are shown. Total network length (b) and average vessel width (c) were calculated and normalized to the vehicle control. ***, **, and * refer to p ≤ 0.001, p ≤ 0.01, and p ≤ 0.05 in comparison to the vehicle control, respectively. # refers to p ≤ 0.05 in comparison to the EACA condition. Scale bar = 100 μm
Fig. 2.

Inhibition of MMPs with increasing concentrations of GM6001 does not mitigate ASC-mediated vessel formation. (a) Day 7 images of fluorescently-transduced HUVECs on microcarrier beads cocultured with ASCs within 2.5 mg/mL fibrin gels in the presence of DMSO vehicle control, 10 μM, 20 μM, and 50 μM GM6001. Total network length was normalized to the vehicle control (b). Scale bar = 100 μm
The endogenous MMP inhibitors, TIMP-1 and TIMP-2 also did not affect vessel formation (Figure 3). Previous studies have shown that the membrane-anchored MT1-MMP is essential for vessel morphogenesis in collagen gels [10, 24]. Treatment of cocultures with a function-blocking antibody against MT1-MMP also did not affect vessel formation in our fibrin-based system (Figure 3).
Fig. 3.
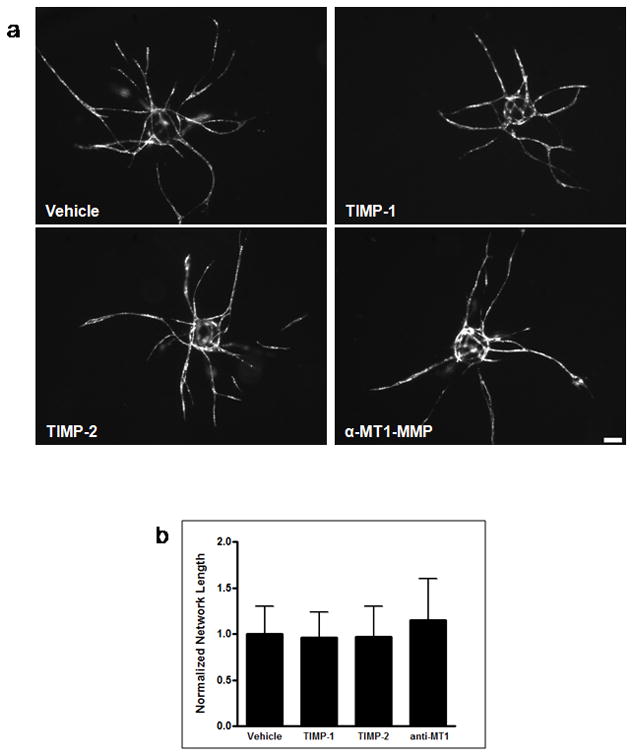
Endogenous inhibitors confirm that ASC-mediated vessel invasion through fibrin is not MMP-dependent. (a) Representative day 7 images of vessel formation in the presence of PBS vehicle control, 5 μg/mL TIMP-1, 5 μg/mL TIMP-2, and 5 μg/mL of a function-blocking MT1-MMP antibody. Total network length was calculated and normalized to the vehicle control (b). Scale bar = 100 μm
MMP and Plasmin Inhibition Correlates with Decreased Matrix Remodeling
A recent study has shown that endothelial cells create vascular guidance channels within the ECM during tubulogenesis [25]. These conduits allow endothelial cells to migrate through a pseudo-two dimensional space in a protease-independent manner. This process is essential for lumen formation and establishing basolateral polarity in the vessel’s stalk region—a region shown to be less proteolytically active than the vessel tip [26]. Confocal reflectance imaging of fibrin tissue co-cultures illustrates the presence of these guidance channels around the invading vessel tip (Figure 4). Cultures treated with the vehicle control at four days post assembly show areas devoid of ECM within the plane of the vessel (Figure 4, arrowheads). We assume these areas to have been proteolytically degraded by the sprouting tip as a means for further vessel elongation. Guidance channels are visible in the planes above vessels (Figure 4, middle column, asterisks). Inhibition of MMPs with GM6001 did not produce any notable differences compared to the vehicle control, whereas inhibition of plasmin with EACA reduces the amount of matrix degradation, as evidenced by the residual fibers within the guidance channels (Figure 4, magnified in right column). As expected, neither matrix voids nor guidance channels were observed when MMPs and plasmin were inhibited simultaneously.
Fig. 4.

Combinatorial inhibition of plasmin and MMPs abrogates the formation of vascular guidance channels. Shown are confocal reflectance images of the tip region of sprouting vessels in the ASC-HUVEC fibrin tissue co-culture. The left column represents a plane encompassing the vessel (rough outline of vessel edge in red), whereas the middle column is that of a plane 20–30 μm above the vessel, used to illustrate the vascular guidance channel (asterisks). The right column shows magnified images of the respective guidance channels to detail matrix degradation. Arrowheads represent areas of ECM degradation which serve as possible conduits for vessel elongation. Images are overexposed to more easily visualize guidance channels. Scale bar = 20 μm
ASC-Mediated and NHLF-Mediated Vessel Morphogenesis Display Similar Gene and Protein Profiles
We have previously shown that two different mesenchymal cell types promote upregulation of a different set of endothelial cell proteases used in vessel morphogenesis [13]. In addition to MT1-MMP, urokinase plasminogen activator (uPA) is another factor thought to be essential in vessel infiltration of the provisional fibrin clot during wound healing [7, 9, 27]. To determine which proteases are upregulated in this angiogenesis model, we examined HUVEC mRNA transcript levels at day 3—a time point which we have observed to be a critical period for protease upregulation [18]. Expression of uPA in ASCs appears to remain relatively constant whether they are alone or in coculture with HUVECs (Figure 5a). However, there is a marked increase in endothelial uPA expression when cocultured with ASCs compared to when they are cultured without ASCs. MT1-MMP expression in ASCs decreases upon coculture with HUVECs, whereas there is minimal expression in HUVECs whether alone or in coculture. Quantitative PCR confirms these observations, showing a 34% increase in endothelial cell-derived uPA when cocultured with ASCs (Figure 5b). ASC-derived uPA and MT1-MMP (Figure 5b–c) decreased 25% and 31%, respectively when cocultured with HUVECs. These data follow the same trend observed in NHLF-mediated vessel morphogenesis (Figure 5b–c). HUVEC-derived uPA increased over twofold when cocultured with NHLFs, whereas NHLF-derived uPA and MT1-MMP decreased 25% and 22%, respectively when cocultured with HUVECs. The greater increase in magnitude of endothelial uPA expression when cultured with NHLFs, as compared to ASCs, may be a contributing factor to the faster kinetics of NHLF-mediated vessel morphogenesis that we have observed in previous studies [13, 28]. As reported previously [13], BMSC-mediated vessel morphogenesis is not dependent upon the plasmin axis, and accordingly, qPCR data showed no appreciable differences in the levels of endothelial cell uPA when cultured with BMSCs versus alone (Figure 5b). BMSC expression of MT1-MMP decreased 26% when cocultured with HUVECs (Figure 5c), which followed the same pattern as ASCs and NHLFs. Endothelial uPA was not detectable on the protein level when cultured alone within fibrin (Figure 5d). Culture with each of the three mesenchymal cell types showed a significant increase in uPA protein, with the most significant being when HUVECs were cultured with ASCs.
Fig. 5.
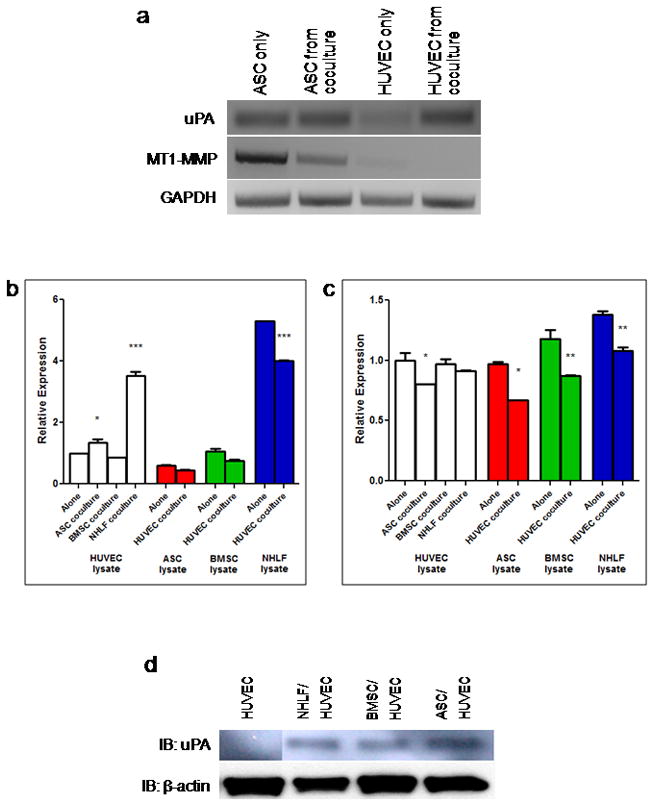
ASCs upregulate the expression of endothelial cell-derived uPA in coculture. (a) Day 3 transcript levels of uPA, MT1-MMP, and GAPDH from ASCs and HUVECs cultured either alone or together. Relative expression levels of (b) uPA and (c) MT1-MMP obtained from qPCR in ASC/HUVEC, NHLF/HUVEC, and BMSC/HUVEC cultures. Quantitative PCR data are normalized to the EC only condition for the respective gene. In (b and c), *, **, and *** refer to p ≤ 0.05, p ≤ 0.01, and p ≤ 0.001, respectively, for the “Alone” condition of the respective lysate group. (d) Immunoblotting results for uPA protein expression at day 3 in HUVEC only, NHLF/HUVEC, BMSC/HUVEC, and ASC/HUVEC cultures. Equal protein loading was assessed via immunoblotting for β-actin. Lanes were reordered without distorting protein expression levels.
Tumor necrosis factor alpha (TNFα) has previously been implicated in increasing the expression of uPA as well as the uPA receptor in endothelial cells [8, 29]. To understand how interstitial cells may regulate protease expression, ASCs, BMSCs, and NHLFs were assayed for TNFα expression alone and in the presence of HUVECs. ASCs and NHLFs showed a noticeable upregulation of TNFα when cultured with HUVECs (Figure 6a). Interestingly, BMSCs downregulated TNFα when cultured with HUVECs, in direct contrast to ASCs and NHLFs. Hepatocyte growth factor (HGF) is another cytokine known to enhance production of uPA and uPAR in some invasive tumors [30–32]. ASC/HUVEC and NHLF/HUVEC cultures expressed similar levels of HGF as assayed via antibody array, whereas the expression level in BMSC/HUVEC cultures was markedly lower (Figure 6b).
Fig. 6.
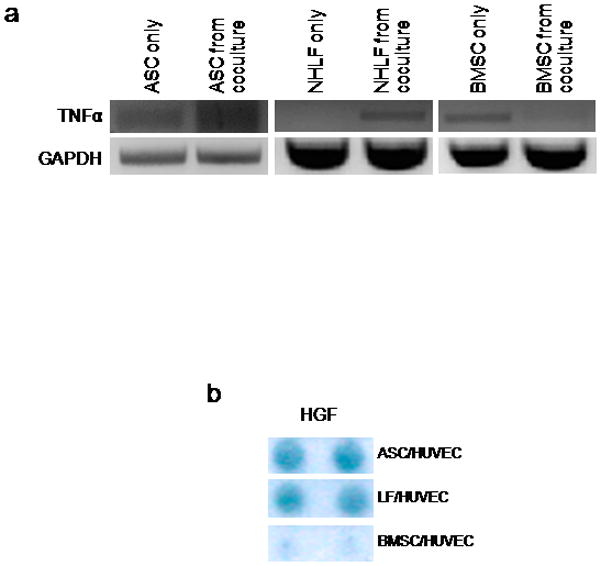
ASC- and NHLF-mediated angiogenesis share a cytokine expression profile different from BMSC-mediated angiogenesis. (a) TNFα transcript expression by mural cells alone and in the presence of HUVECs. (b) HGF positive expression by mural cell/HUVEC cocultures as assayed by an antibody array. Test returns duplicate positive readings for each cytokine.
ASCs Retain their Differentiation Potential
Since NHLFs and ASCs seemingly promote vessel morphogenesis through similar means, it is possible that our ASCs may have simply differentiated to a more terminal fibroblast phenotype during the course of two dimensional expansion and in vitro culture. To assess the multipotency of our ASCs, we compared their differentiation potential against that of NHLFs and BMSCs. NHLFs, BMSCs, and ASCs age-matched to those used in our angiogenesis model, were subjected to culture in osteogenic medium for a period of 14 days. Subsequent Alizarin Red S staining revealed high amounts of calcium deposition by ASCs and BMSCs cultured in osteogenic medium, indicative of differentiation towards an osteogenic lineage. In contrast, staining of NHLF cultures revealed minimal, if any, mineralization (Figure 7). In addition, Oil Red O staining of NHLFs, BMSCs, and ASCs cultured in adipogenic medium revealed the presence of lipids in only the ASC and BMSC cultures (Figure 8). These data affirm that ASCs retain their multipotency and remain phenotypically distinct from NHLFs despite being able to promote vessel morphogenesis in a similar fashion.
Fig. 7.
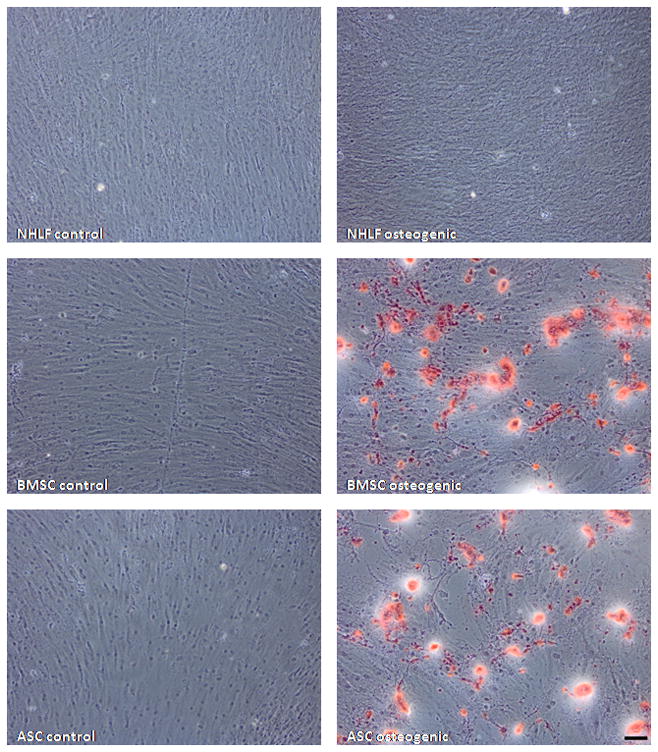
ASCs retain osteogenic differentiation potential. NHLFs, BMSCs, and ASCs were cultured in either DMEM control or osteogenic media for 14 days. Subsequent Alizarin Red S staining shows calcium deposition (red) by ASCs and BMSCs but not by NHLFs. Scale bar = 100 μm
Fig 8.
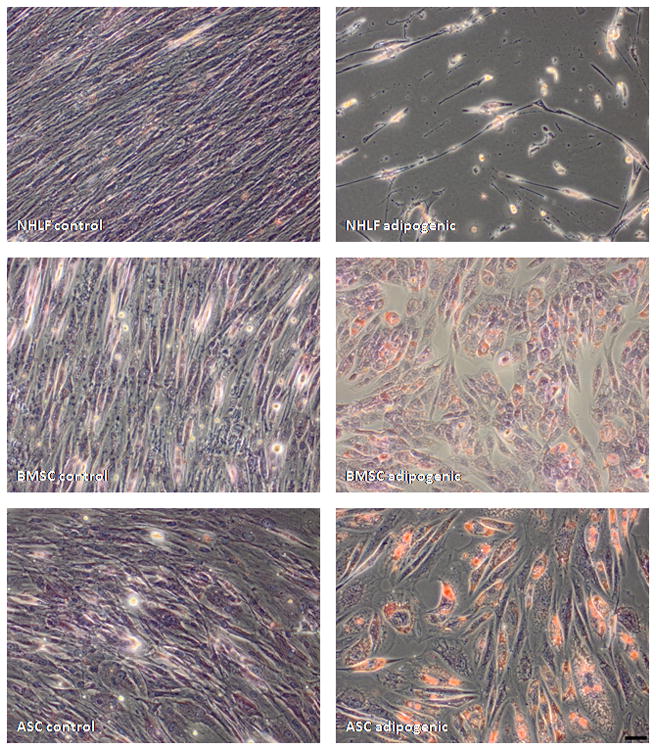
ASCs retain adipogenic differentiation potential. NHLFs, BMSCs, and ASCs were cultured in either DMEM control or adipogenic media for 14 days. Subsequent Oil Red O staining revealed lipid-positive staining (red) in ASC cultures but not in NHLF cultures. Cultures were counterstained with hematoxylin. Scale bar = 50 μm
Discussion
This study explored the mechanisms that modulate ECM proteolysis in a fibrin-based angiogenesis model. In a previous study, we demonstrated that heterotypic cellular interactions dictate the guiding mechanisms during angiogenesis [13]. Particularly, endothelial cells will express and utilize a different set of proteases depending on the identity of the interstitial cells providing the pro-angiogenic cues. Similar to what we previously reported with fibroblasts [13], ASC-mediated vessel morphogenesis relied primarily on the plasmin family of proteases for EC invasion and elongation; however, MMP activity was necessary in establishing a normal vessel phenotype. Furthermore, the expression profiles of key proteases and angiogenic cytokines in ASC-mediated angiogenesis paralleled that of fibroblast-mediated angiogenesis more so than BMSC-mediated angiogenesis.
Broad spectrum inhibition of two separate protease families suggested that ASC-mediated vessel sprouting and elongation is highly dependent on plasmin and to a lesser degree on MMPs, while luminal diameter regulation and possibly vessel stabilization depend on the catalytic function of MMPs. Inhibiting MMPs in our ASC-HUVEC model with a broad spectrum inhibitor did not significantly affect the total vessel network length, but did result in a phenotype characterized by larger diameter vessels. MT1-MMP, in particular, has been shown to be necessary in recruiting mural cells to the vasculature to prevent vessel distention [33]. Another recent study has found that mice overexpressing MT1-MMP do not exhibit vascular permeability and vasodilation upon treatment with mustard oil [34]. Therefore, it is likely that our phenomenon is a direct consequence of MMP inhibition. Further supporting this theory is the fact that sole inhibition of the plasmin axis did not affect vessel diameter. Combinatorial inhibition of both the MMP and plasmin proteases resulted in minimal vessel sprouting, but the diameter of these vessels was similar to that of vessels when only MMPs were inhibited. We hypothesize that, within fibrin, there exists a balance mechanism between MMPs and plasmin, in which plasmin is used for vessel outgrowth and MMPs are necessary for regulating vessel diameter. TNFα signaling has been shown to drive vascular and lymphatic remodeling to result in larger vessels in an in vivo inflammation model [35]. Whether or not this signaling affects the uPA/uPAR/plasmin system, as it has in other studies [7, 8], remains to be seen. We are continuing to explore the possibility of a proteolytic balance using RNA interference against specific MMPs as well as uPA/uPAR.
The coculture of ASCs with HUVECs within fibrin resulted in the upregulation of the pro-angiogenic factors HGF and TNFα. In conjunction, the coculture led to the upregulation of uPA by the ECs. The level of MT1-MMP produced by ECs was unaffected by the presence of ASCs. The data show that these expression patterns mimic those of NHLF/HUVEC cocultures more so than that of BMSC/HUVEC cocultures. This is evident even as the ASCs retain their multipotency. Recent work demonstrated that MSCs reside within perivascular locations irrespective of the tissue origin [14]. MSCs isolated from different tissues displayed similar surface markers and multipotency. We must be careful however, to not assume that these cells are identical in every facet. The microenvironment that constitutes these different tissues varies drastically in terms of mechanical and biochemical properties. Even the vasculature within these different tissues can vary, as seen by the ability to target different tissue by means of specific vessel surface markers [36, 37]. The unique interactions between the microenvironment and the MSCs of a particular tissue could be responsible for the differences that are seen between ASCs and BMSCs.
Recent efforts have been made to engineer synthetic biomaterials which promote vascularization [38, 39]. A key component of these constructs is the addition of biomimetic protease-cleavable peptide sequences which allow for implanted cells to remodel and invade the otherwise proteolytic-resistant matrix. Most of these approaches to date have incorporated an MMP-sensitive crosslink within their hydrogel construct to facilitate vascularization. Based on our findings, progression of this technology towards therapeutic purposes will require an understanding of the heterotypic interactions between the specific interstitial cells and ECs in deciding on which protease-sensitive peptide sequences to embed.
A final point of discussion regards the efficiency with which different populations of MSCs can be used to generate tissue. For cell-based therapies, the source of cells is typically considered in terms of isolation yield, expansion potential, and immunologic tolerance. With respect to MSCs, the yield of ASCs obtained from liposuctions surpasses that of BMSCs obtained from marrow extraction [40, 41]. Furthermore, acquiring lipoaspirate is far easier than extracting bone marrow; thus, ASCs would seem to be the more promising option in these terms. However, here we have shown that these different MSCs, similar in terms of their differentiation potentials, guide angiogenesis via distinct mechanisms. Understanding these mechanistic distinctions will be beneficial not only for promoting vascularization of engineered tissue, but will also be a critical aspect of the ongoing cell source debate in the regenerative medicine literature.
Acknowledgments
We are grateful to Dr. Alicia Garcia Arroyo for graciously providing us the MT1-MMP function-blocking antibody, Dr. Christopher Hughes and Linda Him for providing us with fresh umbilical cords, and Dr. Cyrus Ghajar and Ekaterina Kniazeva for providing insightful discussions. We would also like to thank Dr. Enrico Gratton and Dr. Michelle Digman for assistance with confocal reflectance imaging, along with Dr. Jan Stegemann and Dr. Michael Mayer for providing us access to their instrumentation. This work was supported by a grant from the National Institutes of Health (R01 HL085339).
Footnotes
Ethical Standards Statement
The authors declare that the experiments described within this manuscript comply with the current laws of the United States of America.
Author Disclosure Statement
The authors declare no competing financial interests.
References
- 1.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–57. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 2.Zandonella C. Tissue engineering: The beat goes on. Nature. 2003;421(6926):884–6. doi: 10.1038/421884a. [DOI] [PubMed] [Google Scholar]
- 3.Griffith CK, et al. Diffusion limits of an in vitro thick prevascularized tissue. Tissue Eng. 2005;11(1–2):257–66. doi: 10.1089/ten.2005.11.257. [DOI] [PubMed] [Google Scholar]
- 4.Fuchs S, et al. Transendocardial delivery of autologous bone marrow enhances collateral perfusion and regional function in pigs with chronic experimental myocardial ischemia. J Am Coll Cardiol. 2001;37(6):1726–32. doi: 10.1016/s0735-1097(01)01200-1. [DOI] [PubMed] [Google Scholar]
- 5.Rehman J, et al. Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation. 2004;109(10):1292–8. doi: 10.1161/01.CIR.0000121425.42966.F1. [DOI] [PubMed] [Google Scholar]
- 6.Ghajar CM, George SC, Putnam AJ. Matrix metalloproteinase control of capillary morphogenesis. Crit Rev Eukaryot Gene Expr. 2008;18(3):251–78. doi: 10.1615/critreveukargeneexpr.v18.i3.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kroon ME, et al. Role and localization of urokinase receptor in the formation of new microvascular structures in fibrin matrices. Am J Pathol. 1999;154(6):1731–42. doi: 10.1016/S0002-9440(10)65429-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kroon ME, et al. Urokinase receptor expression on human microvascular endothelial cells is increased by hypoxia: implications for capillary-like tube formation in a fibrin matrix. Blood. 2000;96(8):2775–83. [PubMed] [Google Scholar]
- 9.Kroon ME, et al. Vascular endothelial growth factor enhances the expression of urokinase receptor in human endothelial cells via protein kinase C activation. Thromb Haemost. 2001;85(2):296–302. [PubMed] [Google Scholar]
- 10.Chun TH, et al. MT1-MMP-dependent neovessel formation within the confines of the three-dimensional extracellular matrix. J Cell Biol. 2004;167(4):757–67. doi: 10.1083/jcb.200405001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hotary KB, et al. Matrix metalloproteinases (MMPs) regulate fibrin-invasive activity via MT1-MMP-dependent and -independent processes. J Exp Med. 2002;195(3):295–308. doi: 10.1084/jem.20010815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yancopoulos GD, et al. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407(6801):242–8. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 13.Ghajar CM, et al. Mesenchymal cells stimulate capillary morphogenesis via distinct proteolytic mechanisms. Exp Cell Res. 2010;316(5):813–25. doi: 10.1016/j.yexcr.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crisan M, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3(3):301–13. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 15.Caplan AI. All MSCs are pericytes? Cell Stem Cell. 2008;3(3):229–30. doi: 10.1016/j.stem.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Caplan AI. Why are MSCs therapeutic? New data: new insight. J Pathol. 2009;217(2):318–24. doi: 10.1002/path.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakatsu MN, Hughes CC. An optimized three-dimensional in vitro model for the analysis of angiogenesis. Methods Enzymol. 2008;443:65–82. doi: 10.1016/S0076-6879(08)02004-1. [DOI] [PubMed] [Google Scholar]
- 18.Ghajar CM, et al. Mesenchymal stem cells enhance angiogenesis in mechanically viable prevascularized tissues via early matrix metalloproteinase upregulation. Tissue Eng. 2006;12(10):2875–88. doi: 10.1089/ten.2006.12.2875. [DOI] [PubMed] [Google Scholar]
- 19.Ghajar CM, et al. The effect of matrix density on the regulation of 3-D capillary morphogenesis. Biophys J. 2008;94(5):1930–41. doi: 10.1529/biophysj.107.120774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuk PA, et al. Multilineage cells from human adipose tissue: implications for cell- based therapies. Tissue Eng. 2001;7(2):211–28. doi: 10.1089/107632701300062859. [DOI] [PubMed] [Google Scholar]
- 21.Galvez BG, et al. Membrane type 1-matrix metalloproteinase is activated during migration of human endothelial cells and modulates endothelial motility and matrix remodeling. J Biol Chem. 2001;276(40):37491–500. doi: 10.1074/jbc.M104094200. [DOI] [PubMed] [Google Scholar]
- 22.Grobelny D, Poncz L, Galardy RE. Inhibition of human skin fibroblast collagenase, thermolysin, and Pseudomonas aeruginosa elastase by peptide hydroxamic acids. Biochemistry. 1992;31(31):7152–4. doi: 10.1021/bi00146a017. [DOI] [PubMed] [Google Scholar]
- 23.Prentice CR. Basis of antifibrinolytic therapy. J Clin Pathol Suppl (R Coll Pathol) 1980;14:35–40. [PMC free article] [PubMed] [Google Scholar]
- 24.Davis GE, Saunders WB. Molecular balance of capillary tube formation versus regression in wound repair: role of matrix metalloproteinases and their inhibitors. J Investig Dermatol Symp Proc. 2006;11(1):44–56. doi: 10.1038/sj.jidsymp.5650008. [DOI] [PubMed] [Google Scholar]
- 25.Stratman AN, et al. Endothelial cell lumen and vascular guidance tunnel formation requires MT1-MMP-dependent proteolysis in 3-dimensional collagen matrices. Blood. 2009;114(2):237–47. doi: 10.1182/blood-2008-12-196451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yana I, et al. Crosstalk between neovessels and mural cells directs the site-specific expression of MT1-MMP to endothelial tip cells. J Cell Sci. 2007;120(Pt 9):1607–14. doi: 10.1242/jcs.000679. [DOI] [PubMed] [Google Scholar]
- 27.Collen A, et al. Membrane-type matrix metalloproteinase-mediated angiogenesis in a fibrin-collagen matrix. Blood. 2003;101(5):1810–7. doi: 10.1182/blood-2002-05-1593. [DOI] [PubMed] [Google Scholar]
- 28.Kniazeva E, Putnam AJ. Endothelial cell traction and ECM density influence both capillary morphogenesis and maintenance in 3-D. Am J Physiol Cell Physiol. 2009;297(1):C179–87. doi: 10.1152/ajpcell.00018.2009. [DOI] [PubMed] [Google Scholar]
- 29.Niedbala MJ, Picarella MS. Tumor necrosis factor induction of endothelial cell urokinase-type plasminogen activator mediated proteolysis of extracellular matrix and its antagonism by gamma-interferon. Blood. 1992;79(3):678–87. [PubMed] [Google Scholar]
- 30.Kumar R, et al. Regulation of distinct steps of angiogenesis by different angiogenic molecules. Int J Oncol. 1998;12(4):749–57. doi: 10.3892/ijo.12.4.749. [DOI] [PubMed] [Google Scholar]
- 31.Moriyama T, et al. Simultaneous up-regulation of urokinase-type plasminogen activator (uPA) and uPA receptor by hepatocyte growth factor/scatter factor in human glioma cells. Clin Exp Metastasis. 1999;17(10):873–9. doi: 10.1023/a:1006729611241. [DOI] [PubMed] [Google Scholar]
- 32.Nishimura K, et al. Effects of hepatocyte growth factor on urokinase-type plasminogen activator (uPA) and uPA receptor in DU145 prostate cancer cells. Int J Androl. 2003;26(3):175–9. doi: 10.1046/j.1365-2605.2003.00413.x. [DOI] [PubMed] [Google Scholar]
- 33.Lehti K, et al. An MT1-MMP-PDGF receptor-beta axis regulates mural cell investment of the microvasculature. Genes Dev. 2005;19(8):979–91. doi: 10.1101/gad.1294605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sounni NE, et al. Stromal regulation of vessel stability by MMP14 and TGFbeta. Dis Model Mech. 2010;3(5–6):317–32. doi: 10.1242/dmm.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baluk P, et al. TNF-alpha drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. J Clin Invest. 2009;119(10):2954–64. doi: 10.1172/JCI37626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruoslahti E. Drug targeting to specific vascular sites. Drug Discov Today. 2002;7(22):1138–43. doi: 10.1016/s1359-6446(02)02501-1. [DOI] [PubMed] [Google Scholar]
- 37.Ruoslahti E. Vascular zip codes in angiogenesis and metastasis. Biochem Soc Trans. 2004;32(Pt3):397–402. doi: 10.1042/BST0320397. [DOI] [PubMed] [Google Scholar]
- 38.Phelps EA, et al. Bioartificial matrices for therapeutic vascularization. Proc Natl Acad Sci U S A. 2010;107(8):3323–8. doi: 10.1073/pnas.0905447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moon JJ, et al. Biomimetic hydrogels with pro-angiogenic properties. Biomaterials. 2010;31(14):3840–7. doi: 10.1016/j.biomaterials.2010.01.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aust L, et al. Yield of human adipose-derived adult stem cells from liposuction aspirates. Cytotherapy. 2004;6(1):7–14. doi: 10.1080/14653240310004539. [DOI] [PubMed] [Google Scholar]
- 41.Gimble JM, Katz AJ, Bunnell BA. Adipose-derived stem cells for regenerative medicine. Circ Res. 2007;100(9):1249–60. doi: 10.1161/01.RES.0000265074.83288.09. [DOI] [PMC free article] [PubMed] [Google Scholar]