

Abstract
Considering cells as biofactories, we aimed to optimize its internal processes by using the same engineering principles that large industries are implementing nowadays: lean manufacturing. We have applied reverse engineering computational methods to transcriptomic, metabolomic and phenomic data obtained from a collection of tomato recombinant inbreed lines to formulate a kinetic and constraint-based model that efficiently describes the cellular metabolism from expression of a minimal core of genes. Based on predicted metabolic profiles, a close association with agronomic and organoleptic properties of the ripe fruit was revealed with high statistical confidence. Inspired in a synthetic biology approach, the model was used for exploring the landscape of all possible local transcriptional changes with the aim of engineering tomato fruits with fine-tuned biotechnological properties. The method was validated by the ability of the proposed genomes, engineered for modified desired agronomic traits, to recapitulate experimental correlations between associated metabolites.
Author Summary
Considering cells as biofactories, we aimed to optimize their internal processes by using existing design principles acquired from engineering. Herein, we present a synthetic biology approach based on experimental and computational methodology that integrates genomic, transcriptomic, metabolomic and phenomic data to formulate a kinetic and constraint based model of tomato agronomic and fruit quality characteristics. The model has been used for exploring the landscape of all possible local transcriptional changes with the aim of engineering tomato fruits with improved biotechnological properties. The methodology was validated by the ability of the proposed engineered genomes with modified desired agronomic traits, to recapitulate correlations between associated metabolites that are found experimentally in a number of examples.
Introduction
Considering a cell as a DNA-based molecular factory [1] and applying principles drawn from industrial engineering provides new approaches to optimize cellular performance (Figure S1). This approach adopts the new philosophy implemented nowadays by large industries that is known as Lean Manufacturing (LM). LM consists in the implementation of standards based on elimination of bottlenecks and processes without mark-up and minimization of pathways and excessive costs. This approach can be applied to the emerging fields of systems and synthetic biology, and allows translating engineering concepts into biotechnology [2]–[4]. Our main goal is to optimize the phenotypic response of a natural plant biofactory, exemplified here by the edible tomato fruit, by using a combined experimental and computational synthetic biology approach. The approach involves re-designing the fruit factory from within; i.e., by modeling and identifying the important genes and intermediates for a given trait of agronomical interest.
Previous works have considered modeling the global metabolism [5], transcription [6]–[11] or the integration of both in microbial organisms [12]–[14] from the point of view of systems biology. Many groups, using a re-designing strategy that is characteristic of synthetic biology, have implemented genome-scale re-designs and explorations of the gene knockout landscape both in prokaryotes [15]–[17] and eukaryotes [18]. More recent reports have tackled the prediction of phenotypes from metabolic data based on statistical models for microbes [12] and plants [18]–[20]. The next logical and desirable development should consist in modeling phenotypes of interest in a complex organism from metabolic and gene expression data. For that purpose we have chosen tomato: a model plant for fleshy fruit -this being a natural biofactory of nutrients and healthy compounds, and a plant of agronomic interest with well-developed genetics and genomics (http://solgenomics.net) and with extensive work on metadata analysis [21]–[23]. We have assumed that at least in part the genetic program of the fruit at the ripe stage should have an impact on the metabolite content and also in other high order fruit traits. In this study, we have used omic data that have been experimentally obtained by means of transcriptomics, metabolomics and phenomics for a large number of recombinant inbred lines (RILs) derived from a cross of Solanum lycopersicum×S. pimpinellifolium. Following the LM approach, we have developed here a novel in silico optimization method that extensively explores single and multiple genetic perturbations to render a series of desired tomato phenotypes; i.e., show agronomical properties of biotechnological interest. Recently, large efforts in genome-scale modeling have been reported [24], [25] (e.g., genome wide selection methods). Herein, techniques based on reverse engineering were applied to a large set of experimental omics data to obtain a kinetic model based on ordinary differential equations (ODEs) that describe the steady state concentration of mRNAs. This model has the advantage of quantitatively characterizing the kinetic parameters describing molecular interactions that are essential for simulating the genetic perturbations involved in redesigning genomes. Hence, this model describes the fruit metabolic profile from gene expression data for an autonomous subset of genes with potential effect on transcription regulation. By capturing relationships between metabolic profiles and high-throughput phenomic data, our model was extended to predict changes in agronomic properties that would be produced by specific changes in genetic expression (Figure S2).
Finally, in order to close the design cycle imposed by LM, the genetic modifications suggested by our computational approach were experimentally verified. This was done by demonstrating the predicted ability of the in silico modified fruit genomes to reproduce the correlations between metabolites empirically found in the fruit. We propose that the principles and practices learned from these engineering success cases can help to formulate a model to guide the design of new organisms with biotechnological applications.
Results
A genome-wide transcriptional model allows the integration of tomato fruit metabolism
We have extended our recently developed inference methodology, InferGene [7], to obtain a gene regulatory model coupled to metabolism that allows us analyzing optimality in terms of specified agronomic and organoleptic properties of the tomato fruit (Figure 1). For this, we have taken advantage of an experimentally characterized subset of the metabolome of 169 tomato RILs, which includes the accumulation levels of 67 metabolites in the fruit and that contribute to the flavor (sugars, acids and some volatiles), aroma (volatiles) and other quality traits (such as color and healthy carotenoids and vitamins). Moreover, we have also used the information on transcript levels from fruits for a subset of the 50 RILs analyzed at the metabolic level, to select 5592 non-redundant genes that were consistently expressed in those fruit samples (see Methods).
Figure 1. Lean Manufacturing as a model applied in systems and synthetic biology.

From omic data (transcriptomics, metabolomics and phenomics), a quantitative global model was constructed using reverse engineering methods. The predictive model was used to propose genome perturbations, to improve desired phenotypes with relevant biotechnological applications. The genome perturbations were guided by an in silico optimization that imposed the desired selective pressure.
Transcriptomic and metabolomic data from these 50 RILs were normalized by the LOWESS method [26] and used to construct a model that predicts components of the fruit quality metabolome from transcriptome data; i.e., level of a given metabolite is effectively determined by the expression of a minimal set of genes. The size of the space of possible gene-predictors was reduced in one order of magnitude by using a CLR method (Dataset S1). After that, LASSO method was used to find a minimal set of potential predictor genes for each metabolite; subsequently, multiple regressions were obtained to estimate the effective kinetic parameters of a linear model based on ODEs that integrates transcription and metabolism processes in steady state (Figure 2) [7]. Values 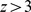 were used as optimal threshold in order to limit the number of possible gene-metabolite interactions and minimize the distance between the predicted and measured metabolic profiles over the training set in terms of average Pearson correlations (blue bars in Figure 2C; r = 0.85, 167 d.f.,
were used as optimal threshold in order to limit the number of possible gene-metabolite interactions and minimize the distance between the predicted and measured metabolic profiles over the training set in terms of average Pearson correlations (blue bars in Figure 2C; r = 0.85, 167 d.f., 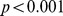 ). Hence, on average, each metabolite required 18 genes for explaining its behavior, thus a total of 959 genes was required to describe our tomato fruit metabolome. This subset of genes constitutes the effective transcription network. We performed a 5-fold cross-validation test to rule out dependence of the testing set, this reducing the metabolite average prediction (red bars in Figure 2C; r = 0.42, 167 d.f., p = 0.067 with a mean false positive rate (FPR) of 14% and a 56% mean positive predictive value (PPV) of predictors (bootstrap test,
). Hence, on average, each metabolite required 18 genes for explaining its behavior, thus a total of 959 genes was required to describe our tomato fruit metabolome. This subset of genes constitutes the effective transcription network. We performed a 5-fold cross-validation test to rule out dependence of the testing set, this reducing the metabolite average prediction (red bars in Figure 2C; r = 0.42, 167 d.f., p = 0.067 with a mean false positive rate (FPR) of 14% and a 56% mean positive predictive value (PPV) of predictors (bootstrap test, 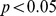 and
and 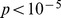 , respectively).
, respectively).
Figure 2. Predictive power and statistical significance of the effective global model of tomato fruit.

(A) Prediction of the agronomic properties experimentally measured over the 169 RILs. The straight line represents the exact prediction. (B) Distance between distributions of Pearson correlations for the fruit agronomic properties, metabolites and genes (green, red and blue points, respectively) over training sets and in random permutations of them with different noise levels. (C, D) Histogram of Pearson correlations between the measured and predicted metabolite and gene levels over their training sets (blue bars) and over sets with a 10- and 5-fold cross validation tests (red bars), respectively.
The next step was to construct an effective gene regulatory model able to predict autonomously the transcriptional processes that, by means of the model previously described, would generate a quantitative metabolic response. In this way changes at the transcriptional level resulting from the proposed genetic perturbations could be translated and predicted effectively into metabolic changes. For doing that, we used the microarray data obtained from fruits of 50 of the RILs to infer a network of gene-gene interactions. The CLR method provided the first sets (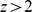 ) of predictor genes for each gene considered. Afterwards, LASSO method reduced the number of regulations per gene to a scale-free space following a power-law with exponent
) of predictor genes for each gene considered. Afterwards, LASSO method reduced the number of regulations per gene to a scale-free space following a power-law with exponent 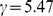 (
(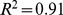 ) and an average of 26 interactions per gene. High values of similarity between the predicted and measured gene expression (blue bars in Figure 2D) were computed for the whole training set (
) and an average of 26 interactions per gene. High values of similarity between the predicted and measured gene expression (blue bars in Figure 2D) were computed for the whole training set (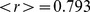 , 48 d.f.,
, 48 d.f., 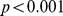 ) while for a 5-fold cross validation the average similarity (red bars in Figure 2D) was r = 0.59 (48 d.f.,
) while for a 5-fold cross validation the average similarity (red bars in Figure 2D) was r = 0.59 (48 d.f., 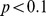 ) with a mean FPR of the 25% and a 63%mean PPV of predictors (bootstrap test,
) with a mean FPR of the 25% and a 63%mean PPV of predictors (bootstrap test, 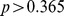 and
and 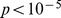 , respectively).
, respectively).
Specific metabolic combinations can reliably model different aspects of the fruit phenotype
We addressed the question of whether the agronomic/phenotypic properties of the tomato fruit could be determined by their metabolite composition. For that, we studied the relationship between agronomic properties and metabolic composition across 169 tomato RILs. We applied LASSO method to select a set of metabolites that may act as predictors for each agronomic property (Dataset S1). Our model included 47 metabolites observing considerably high Pearson correlations between the measured and predicted phenotypic responses over the 169 RILs for number of fruits per plant and fruit harvested across two different seasons, (Figure 2A; r = 0.62 and r = 0.73 respectively, 167 d.f., 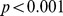 in both cases). A reduction to r = 0.46 (167 d.f.,
in both cases). A reduction to r = 0.46 (167 d.f., 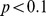 ) and r = 0.62 (167 d.f.,
) and r = 0.62 (167 d.f., 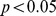 ) in the median correlation was computed in a 10-fold cross validation, with 84% mean PPV in both cases (bootstrap test,
) in the median correlation was computed in a 10-fold cross validation, with 84% mean PPV in both cases (bootstrap test, 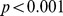 ), and mean FPR of 33% and 35% (bootstrap test,
), and mean FPR of 33% and 35% (bootstrap test,  in both cases), respectively. Average fruit weight and pH required as many as 44 metabolites as potential predictors with high reliability levels. Reliability was assessed by comparing the corresponding predicted and measured values for the 169 RILs (Figure 2A; r = 0.85 and r = 0.80, 167 d.f.,
in both cases), respectively. Average fruit weight and pH required as many as 44 metabolites as potential predictors with high reliability levels. Reliability was assessed by comparing the corresponding predicted and measured values for the 169 RILs (Figure 2A; r = 0.85 and r = 0.80, 167 d.f., 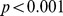 in both cases). A 10-fold validation only reduced those similarities to r = 0.73 and r = 0.63 (167 d.f.,
in both cases). A 10-fold validation only reduced those similarities to r = 0.73 and r = 0.63 (167 d.f., 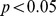 in both cases), with mean FPRs of 37% and 22% (bootstrap test,
in both cases), with mean FPRs of 37% and 22% (bootstrap test,  and
and 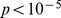 , respectively), and mean PPVs of 81% and 88% (bootstrap test,
, respectively), and mean PPVs of 81% and 88% (bootstrap test, 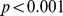 and
and 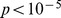 ), respectively. Additionally, to test how the metabolome contributed to an accurate prediction of tomato phenotype, we studied the relationship between agronomic properties and gene expression of the core of 959 genes across the 50 tomato RILs [27]. Note that we select this reduced set of genes as a core of potential predictors to avoid model over-fitting due to the low number of RILs with the transcript levels measured. Imposing the same criteria that was used to select metabolites as predictors, we observed that similarities between predicted and measured values of number of fruits per plant and harvested fruits increased (r = 0.80 and r = 0.81, 48 d.f.,
), respectively. Additionally, to test how the metabolome contributed to an accurate prediction of tomato phenotype, we studied the relationship between agronomic properties and gene expression of the core of 959 genes across the 50 tomato RILs [27]. Note that we select this reduced set of genes as a core of potential predictors to avoid model over-fitting due to the low number of RILs with the transcript levels measured. Imposing the same criteria that was used to select metabolites as predictors, we observed that similarities between predicted and measured values of number of fruits per plant and harvested fruits increased (r = 0.80 and r = 0.81, 48 d.f., 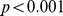 in both cases) while average of fruit weight and pH decreased (r = 0.79 and r = 0.73, 48 d.f.,
in both cases) while average of fruit weight and pH decreased (r = 0.79 and r = 0.73, 48 d.f., 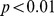 in both cases) (see dashed line in Figure S3A–B). Moreover, relaxing the threshold (
in both cases) (see dashed line in Figure S3A–B). Moreover, relaxing the threshold (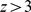 ) to include possible interactions agronomic variable-genes in the LASSO method, surprisingly similarities for all agronomic variables highly decreased (r<0.65, 48 d.f.,
) to include possible interactions agronomic variable-genes in the LASSO method, surprisingly similarities for all agronomic variables highly decreased (r<0.65, 48 d.f., 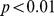 ; see Figure S3C–D). Hence, we illustrated an alternative way to described accurately phenotypic properties of tomato fruit by using gene expression profile of the reduced set of RILs.
; see Figure S3C–D). Hence, we illustrated an alternative way to described accurately phenotypic properties of tomato fruit by using gene expression profile of the reduced set of RILs.
Next, to test the specificity of the inferred model parameters, we perturbed the target phenotypic profile for each RIL adding different levels of noise. Figure 2B shows the distance between predicted and measured values (green points) and mean correlations for different noise levels. A similar approach was performed by using the metabolic and gene expression profiles (red and blue points, respectively). Correlations with significance levels higher than the indicated above were not considered in the cross-validations. In addition, we estimated a very low mean error in predicting the agronomic properties across the training set (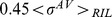 , see Methods).
, see Methods).
Genome design based on single perturbations results in discrete but consistent improvements in agronomic properties
Here, our main goal is to redesign the genome of tomato to generate an engineered surrogate that, if viable, would be easier to study and of greater potential biotechnological interest. Our design approach was inspired by the practice of in silico optimization over a predictive global model. Our next step was to test the possibility of improving agronomical properties of interest. We tested several scoring functions that fall into two global types: on the one hand, agronomical variables measured experimentally such as the number of fruits harvested per plant, the average fruit weight or its pH; and on the other hand, more complex fruit attributes that could be defined according to some of the components of the metabolic profile and are related to organoleptic properties of the fruit. In this later case, we first evaluated as proof of concept: fruit acceptability according to criteria based on acidity and sugars [28], quality as defined by the contribution of specific volatiles to aroma and by a reported [28] panel assessments of the tomato fruit and consequently on organoleptic acceptance. For this latter case we assumed a strong influence of a set of metabolites to be either maximized (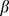 -ionone,
-ionone, 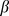 -damascenone, 2-phenylethanol and benzaldehyde) or minimized (methyl salicylate, guaiacol, hexanal, 1-penten-3-one and (E)-2-hexenal) using balanced weighting factors to account for their positive or negative contribution to quality. Moreover, all single metabolites were also optimized in single target analyses. Finally, a bi-objective function that included a high trade-off was proposed to optimize fruit quality and its production. As a first approach, we re-engineered tomato genome by perturbing independently the 959 genes included in the model, then we re-computed the scoring functions for all RILs enumerating all single knockouts and finally, all gene over-expression models were obtained.
-damascenone, 2-phenylethanol and benzaldehyde) or minimized (methyl salicylate, guaiacol, hexanal, 1-penten-3-one and (E)-2-hexenal) using balanced weighting factors to account for their positive or negative contribution to quality. Moreover, all single metabolites were also optimized in single target analyses. Finally, a bi-objective function that included a high trade-off was proposed to optimize fruit quality and its production. As a first approach, we re-engineered tomato genome by perturbing independently the 959 genes included in the model, then we re-computed the scoring functions for all RILs enumerating all single knockouts and finally, all gene over-expression models were obtained.
Hence, mimicking the optimization patterns typical from LM, the landscape of desired agronomic properties of tomato fruit was exhaustively explored perturbing its effective transcriptional regulatory network (TRN) with single-gene alterations. Figure 3A shows the improvement of two of the agronomic properties mentioned above (fruit acceptability and quality vs production) as result of single gene perturbations according to our model. The success of the approach is shown by the efficiency function obtained for each transcriptional perturbation computed and which is defined by the normalized ratio between the agronomic property obtained for the re-engineered TRN and that for the wild-type TRN. Both agronomic properties and efficiencies in the case of single-perturbations were computed for each of the 169 RILs, resulting in a high variability between the lineages for all knockouts and over-expressed gene re-engineered TRN cases. We corroborated that there is a highly significant linear correlation (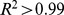 ,
, 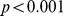 for fruit acceptability and quality vs production) between the average value of the improved agronomic properties and the efficiencies reached across the set of RILs for all transcriptional perturbations. Both gene knockout and over-expression models resulted in similar linear regression slopes when considering acceptability and quality vs production together (0.05 and 0.24, respectively, Figure 3A). In addition, we also explored the possibility of tuning a given agronomic property towards a defined value, as it is desired for some biotechnological applications (see Text S1); achieving also in this case high efficiency values (Figure S4 and Tables S1 and S2).
for fruit acceptability and quality vs production) between the average value of the improved agronomic properties and the efficiencies reached across the set of RILs for all transcriptional perturbations. Both gene knockout and over-expression models resulted in similar linear regression slopes when considering acceptability and quality vs production together (0.05 and 0.24, respectively, Figure 3A). In addition, we also explored the possibility of tuning a given agronomic property towards a defined value, as it is desired for some biotechnological applications (see Text S1); achieving also in this case high efficiency values (Figure S4 and Tables S1 and S2).
Figure 3. Exploration and statistical significance of the landscape of multiple agronomic properties of interest for tomato fruit applying local perturbations in its effective TRN.

(A) Agronomic properties improved by perturbing a single gene as function of efficiency reached by that transcriptional perturbation with respect to the wild-type scenario; only perturbations causing positive mean efficiencies are plotted. Both agronomic properties and efficiencies of a single perturbation are tested on the 169 RILs and error bars represent their minimum and maximum values in both axis. (B) Relationship between agronomic properties in the wild-type genome and the average of the agronomic properties resulting of all single perturbations in the wild-type TRN for each RIL; vertical error bars represent the best and worst optimized re-engineered TRN for a given RIL. (C) Average number of single gene perturbations that overcome a given efficiency threshold in the 169 RILs (light bars; error bars represent standard deviation for the 169 RILs) and average probability of selecting the same gene-perturbation in a set of RILs (dark bars; error bars show standard deviation for all genes of the TRN). Left and right columns represent perturbations of single gene in case of knockout or over-expression, respectively. (A, B) show fitness as related to the acceptability of tomato fruit (blue) and production vs. quality (red); (C) and fitness values associated to maximize only fruit quality (green). Agronomic properties are plotted in arbitrary units.
After this, we ranked the list of knockout/over-expressed genes of the TRN according to two criteria directed to maximize: (i) the mean efficiency across all lineages in the case of goals such as acceptability and quality vs production; and (ii) the average of the maximum agronomic property reached by all possible TRN reconfigurations in the case of fruit quality (Dataset S2). Specifically, Table 1 shows the top 5 genes proposed for knockouts or over-expressed depending on the fitness evaluated. Fruit acceptability could be improved to 2.91% or 8.84% using gene knockout (i.e., LE24K20) or over-expression (i.e., LE13M10) in all lineages, respectively. By contrast, quality was highly increased achieving improvement ratios of 43.34% by gene knockout (i.e., LE24K20) and 227.31% by over-expression of LE15D07. Finally, taking into account not only the quality but also fruit production, ratios decreased to 15.32% (i.e., LE13F23) and 35.94% (i.e., LE14B20) using the two types of perturbations, respectively. Notice that all these rates of improvement were achieved in the lineages that provided maximum fitness in the wild-type TRN.
Table 1. The top 5 single-gene knockouts and over-expressions that maximize the agronomic properties of the tomato fruit resulting of optimize several objectives.

Notice that the first five genes is the top 5 of single-gene knockouts and the following five is the top 5 in over-expression.
Efficiencies were selected in the RIL where the perturbation maximizes the fitness.
Probability of selecting the given perturbation across the set of RILs at the maximum level of efficiencies.
Lineages exhibited variability in their resistance to be optimized and this resistance changed with each target agronomic property. Figure 3B shows a strong linear dependence between the level of the agronomic property in the wild-type TRN and the average level of the agronomic properties resulting from all single perturbations in the TRN for each RIL (linear regression slope in the range 0.99–1.12 and 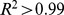 ,
, 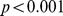 ). Interestingly, we observed that the effect of predicting agronomic properties under genetic perturbations was not dependent on the lineage selected. This provided a high level of robustness when we selected the lineages to implement experimentally re-designed TRN.
). Interestingly, we observed that the effect of predicting agronomic properties under genetic perturbations was not dependent on the lineage selected. This provided a high level of robustness when we selected the lineages to implement experimentally re-designed TRN.
We computed the average number of single-gene perturbations to overcome an efficiency threshold given in the 169 RILs and the average probability of selecting the same gene-perturbation commonly for the whole set of RILs. The right panel in Figure 3C shows that only a few gene knockouts were able to improve fruit acceptability with a high probability in all lineages whereas, on the other hand, tens of gene knockouts could be proposed for increasing fruit quality and for the quality and production. On the other hand, the left panel in Figure 3C allowed re-asserting that re-engineering the TRN by gene over-expression could result in higher increments in the agronomic properties and with a higher density of suggested perturbations across the RILs.
A sub-optimal design landscape can be proposed using multiple genetic perturbations
The next step in our study was to propose new genome re-designs including multiple perturbations. To do this, we sampled widely the landscape of the acceptability, quality and quality vs production of tomato fruits by introducing two-gene perturbations either by knockouts and over-expressions (Dataset S3). Figure 4A shows the median efficiencies reached by two-gene transcriptional perturbations based on knockouts and over-expression in order to improve the agronomic properties defined as multiple-objective. As expected, we corroborated that multiple perturbations, located in different pathways (Table 2), could improve the agronomic properties significantly better than single perturbations. Table 2 lists the best gene-pairs to be used in perturbations that maximize such agronomic properties of the fruit. Figure 4B shows the average number of single gene perturbations that are able to overcome a given efficiency threshold for the top 5 RILs when ranked for single perturbations as well as the average probability of selecting the same multiple-perturbation commonly in a set of RILs.
Figure 4. Experimental validation of the landscape of tomato agronomic properties by using genetic perturbations.

Heuristic exploration (A) and statistical significance (B) of the landscape of multiple desired agronomic properties of tomato fruit perturbing its effective TRN adding multiple genetic changes and, predictive power (C–F) for optimizing the levels of volatile compounds and identifying compounds in closed metabolic pathways. (A) Median efficiencies reached by transcriptional perturbation based in gene knockouts or over-expression to improve agronomic properties. (B) Average number of single gene perturbations that overcome an efficiency threshold in the top 5 RILs scored by single perturbation (light bars; error bars represent standard deviation for the selected RILs) and average probability of selecting the same multiple-perturbation commonly in a set of RILs (dark bars; error bars show standard deviation for all genes of the TRN). Precision, recall and F-score (green, red and blue lines, respectively) compare observed experimentally volatile compound correlations vs inferred set of potential genetic perturbations (gene knockout (C, D) or over-expression (E, F)) shared to optimize each compound independently. Note that experimental metabolite correlations r<0.5 were not considered in (D, F).
Table 2. The top 10 pairs-gene knockouts or over-expressions that maximize the agronomic properties of the tomato fruit.
| Gene | Gene Annotation | Efficiency 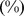 1
1
|
| Acceptability | ||
| LE33G22; LE28J07 | Adenylate kinase, putative; vesicle-associated membrane protein, putative | 16.54 |
| LE15D09; LE33G22 | Vesicle-associated membrane protein, putative; adenylate kinase, putative | 16.54 |
| LE17M21; LE33G22 | Selenoprotein O, putative; adenylate kinase, putative | 16.40 |
| LE17D17; LE33G22 | F-box family protein; adenylate kinase, putative | 16.37 |
| LE15E19; LE33G22 | Ribosomal protein; adenylate kinase, putative | 16.07 |
| LE7I21; LE33G22 | Proline-rich cell wall protein-like; adenylate kinase, putative | 15.90 |
| LE33G22; LE23K21 | Adenylate kinase, putative; amino acid transporter, putative | 15.87 |
| LE33G22; LE2C08 | Adenylate kinase, putative; chloroplast lumen common family protein | 15.85 |
| LE33G22; LE25J09 | Adenylate kinase, putative; AT-HSFA6B, DNA binding/transcription factor | 15.84 |
| LE33G22; LE24D10 | Adenylate kinase, putative; not found | 15.78 |
| Quality (aroma and taste) | ||
| LE27F15; LE29L05 | Protein kinase family protein; branched-chain amino acid aminotransferase | 422.60 |
| LE16D08; LE6G08 | Similar to 60S ribosomal protein L35; sucrose phosphate synthase | 360.63 |
| LE9A08; LE15E23 | GRAM domain-containing protein/ABA-responsive protein-related; putative threonyl-tRNA synthetase | 303.04 |
| LE18E13; LE8A19 | MYB transcription factor; putative glycerophosphoryl diester phosphodiesterase family protein | 263.91 |
| LE32B05; LE4D06 | YABBY2-like transcription factor YAB2; tRNA-dihydrouridine synthase A, putative | 253.33 |
| LE15L08; LE4J06 | Putative rac protein; 50S ribosomal protein L27, chloroplastic | 244.32 |
| LE29E13; LE13F06 | Fyve finger-containing phosphoinositide kinase, fyv1, putative; transmembrane protein, putative | 242.56 |
| LE13F06; LE15J03 | Transmembrane protein, putative; ankyrin-like protein | 240.19 |
| LE17G02; LE15D07 | Pantothenate kinase, putative; polynucleotide kinase- 3′-phosphatase, putative | 239.10 |
| LE15D07; LE20I03 | Polynucleotide kinase- 3′-phosphatase, putative; DEX1, calcium ion binding | 239.03 |
| Quality vs production | ||
| LE13F06; LE15J03 | Transmembrane protein, putative; ankyrin-like protein | 49.79 |
| LE12O13; LE33G22 | Prefoldin subunit, putative; adenylate kinase, putative | 49.16 |
| LE2C24; LE29J02 | ATAB2; RNA binding; GTP-binding protein LepA homolog | 49.15 |
| LE12P11; LE2C24 | Not found; ATAB2; RNA binding | 48.81 |
| LE2C24; LE21J01 | ATAB2; RNA binding; Dolichyl-phosphate beta-glucosyltransferase, putative | 48.28 |
| LE12O13; LE25M06 | Prefoldin subunit, putative; Pre-mRNA-processing protein prp39, putative | 46.63 |
| LE12O13; LE14B20 | Prefoldin subunit, putative; clathrin adaptor complexes medium subunit family protein | 46.18 |
| LE14B20; LE21J01 | Clathrin adaptor complexes medium subunit family protein; dolichyl-phosphate beta-glucosyltransferase, putative | 44.86 |
| LE33B09; LE2C24 | Not found; ATAB2; RNA binding | 44.64 |
| LE18M21; LE14B20 | Cysteine protease; clathrin adaptor complexes medium subunit family protein | 44.05 |
Efficiencies were selected in the RIL where the perturbation maximizes the fruit acceptability, quality and, quality vs production (RILs 103, 142, and 135, respectively).
Knockout genes were showed in bold type and the others were gene over-expressed.
Model validation: the proposed genetic perturbations in re-engineered fruits with modified aroma reconstruct the correlation matrix found experimentally between aroma volatile compounds
After generating our predictive model for the TRN and metabolism of tomato fruit, we use it to automatically design tomato genomes with extreme alterations for each of the 56 volatile compounds by introducing a set of genetic perturbations. We compared sets of genetic perturbations for all pairs of volatile compounds and then inferred their levels of correlations (see Methods). Hence, these predicted correlations were compared to the levels of correlations obtained from the experimental values for each volatile pair that often reflects their belonging or not to the same metabolic/regulatory pathway or to be or not structurally related. Figure 4C–4F shows the predictive power of our model to determine correlations between all the volatile compounds. Interestingly, selecting a correlation cut-off between 0.5 and 0.8 we obtained high performance 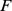 -scores (see Methods section) ranging between 0.32 and 0.91 (Figure 4D) for gene knockouts and between 0.31 and 0.80 when model selected genes by over-expression (Figure 4F). Notice that only pairs of experimental volatile compounds with
-scores (see Methods section) ranging between 0.32 and 0.91 (Figure 4D) for gene knockouts and between 0.31 and 0.80 when model selected genes by over-expression (Figure 4F). Notice that only pairs of experimental volatile compounds with 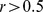 were considered. Predictions decreased when we incorporated all pairs of compounds (Figure 4E–4F) indicating that our model captured high correlations observed experimentally with more precision. Figure S5 shows the dendograms of the volatile compound obtained from the correlation of experimentally obtained volatiles levels and the dendograms obtained using as distance between volatile compounds the number of common genetic perturbations proposed by the model. We observed that perturbations proposed by gene over-expression were pivotal to predict computationally significant distances between volatile compounds (Mantel test: r = 0.54, 1540 d.f.,
were considered. Predictions decreased when we incorporated all pairs of compounds (Figure 4E–4F) indicating that our model captured high correlations observed experimentally with more precision. Figure S5 shows the dendograms of the volatile compound obtained from the correlation of experimentally obtained volatiles levels and the dendograms obtained using as distance between volatile compounds the number of common genetic perturbations proposed by the model. We observed that perturbations proposed by gene over-expression were pivotal to predict computationally significant distances between volatile compounds (Mantel test: r = 0.54, 1540 d.f., 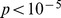 ) thus providing high support to our model. By contrast, predicted perturbations based on gene knockout could only identify a small fraction of the entire dendogram (Mantel test: r = 0.38, 1540 d.f.,
) thus providing high support to our model. By contrast, predicted perturbations based on gene knockout could only identify a small fraction of the entire dendogram (Mantel test: r = 0.38, 1540 d.f., 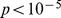 ).
).
To give further support to our model we constructed experimentally two inbred lines (ILs) derived from another interspecific cross whose transcriptome and metabolome were also experimentally measured. Parents of these ILs are a different cultivar of tomato M82 and a S. pennelli accession and therefore represent a completely different set of gene alleles from those in RILs used to construct the model. These ILs can be used as independent and useful test case to evaluate the validity of the model. We corroborated that a significant set of genetic perturbations suggested by computational design to optimize the phenotype observed were identified as genes differentially altered in the target phenotype (Text S1 and, Figure S6).
Discussion
LM is a methodology that is being implemented by large industries to optimize their production. In the process of decision making applied to the redesign of production systems, firstly, engineers evaluate systematically the addition or elimination of resources in each of the participating single processes; afterwards, multiple changes are considered trying to achieve maximum quality and production [29]. Translating this engineering approach to a cellular molecular factory and identifying the basic functional elements has allowed us to develop a design methodology that optimizes the genome, resulting in a more desirable phenotypic properties. In addition, by mimicking the methodology from LM we have provided a first robust optimization to redesign an optimal genetic network based on the systemic exploration of the effects of a large number of single gene knockout and over-expression genotypes; then, a second multiple-optimization of random paths allowed improving substantially the desired agronomical properties. The success of this approach indicates that despite the existence of molecular interactions, the model is able to overcome this limitation and results in a good predictor.
We have proposed several re-engineered genomes that improve desired agronomic properties of the fruit by targeting single or multiple genetic modifications. It has been previously reported that single under-/over-expressed of certain genes may affect fruit quality traits, being these key genes involved in the biosynthesis of a product of fruit metabolism or to a general ripening regulators (i.e., carotenoids [30]). We have explored single perturbations by gene knockout or over-expression and our results indicated that a significantly better fine-tuning could be obtained by using over-expression approaches. We observed that improvement ratios could reach even more than 4-fold the wild-type value of most of phenotypes desired by designing genomes with only two genetic perturbations (Figure 4A and Table 2). The magnitude of the predicted change sometimes may appear low but an improvement in a quantitative trait, if consistent and predictable, maybe economically important. Indeed, a good combination of high yield with even slightly increased solid solids content is a major breeding goal for processing tomatoes that it is difficult to be achieved [31] because of polygenic nature and pleiotropic relationships of both traits [32].
Although it is not the objective of this paper, it does not escape our attention that some of the perturbations proposed are consistent with the biological processes associated to the trait and therefore the model could be used to reveal the molecular underpinnings of quality traits (see experimental evidences of each gene perturbation proposed by the model in the Dataset S4). For instance the role of YABBY (a gene proposed by our model to affect quality) in controlling fruit size probably through the auxin pathway and the effect of auxin in altering fruit growth and ripening has been previously reported [33], [34]. Similarly the importance of phytoene desaturase to affect carotenoids and carotenoid derived volatiles has been reported [35]. Most of the genes proposed by the models however are new, therefore opening new avenues of research either by targeting in transgenic plants, identification of mutants in those genes by TILLING [36] or by TAL engineering [37], as well as to be used as an additional guide during plant breeding. In principle these modifications are to be implemented in red fruit or around red fruit stage either genetically or by the use of external elicitors (physical or chemical) and our model provides roadmap for those approaches. Our methodology takes advantage of our ability to predict variations in fruit cell phenotype based on changes in the transcriptome. The linear relationships shown in Figure 3 (A, C, and D) guarantees that by optimizing our effective transcriptomic, metabolic or phenotypic fitness we are also optimizing the phenotype measured experimentally of the tomato fruits. While it is true that complex multi-organ organism such as tomato rely on the coordination and transport of multiple signals and nutrients from different parts of the plants to achieve the final phenotype, and this is especially true for the fruit [19], [38], it not less true that the most important part of the fruit characteristics at ripening depends basically on the fruit program before around the ripening stage [39], [40].
The ability to target redesign crops for enhanced content of metabolites of interest has been experimentally achieved in a number of cases (for instance vitamins C [41] and E [42]) using transgenic approaches and the information of bottlenecks or limiting steps for the biochemical pathways of the compounds of interest. The most dramatic examples of this have been introducing the new trait in a genetic background with very low value for it (i.e., golden rice [43]) using ectopic expression of one or several foreign genes. The use of natural genetic variability in combination with our nonbiased (hypothesis-free) modeling approach allows us to identify new candidate genes as potential targets to engineer the plant (although the biotechnological use of more active orthologs from other organisms is not discarded in our approach). The existence of regulatory networks connecting primary and secondary metabolism in plants should also be taken into consideration in future attempts to metabolically engineer the various classes of plant secondary metabolites [44]. It is interesting that known genes in the biosynthesis path often do not co-localize with quantitative trait locus for the metabolites in the path [35] indicating that there is ample of opportunities to be explored for metabolite and quality improvement, and our model fits nicely in this gap.
Materials and Methods
Plant material, transcriptomic, metabolomic and phenomic data
The construction of the tomato RILs used in this study has been described elsewhere [45]. Triplicate samples of red ripe fruits (each representing at least 5 fruit) from each of 169 RILs were harvested and analyzed for volatile compounds as described in [46]. For method validation, red ripe fruits from five ILs with a different genetic background [47] were used. Transcript profile datasets (11876×3×50 data points) were obtained from triplicate fruit samples of 50 selected RILs using TOM2 microarray, as previously reported [48]. Data sets corresponding to the rest of metabolites and phenomic data were obtained as in [46] from triplicate samples of the 169 RILs. To decrease experimental variability, the same fruits representing each RIL were homogenized and divided in different aliquot samples for the different metabolite or transcript profiling techniques. Before use all transcriptomic, metabolomic and phenomic data were normalized and transformed to log-scale. The ILs used for model validation have been described previously [21].
Mathematical model
An effective linear model based on ODEs each providing the steady states of tomato fruit mRNA was used to describe transcriptional gene regulations [7]. Thus, the mRNA steady state from the 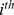 gene,
gene, 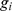 , is given by
, is given by  , where
, where 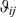 represents the regulatory effect that gene
represents the regulatory effect that gene 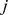 has on gene
has on gene  . Each gene expression value is contained
. Each gene expression value is contained 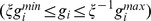 in a range interval defined by the minimum
in a range interval defined by the minimum  and maximum
and maximum 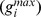 value of all its experimental measurements obtained from the subset of 50 RILs used for transcript profiling.
value of all its experimental measurements obtained from the subset of 50 RILs used for transcript profiling. 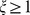 is a tunable parameter that decreases the gene expression range to improve the predictive capacity of the presented model under genetic predictions. The dynamics of metabolic profile was computed by
is a tunable parameter that decreases the gene expression range to improve the predictive capacity of the presented model under genetic predictions. The dynamics of metabolic profile was computed by 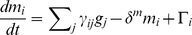 , where
, where 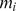 is the steady-state concentration from the
is the steady-state concentration from the 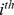 metabolite,
metabolite, 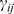 is the regulatory strength that gene
is the regulatory strength that gene 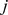 has on metabolite
has on metabolite  . Hence, agronomic variables (
. Hence, agronomic variables (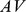 ) were predicted by means of a linear combination of the metabolic profile,
) were predicted by means of a linear combination of the metabolic profile, 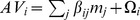 , where
, where  is the regulatory effect that metabolite
is the regulatory effect that metabolite 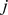 has on agronomic variable
has on agronomic variable  .
. 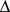 ,
,  and
and  are the perturbation terms that allow to calibrate gene expression, metabolic profiles and predicted agronomic properties, respectively, for all RILs. Notice that degradation coefficients of genes and metabolites (
are the perturbation terms that allow to calibrate gene expression, metabolic profiles and predicted agronomic properties, respectively, for all RILs. Notice that degradation coefficients of genes and metabolites (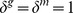 , respectively) scaled time conveniently and that we assumed the model in steady state (
, respectively) scaled time conveniently and that we assumed the model in steady state (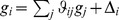 and
and 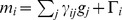 ).
).
Construction of an effective transcriptional regulatory network connected with metabolism to explain agronomic properties
Our global model consists of three blocks of algebraic equations covering respectively from gene expression, through metabolic profile until agronomic properties, and in all three cases the same methodology was applied. The inference procedure consisted of two nested steps. Firstly, the network connectivity was inferred by using the InferGene algorithm [7]. This method uses mutual information with a local significance value (z-score computation) to obtain the effective regulations. Hence, the potential interaction between a predictor and a target is z-scored, constituting an estimator of the likelihood of mutual information. Subsequently, we selected a z-score threshold for a predictor cutoff. In a second step, LASSO method was used to avoid over-fitting and to estimate the kinetic parameters of each effective model. Notice that the 8.7% of the selected genes in the TRN were annotated as TFs and 16.2% as encoding enzymatic activities and, in neither case, they were over-represented since both the tomato genome and the whole array contain similar fractions of TFs (8.8%) and enzymes (17.1%).
For the construction of the effective TRN model and its later integration with the metabolism, we used steady-state mRNA expression profiles derived from RILs transcriptionally and metabolically characterized. The dataset contains pre-processed expression data from 50×3 = 150 hybridization experiments using an array with 11876 probe sets spotted, and data for levels of 67 metabolites that were quantified over the same sample set. For this study, we only considered the 5592 genes whose expression values could be consistently found in more than 80% of the microarrays. We found 1057 TFs and 1962 genes with enzymatic activity after searching for the motifs transcription regulator and enzyme activity respectively in the functionally annotated tomato genome (Tom2). Moreover, all 169 RILs (including the previous 50 ones) for which we had metabolite and phenotype data were used to train a linear model able to predict agronomic properties of the fruit from potentially predictor metabolites. In all cases transcriptomic and metabolomic data were first normalized using the LOWESS procedure [26] and subsequently converted into z-scores across the RILs. In order to calibrate gene expression and metabolite concentration, both models included a perturbation term (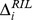 and
and 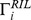 , respectively) to fit all their
, respectively) to fit all their  -genes and
-genes and 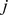 -metabolites for a given RIL. We assumed a constant perturbation in the gene expression prediction because of its low variation across the training set (standard deviation of
-metabolites for a given RIL. We assumed a constant perturbation in the gene expression prediction because of its low variation across the training set (standard deviation of 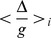 for all RILs is 0.072-fold the standard deviation of gene expression,
for all RILs is 0.072-fold the standard deviation of gene expression, 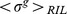 ) with respect to the mean value,
) with respect to the mean value, 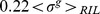 . Similarly, the average error to predict the metabolic profile across the training set was increased to
. Similarly, the average error to predict the metabolic profile across the training set was increased to 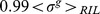 .
.
Three plain text files containing the transcriptional, metabolic and phenotypic model for tomato were constructed and are available in Dataset S1. A directed network was constructed which places genes and metabolites as nodes and effective transcriptional and gene-metabolite interactions as edges. For the transcriptional interactions, edges link genes (including TFs, enzymes and genes without ability to regulate) to other genes or to a metabolite, in the case of metabolism.
Genome-wide multiple-optimization
Our algorithm searches possible reconfigurations of the global effective transcription regulatory network of tomato such as that the specified agronomic properties are improved (maximized or minimized) with respect to the properties of interest obtained in a given RIL. Different properties of interest have been optimized, ranging from single metabolites defining the sweetness or sourness of the fruit, to linear combinations of a set of metabolites determining the quality in terms of flavor and taste and even further to include objective functions that try to integrate two of those goals with a trade-off and balanced weighting factors such as fruit quality and yield.
We have addressed this optimization problem using two approaches. Firstly, we exhaustively enumerated all possible single gene knockouts and over-expression for each case to be optimized under a given selective pressure of interest. Second, we ranked all possible perturbations according to the new agronomic properties they would generate. The third step was to suggest genome reconfigurations that include multiple actions: gene knockouts, over-expressed genes, or both, in order to enlarge the combinatorial space of perturbed genomes. To do that, we have used an exhaustive method aimed at finding the global optimum in the space of all possible synthetic TRN. We started from the inferred model (see Mathematical model above) and applied an optimization scheme. At each step of the optimization process, we selected each gene among the ones involved in the transcriptomic model to evaluate the effect of three possible approaches (knockout, over-expression or wild-type scenario); we updated the model with the genetic perturbation that provided the best score. Note that to simulate knockout or over-expression in the gene  , we substituted its ODE by the minimum (
, we substituted its ODE by the minimum ( ) or maximum (
) or maximum (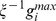 ) values respectively observed in the range of diversity of the 50 RILs.
) values respectively observed in the range of diversity of the 50 RILs.
Experimental and computational metabolite correlation
We computed the sets of single-gene perturbations, ?, by gene knockout or over-expression that alter significantly the levels of the 56 volatile metabolites representing the volatile compounds taking into account the global model. For the sake of the model we considered only those gene perturbations that would cause significant changes in the metabolite concentration higher than 1% (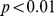 ).
). 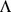 can be divided into genetic modifications that increase (
can be divided into genetic modifications that increase ( ) or decrease (
) or decrease ( ) the metabolite concentrations, respectively. Hence, correlations between metabolite pairs
) the metabolite concentrations, respectively. Hence, correlations between metabolite pairs  and
and 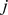 (
(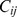 ) were calculated as the difference between
) were calculated as the difference between 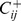 and
and 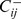 by using the set of single-gene perturbations proposed by the model
by using the set of single-gene perturbations proposed by the model
where 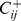 and
and 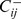 is the maximum normalized intersection predicted between the set of gene perturbations proposed by altering positively or/and negatively, respectively. We used these correlations to compute dendograms of all volatile compounds by using the distance inferred by the model (
is the maximum normalized intersection predicted between the set of gene perturbations proposed by altering positively or/and negatively, respectively. We used these correlations to compute dendograms of all volatile compounds by using the distance inferred by the model (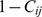 ) depending on the
) depending on the  selected by gene knockout or over-expression.
selected by gene knockout or over-expression.
The performance of the inferred metabolite correlations was evaluated using as a reference a set of empirical correlations previously obtained among these metabolites. We used different cut-offs, k, to identify metabolite correlations (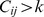 ). The fraction of metabolite pairs that were correctly predicted by the model (precision,
). The fraction of metabolite pairs that were correctly predicted by the model (precision,  ) and the fraction of all known correlations that were discovered by the model (sensitivity,
) and the fraction of all known correlations that were discovered by the model (sensitivity, 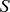 ) were used to compute a performance statistic defined as
) were used to compute a performance statistic defined as 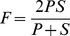 .
.
Robustness of statistical inferences in the model construction
To estimate the range of 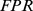 and
and 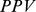 statistics computed in the different cross-validations of the model, a bootstrap method was used. To this end, we generated 10000 random lists (with replacement) of metabolites/genes of size equal than the set of metabolites/genes proposed by the model as predictors of agronomic properties/metabolites/genes. Each of these random lists was then compared to the actual list of predictors proposed by the model and the corresponding
statistics computed in the different cross-validations of the model, a bootstrap method was used. To this end, we generated 10000 random lists (with replacement) of metabolites/genes of size equal than the set of metabolites/genes proposed by the model as predictors of agronomic properties/metabolites/genes. Each of these random lists was then compared to the actual list of predictors proposed by the model and the corresponding 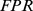 and
and 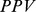 values computed to construct their expected null distributions. The observed
values computed to construct their expected null distributions. The observed  and
and 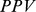 values were contrasted against these distributions and their significance assessed.
values were contrasted against these distributions and their significance assessed.
Supporting Information
Transcriptional, metabolic and phenotypic models of tomato fruit.
(XLS)
Single knockout and over-expressed genes to improve desired agronomic properties (acceptability, quality and quality vs production of tomato fruits; four volatile compounds; vitamin C, and different types of sugars and acids) and functional categorization of genes that induced high degree of improvement in those agronomic properties; notice that functional enrichment of all genes involved in the TRN was included. Gene ontology enrichment analyses were performed using the TFGD tool [TFGD]. It is also showed the functional categories significantly represented among those genes that were selected to describe the TRN of tomato fruit. A total of 19 cellular processes and 45 biological components were represented. Among these, genes related to cellular metabolic processes were the most abundant (p<0.0001), what makes sense since they were selected to predict cellular metabolism; whereas genes related to response to nutrient stimulus were present but the least common (p<0.1).
(XLS)
Multiple combinations of knockout and over-expressed gene sets to improve desired agronomic properties (acceptability, quality and quality vs production of tomato fruits).
(XLS)
Experimental evidences of each gene perturbation proposed by the model to optimize the different scoring function used.
(XLS)
Synthetic biology of tomato fruit vs computer science.
(PDF)
From data to global models to redesign using an approach based on synthetic biology.
(PDF)
Phenotype prediction (number of fruits per plant, fruit harvested, average fruit weight and pH) by using the genotype described in the 50 RILs in which transcript levels were measured. Pearson coefficient correlation (A,C) between the predicted and measured phenotypic profile and number of genes (B,D) selected by LASSO method as predictors for different thresholds of the fitting parameter (tLASSO). Note that we used two different z-score levels (z = 2, (A,B); and z = 3 (C,D)) to included genes as possible predictors to be selected by LASSO. The dashed line plotted in (A,B) shows the parameter, tLASSO, and the level of z-score used to constructed the relationship between phenotype and metabolome.
(PDF)
Exhaustive exploration and statistical significance of the landscape of single desired agronomic properties of tomato fruit (vitamin C, blue; fructose and glucose, red; and citric and malic acids, green) perturbing its effective TRN locally. (A) Agronomic properties improved by perturbing a single gene as function of efficiency reached by that transcriptional perturbation with respect to the wild-type scenario; notice that only perturbations with positive mean efficiencies are plotted. Both agronomic properties and efficiencies of a single perturbation are average variables tested on the 169 RILs and error bars represent their minimum and maximum values in both axis. (B) Dependence between agronomic properties in the wild-type genome and the average of the agronomic properties resulting of all single perturbations in the wild-type TRN for each RIL; vertical error bars represent the best and worst optimized re-engineered TRN for a given RIL. (C–D) Average number of single gene perturbations that overcome an efficiency threshold in the 169 RILS (light bars; error bars represent standard deviation for the 169 RILs) and average probability of selecting the same gene-perturbation commonly in a set of RILs (dark bars; error bars show standard deviation for all genes of the TRN). Left and right columns represent perturbations in terms of single gene knockout or overexpression, respectively.
(PDF)
(A) Dendogram of the volatile compound correlations observed experimentally. (B, C) Dendograms inferred by the model defining the distance between volatile compound as the number of common genetic perturbations predicted to optimize the levels of each volatile compound.
(PDF)
Percentage of altered genes (via gene knockout or over-expression; blue bars) proposed by the model to minimize the levels of volatile compounds (linalool (A) or, 1-nitro-2-phenylethane, 2-isobutylthiazole and benzylnitrile (B)) that were found significantly over-/under-expressed in the transcriptome of two ILs characterized experimentally with extremely low levels of those volatile compounds. The cut-off of the coefficient of variation between replicates was 75%. The Mann-Withney's U-test significance using random selection of gene perturbations (red bars) is shown (***statistically significant). Error bars represent the standard deviations of scores obtained from three ILs. 16.7% of the over-expressed genes proposed by the model to minimize the level of linalool were significantly recovered in gene expression (Figure S4A). In addition, 1.89% and 3.33% of genes candidates to be knockout or over-expressed (Figure S4B), respectively, also were identified significantly altered in the gene expression of the IL in which the three volatile compounds were found in minimum amount indicating this part of the transcriptome is relevant and associated to this volatile sub-phenotype among the other differential traits in these ILs.
(PDF)
Correlations observed between agronomic variables and metabolites of different fruit genotypes generated by simulating all possible single gene knockout (A–E) or over-expression (F–J) in the wild-type genome model of the tomato fruit. Standard deviations of all metabolites or agronomic variables show the diversity generated by implementing each genetic perturbation in the 169 RILs. Note that we only plotted re-engineered genomes whose transcriptome predicted showed errors lower than 1% (241 d.f. and 25 d.f. for knockout and over-expressed genes, respectively).
(PDF)
The top 5 single-gene knockouts and over-expressions that maximize the agronomic properties of the tomato fruit based on improve only one objective.
(PDF)
The top 5 single-gene knockouts and over-expressions that minimize the agronomic properties of the tomato fruit based on improve only one objective.
(PDF)
Genome design based on single perturbations to fine-tuning phenotypes with biotechnological interests. Model validation: fine-tuning tomato phenotype of two experimental inbred lines by computational genome design. Prediction of phenotypic correlations in re-engineered tomato fruits.
(PDF)
Acknowledgments
We thank Sophie Mirabel for excellent technical skills in microarray hybridization, J. Forment for help with computer resources and, G. Rodrigo and F. Heras for his fruitful comments.
Footnotes
The authors have declared that no competing interests exist.
This work was supported by grant TIN2006-12860 from the Spanish Ministerio de Ciencia e Innovación), the Structural Funds of the European Regional Development Fund (ERDF), FP7-ICT-043338 (BACTOCOM), the FP7-ICT-265505 (CADMAD), the ATIGE-Genopole, and the Fondation pour la Recherche Medicale grants to AJ, and by grant BFU2009-06993 from the Spanish Ministerio de Ciencia e Innovación to SFE and ESPSOL Fundación Genoma España and EUSOL European Commission Contract number: FOOD-CT-2006-016214, to AG. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Baker D, Church G, Collins JJ, Endy D, Jacobson J. Engineering life: building a fab for biology. Nature. 2006;296:44–51. doi: 10.1038/scientificamerican0606-44. [DOI] [PubMed] [Google Scholar]
- 2.Endy D. Foundations for engineering biology. Nature. 2005;438:449–453. doi: 10.1038/nature04342. [DOI] [PubMed] [Google Scholar]
- 3.Knight TF. Engineering novel life. Mol Syst Biol. 2005;1:20. doi: 10.1038/msb4100028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andrianantoandro E, Basu S, Karig D, Weiss R. Synthetic biology: new engineering rules for an emerging discipline. Mol Syst Biol. 2006;2:28. doi: 10.1038/msb4100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feist AM, Herrgard MJ, Thiele I, Reed JL, Palsson B. Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol. 2008;7:129–143. doi: 10.1038/nrmicro1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernardo DD, Thompson MJ, Eastwood TSGSECEL. Chemogenomic profiling on a genome-wide scale using reverse-engineered gene networks. Nat Biotech. 2005;23:377–383. doi: 10.1038/nbt1075. [DOI] [PubMed] [Google Scholar]
- 7.Carrera J, Rodrigo G, Jaramillo A. Model-based redesign of global transcription regulation. Nucleic Acids Res. 2009;37:e38. doi: 10.1093/nar/gkp022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrera J, Rodrigo G, Jaramillo A, Elena SF. Reverse-engineering the arabidopsis thaliana transcriptional network under changing environmental conditions. Genome Biol. 2009;10:R96. doi: 10.1186/gb-2009-10-9-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Faith J, Hayete B, Thaden J, Mogno I, Wierzbowski J. Large-scale mapping and validation of escherichia coli transcriptional regulation from a compendium of expression proles. PLoS Biol. 2007;5:e8. doi: 10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonneau R. A predictive model for transcriptional control of physiology in a free living cell. Cell. 2007;131:1354–1365. doi: 10.1016/j.cell.2007.10.053. [DOI] [PubMed] [Google Scholar]
- 11.Tagkopoulos I, Liu Y, Tavazoie S. Predictive behavior within microbial genetic networks. Science. 2008;320:1313–1317. doi: 10.1126/science.1154456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO. Integrating high-throughput and computational data elucidates bacterial networks. Nature. 2004;429:92–96. doi: 10.1038/nature02456. [DOI] [PubMed] [Google Scholar]
- 13.Endy D, Brent R. Modelling cellular behaviour. Nature. 2001;409:391–395. doi: 10.1038/35053181. [DOI] [PubMed] [Google Scholar]
- 14.Joyce AR, Palsson BO. The model organism as a system: integrating ‘omics’ data sets. Nat Rev Mol Cell Biol. 2006;7:198–210. doi: 10.1038/nrm1857. [DOI] [PubMed] [Google Scholar]
- 15.Burgard AP, Pharkya P, Maranas CD. Optknock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng. 2003;84:647–657. doi: 10.1002/bit.10803. [DOI] [PubMed] [Google Scholar]
- 16.Segre D, Vitkup D, Church GM. Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci U S A. 2002;99:15112–15117. doi: 10.1073/pnas.232349399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rocha M, Maia P, Mendes R, Pinto JP, Ferreira EC. Natural computation metaheuristics for the in silico optimization of microbial strains. BMC Bioinfo. 2008;9:499. doi: 10.1186/1471-2105-9-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyer RC, Steinfat M, Lisec J, Becher M, Witucka-Wall H. The metabolic signature related to high plant growth rate in arabidopsis thaliana. Proc Natl Acad Sci U S A. 2007;104:4759–4764. doi: 10.1073/pnas.0609709104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mounet F, Moing A, Garca V, Petit J, Maucourt M. Gene and metabolite regulatory network analysis of early developing fruit tissues highlights new candidate genes for the control of tomato fruit composition and development. Plant Physiol. 2009;149:1505–1528. doi: 10.1104/pp.108.133967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garca V, Stevens R, Gil L, Gilbert L, Gest N. An integrative genomics approach for deciphering the complex interactions between ascorbate metabolism and fruit growth and composition in tomato. C R Biol. 2009;32:1007–1021. doi: 10.1016/j.crvi.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 21.Schauer N, Semel Y, Roessner U, Gur A, Balbo I. Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat Biotech. 2006;24:447–454. doi: 10.1038/nbt1192. [DOI] [PubMed] [Google Scholar]
- 22.Osorio S, Alba R, Damasceno CM, Lopez-Casado G, Lohse M. Systems biology of tomato fruit development: combined transcript, protein, and metabolite analysis of tomato transcription factor (nor, rin) and ethylene receptor (nr) mutants reveals novel regulatory interactions. Plant Physiol. 2011;157:405–425. doi: 10.1104/pp.111.175463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rohrmann J, Tohge T, Alba R, Osorio S, Caldana C. Combined transcription factor profiling, microarray analysis and metabolite profiling reveals the transcriptional control of metabolic shifts occurring during tomato fruit development. Plant J. 2011;68:999–1013. doi: 10.1111/j.1365-313X.2011.04750.x. [DOI] [PubMed] [Google Scholar]
- 24.Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daetwyler HD, Villanueva B, Bijma P, Woolliams JA. Inbreeding in genome-wide selection. J Anim Breed Genet. 2007;124:369–376. doi: 10.1111/j.1439-0388.2007.00693.x. [DOI] [PubMed] [Google Scholar]
- 26.Magniette F, Renou JP, Daudin JJ. Normalization for triple-target microarray experiments. BMC Bioinf. 2008;9:216. doi: 10.1186/1471-2105-9-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riedelsheimer C, Czedik-Eysenberg A, Grieder C, Lisec J, Technow F. Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat Genet. 2012;44:217–220. doi: 10.1038/ng.1033. [DOI] [PubMed] [Google Scholar]
- 28.Buttery RG, Teranishi R, Flath RA, Ling LC. Teranishi r, buttery rg, shahidi f., editors. Fresh tomato volatiles: Composition and sensory studies. Washington DC: American Chemical Society. 1989. pp. 213–222. favor chemistry: Trends and developments.
- 29.Shaha R, Ward PT. Lean manufacturing: context, practice bundles, and performance. J Oper Manag. 2003;21:129–149. [Google Scholar]
- 30.Rosati C, Diretto G, Giuliano G. Biosynthesis and engineering of carotenoids and apocarotenoids in plants: state of the art and future prospects. Biotechnol Genet Eng Rev. 2010;26:139–162. doi: 10.5661/bger-26-139. [DOI] [PubMed] [Google Scholar]
- 31.EF EFF, Liu YL, Carmel-Goren L, Gur A, Shoresh M. Two tightly linked qtls modify tomato sugar content via different physiological pathways. Mol Genet Genom. 2002;266:821–826. doi: 10.1007/s00438-001-0599-4. [DOI] [PubMed] [Google Scholar]
- 32.Emery GC, Munger HM. Effects of inherited differences in growth habit on fruit size and soluble solids in tomato. J Amer Soc Hort Sci. 1970;95:410–412. [Google Scholar]
- 33.Cong B, Barrero LS, Tanksley SD. Regulatory change in yabby-like transcription factor led to evolution of extreme fruit size during tomato domestication. Nat Genet. 2008;40:800–804. doi: 10.1038/ng.144. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Schauer N, Usadel B, Frasse P, Zouine M. Regulatory features underlying pollination-dependent and -independent tomato fruit set revealed by transcript and primary metabolite profiling. Plant Cell. 2009;21:428–452. doi: 10.1105/tpc.108.060830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klee HJ. Improving the avor of fresh fruits: genomics, biochemistry, and biotechnology. New Phytol. 2010;187:44–56. doi: 10.1111/j.1469-8137.2010.03281.x. [DOI] [PubMed] [Google Scholar]
- 36.Minoia S, Petrozza A, DOnofrio O, Piron F, Mosca G. A new mutant genetic resource for tomato crop improvement by tilling techonology. BMC Res Notes. 2010;12:69. doi: 10.1186/1756-0500-3-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bogdanove AJ, Voytas DF. Tal effectors: customizable proteins for dna targeting. Science. 2011;333:1843–1846. doi: 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- 38.Schauer N, Semel Y, Roessner U, Gur A, Balbo I. Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat Biotech. 2006;24:447–454. doi: 10.1038/nbt1192. [DOI] [PubMed] [Google Scholar]
- 39.Hetherington S, Smillie R, Davies W. Photosynthetic activities of vegetative and fruiting tissues of tomato. J Exp Bot. 1998;49:1173. [Google Scholar]
- 40.Fridman E, Carrari F, Liu YS, Fernie AR, Zamir D. Zooming in on a quantitative trait for tomato yield using interspecific introgressions. Science. 2004;305:1786–1789. doi: 10.1126/science.1101666. [DOI] [PubMed] [Google Scholar]
- 41.Agius F, Gonzalez-Lamothe R, Caballero JL, noz Blanco JM, Botella MA. Engineering increased vitamin c levels in plants by overexpression of a d-galacturonic acid reductase. Nat Biotech. 2003;21:177–181. doi: 10.1038/nbt777. [DOI] [PubMed] [Google Scholar]
- 42.BCahoon E, Hall SE, Ripp KG, Ganzke TS, Hitz WD. Metabolic redesign of vitamin e biosynthesis in plants for tocotrienol production and increased antioxidant content. Nat Biotech. 2003;21:1082–1087. doi: 10.1038/nbt853. [DOI] [PubMed] [Google Scholar]
- 43.Ye X, Al-Babili S, Kloti A, Zhang J, Lucca P. Engineering the provitamin a (betacarotene) biosynthetic pathway into (carotenoid-free) rice endosperm. Science. 2000;87:303–305. doi: 10.1126/science.287.5451.303. [DOI] [PubMed] [Google Scholar]
- 44.Aharoni A, Galili G. Metabolic engineering of the plant primary-secondary metabolism interface. Curr Op Biotechnol. 2011;22:239–244. doi: 10.1016/j.copbio.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 45.Alba JM, Montserrat M, noz RFM. Resistance to the two-spotted spider mite (tetranychus urticae) by acylsucroses of wild tomato (solanum pimpinellifolium) trichomes studied in a recombinant inbred line population. Exp App Acar. 2009;47:35–47. doi: 10.1007/s10493-008-9192-4. [DOI] [PubMed] [Google Scholar]
- 46.Zanor MI, Rambla JL, Chaib J, Steppa A, Medina A. Metabolic characterization of loci affecting sensory attributes in tomato allows an assessment of the inuence of the levels of primary metabolites and volatile organic contents. J Exp Bot. 2009;60:2139–2154. doi: 10.1093/jxb/erp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eshed Y, Zamir D. An introgression line population of lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated qtl. Genetics. 1995;141:1147–1162. doi: 10.1093/genetics/141.3.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lytovchenko A, Eickmeier I, Pons C, Szecowka M, Lehmberg K. Tomato fruit photosynthesis is seemingly unimportant in primary metabolism and ripening but plays a considerable role in seed development. Plant Physiol. 2011;157:1650–1663. doi: 10.1104/pp.111.186874. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transcriptional, metabolic and phenotypic models of tomato fruit.
(XLS)
Single knockout and over-expressed genes to improve desired agronomic properties (acceptability, quality and quality vs production of tomato fruits; four volatile compounds; vitamin C, and different types of sugars and acids) and functional categorization of genes that induced high degree of improvement in those agronomic properties; notice that functional enrichment of all genes involved in the TRN was included. Gene ontology enrichment analyses were performed using the TFGD tool [TFGD]. It is also showed the functional categories significantly represented among those genes that were selected to describe the TRN of tomato fruit. A total of 19 cellular processes and 45 biological components were represented. Among these, genes related to cellular metabolic processes were the most abundant (p<0.0001), what makes sense since they were selected to predict cellular metabolism; whereas genes related to response to nutrient stimulus were present but the least common (p<0.1).
(XLS)
Multiple combinations of knockout and over-expressed gene sets to improve desired agronomic properties (acceptability, quality and quality vs production of tomato fruits).
(XLS)
Experimental evidences of each gene perturbation proposed by the model to optimize the different scoring function used.
(XLS)
Synthetic biology of tomato fruit vs computer science.
(PDF)
From data to global models to redesign using an approach based on synthetic biology.
(PDF)
Phenotype prediction (number of fruits per plant, fruit harvested, average fruit weight and pH) by using the genotype described in the 50 RILs in which transcript levels were measured. Pearson coefficient correlation (A,C) between the predicted and measured phenotypic profile and number of genes (B,D) selected by LASSO method as predictors for different thresholds of the fitting parameter (tLASSO). Note that we used two different z-score levels (z = 2, (A,B); and z = 3 (C,D)) to included genes as possible predictors to be selected by LASSO. The dashed line plotted in (A,B) shows the parameter, tLASSO, and the level of z-score used to constructed the relationship between phenotype and metabolome.
(PDF)
Exhaustive exploration and statistical significance of the landscape of single desired agronomic properties of tomato fruit (vitamin C, blue; fructose and glucose, red; and citric and malic acids, green) perturbing its effective TRN locally. (A) Agronomic properties improved by perturbing a single gene as function of efficiency reached by that transcriptional perturbation with respect to the wild-type scenario; notice that only perturbations with positive mean efficiencies are plotted. Both agronomic properties and efficiencies of a single perturbation are average variables tested on the 169 RILs and error bars represent their minimum and maximum values in both axis. (B) Dependence between agronomic properties in the wild-type genome and the average of the agronomic properties resulting of all single perturbations in the wild-type TRN for each RIL; vertical error bars represent the best and worst optimized re-engineered TRN for a given RIL. (C–D) Average number of single gene perturbations that overcome an efficiency threshold in the 169 RILS (light bars; error bars represent standard deviation for the 169 RILs) and average probability of selecting the same gene-perturbation commonly in a set of RILs (dark bars; error bars show standard deviation for all genes of the TRN). Left and right columns represent perturbations in terms of single gene knockout or overexpression, respectively.
(PDF)
(A) Dendogram of the volatile compound correlations observed experimentally. (B, C) Dendograms inferred by the model defining the distance between volatile compound as the number of common genetic perturbations predicted to optimize the levels of each volatile compound.
(PDF)
Percentage of altered genes (via gene knockout or over-expression; blue bars) proposed by the model to minimize the levels of volatile compounds (linalool (A) or, 1-nitro-2-phenylethane, 2-isobutylthiazole and benzylnitrile (B)) that were found significantly over-/under-expressed in the transcriptome of two ILs characterized experimentally with extremely low levels of those volatile compounds. The cut-off of the coefficient of variation between replicates was 75%. The Mann-Withney's U-test significance using random selection of gene perturbations (red bars) is shown (***statistically significant). Error bars represent the standard deviations of scores obtained from three ILs. 16.7% of the over-expressed genes proposed by the model to minimize the level of linalool were significantly recovered in gene expression (Figure S4A). In addition, 1.89% and 3.33% of genes candidates to be knockout or over-expressed (Figure S4B), respectively, also were identified significantly altered in the gene expression of the IL in which the three volatile compounds were found in minimum amount indicating this part of the transcriptome is relevant and associated to this volatile sub-phenotype among the other differential traits in these ILs.
(PDF)
Correlations observed between agronomic variables and metabolites of different fruit genotypes generated by simulating all possible single gene knockout (A–E) or over-expression (F–J) in the wild-type genome model of the tomato fruit. Standard deviations of all metabolites or agronomic variables show the diversity generated by implementing each genetic perturbation in the 169 RILs. Note that we only plotted re-engineered genomes whose transcriptome predicted showed errors lower than 1% (241 d.f. and 25 d.f. for knockout and over-expressed genes, respectively).
(PDF)
The top 5 single-gene knockouts and over-expressions that maximize the agronomic properties of the tomato fruit based on improve only one objective.
(PDF)
The top 5 single-gene knockouts and over-expressions that minimize the agronomic properties of the tomato fruit based on improve only one objective.
(PDF)
Genome design based on single perturbations to fine-tuning phenotypes with biotechnological interests. Model validation: fine-tuning tomato phenotype of two experimental inbred lines by computational genome design. Prediction of phenotypic correlations in re-engineered tomato fruits.
(PDF)