

Abstract
AIM: To investigated whether sall3 transcription was regulated by promoter CpG island hypermethylation in hepatocellular carcinoma (HCC).
METHODS: The cell lines Huh7, HepG2, SK-HEP1, SMMC7721, Bel7402, QGY7703 and a cohort of 38 HCC tissue specimens and corresponding nontumorous tissues were subjected to analysis for sall3 promoter CpG island methylation and mRNA transcription. sall3 promoter CpG island methylation levels were determined using the MassARRAY platform and mRNA transcription levels of the gene were detected by quantitative real-time polymerase chain reaction.
RESULTS: The levels of sall3 mRNA were decreased by more than twofold in 33 of 38 tumor tissues compared to adjacent noncancerous tissues. Among these 33 tumor tissues with lower levels of sall3 mRNA, 24 showed higher levels of methylation. Based on these results, we hypothesized that the decrease in sall3 mRNA transcription level was likely due to promoter CpG island hypermethylation. Changes in sall3 mRNA transcription and promoter CpG island methylation were determined in the above six cell lines after treatment with 0, 0.1, 0.5 and 2.5 μmol 5-aza-2-deoxycytidine, a demethylating agent. Promoter CpG island methylation levels decreased in a dose-dependent manner in all six cell lines, while the mRNA transcription level increased dose-dependently in Huh7, HepG2, SK-HEP1 and SMMC7721 cells and irregularly in Bel7402 and QGY7703 cells.
CONCLUSION: These results indicated that promoter CpG island hypermethylation contributes to the downregulation of sall3 mRNA transcription in HCC.
Keywords: Hepatocellular carcinoma, sall3, Aberrant methylation, Down regulation mRNA transcription
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors, representing a major public health issue, especially in Asia[1]. The pathogenesis of HCC involves chronic hepatitis virus infection and activation of oncogenes and/or inactivation of tumor suppressor genes by mutations and epigenetic modification[2]. Epigenetic inactivation of tumor suppressor genes by DNA hypermethylation plays an important role in carcinogenesis[3]. Many groups have reported that promoter hypermethylation of CpG islands is associated with development, stage, recurrence, progression and survival in HCC[4,5]. In many cases, aberrant methylation of promoter regions within genes is correlated with a loss of gene expression[6]. Furthermore, in contrast to mutations, epigenetic changes may be reversible, raising the possibility of developing therapeutics based on restoring a normal epigenetic state in cancer-associated genes[7,8].
sal was originally identified as a region-specific homeotic gene in Drosophila[9]. sall3 is one of four mammalian members of the sal-like (sall) gene family (sall1, sall2, sall3 and sall4), which are involved in embryonic development[10]. sall3 is one of several genes deleted in 18q deletion syndrome, characterized by hearing loss, mental retardation, midfacial hypoplasia, delayed growth, and limb abnormalities[11]. Loss of the sall3 gene leads to palate deficiency, abnormalities in cranial nerves, and perinatal lethality[12]. Recently, it was reported that sall3 can interact with DNMT3A and shows the ability to inhibit CpG island methylation in HCC[13]. However, when scanning the nucleotide sequences of sall3, we found a CpG island in the promoter. It has been reported that sall3 gene methylation levels are significantly increased in bladder cancer compared to nontumorous controls, and may be a new biomarker for the sensitive and specific detection of bladder cancer[14]. Furthermore, it has been reported that the sall3 gene CpG island has a higher frequency of hypermethylation in HCC tumors compared with adjacent noncancerous tissues as determined by a qualitative methylation method[15]. However, the decreased sall3 mRNA transcription levels in human HCC tissues and whether this is caused by promoter CpG island hypermethylation have not been fully examined.
Here, we show that sall3 mRNA transcription was downregulated in most (33/38) tumor tissues examined compared with adjacent noncancerous tissues. Most (24/33) downregulation of mRNA transcription was strongly associated with hypermethylation of the promoter CpG island. This association was further confirmed by subsequent cell line experiments; treatment of the cell lines with the DNA methyltransferase inhibitor 5-aza-2-deoxycytidine reversed promoter CpG island hypermethylation and restored sall3 mRNA transcription. These results indicated that promoter CpG island hypermethylation is the main reason for the downregulation of sall3 mRNA transcription in HCC.
MATERIALS AND METHODS
Tissue specimens and cell lines
Thirty-eight paired clinical samples of HCC, including tumor tissues and adjacent noncancerous tissues, were collected from surgical specimens at the Department of Hepatobiliary Surgery, Nanfang Hospital (16 cases), and the Cancer Institute of Sun Yat-sen University (22 cases), both in Guangzhou, China. All specimens were obtained immediately after surgical resection and were stored at -70 °C until DNA/RNA extraction.
Written informed consent was obtained from all patients prior to inclusion in the study. The study protocol was approved by the Nanfang Hospital Ethics Committee at Southern Medical University and the Sun Yat-sen Cancer Center Ethics Committee at Sun Yat-sen University.
Cell culture and 5-aza-CdR treatment
Six HCC cell lines (Huh7, HepG2, SMMC-7721, Bel-7402, SK-HEP1, QGY7703) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL, Gaithersburg, MD), supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin and incubated in a 5% CO2 atmosphere at 37 °C. The demethylating agent, 5-aza-2-deoxycytidine (5-aza-CdR; Sigma, St. Louis, MO), was freshly prepared in ddH2O. HepG2 cells (3 × 105 cells/well) and other hepatoma cells (1 × 105 cells/well) in exponential growth phase were seeded in 6-well plates. After 24 h of culture, cells were treated with 5-aza-CdR at 0, 0.1, 0.5 and 2.5 mol for 3 d. The culture medium was replaced every 24 h with fresh media containing 5-aza-CdR. Total RNA and genomic DNA were extracted for real-time quantitative reverse transcription-polymerase chain reaction (qRT-PCR) and DNA methylation level analysis.
Detection of sall3 CpG Island DNA hypermethylation
Genomic DNA was extracted from cells and HCC samples using a QIAamp DNA Minikit (Qiagen, Valencia, CA). Genomic DNA (1 μg) was modified with sodium bisulfite using the EZ DNA methylation kit (Zymo Research, Orange, CA). DNA methylation levels of clinical samples and cell lines were determined using the MassARRAY platform (Sequenom, San Diego, CA) as described previously[16]. Briefly, two fragments covering 38 CpG sites from sall3 were amplified from bisulfite-modified DNA. A 10-mer tag sequence was added to the forward primer, and a T7-promoter tag was added to the reverse primer to balance the PCR primer length. The primers used were 5’-AGGAAGAGAGGGATTGTTTGGATTTGATTTTAATTT-3’ (sense) and 5’-CAGTAATACGACTCACTATAGGGAGAAGGCTCACAAATAACCTCCTAAAACTTCCC-3’ (antisense); 5’-AGGAAGAGAGTTTTAAGGTTGGTTTTATTTTGTTT-3’ (sense) and 5’-CAGTAATACGACTCACTATAGGGAGAAGGCTTCTCAAAAATAATCTCAAACCCCTA-3’ (antisense). Methylation data for individual units (1-3 CpG sites per unit) were analyzed using the EpiTyper software (Sequenom).
RNA extraction and quantitative real-time PCR analysis
Total RNA was extracted from cell lines and tissue samples using the Trizol reagent (Invitrogen), according to the manufacturer’s protocol. First-strand cDNA was generated using a SYBR PrimeScript RT-PCR Kit (TaKaRa, Kyoto, Japan). sall3 mRNA expression was detected by qRT-PCR using a SYBR Premix Ex Taq Kit (TaKaRa) on an ABI 7500 Real-Time PCR System (Applied Biosystems). β-actin was used as an internal control. The primers used were as follows: sall3 forward primer: 5’-GCTGCCTTCTCAGTTATTTGACC-3’, reverse primer: 5’-TGACCGTTCACTTCCATTTTGA-3’; β-actin forward primer: 5’-TTGTTACAGGAAGTCGCTTGCC-3’, reverse primer: 5’-ATGCTATCACCTCCCCTGTGTGT-3’. Relative levels of sall3 mRNA were calculated and expressed as 2-ΔΔCt[17].
Statistical analysis
qRT-PCR results in different groups were analyzed by Student’s t test. The methylation levels of Oct-6 in HCC tumors and adjacent noncancerous tissues were compared using the Wilcox rank sum test. All tests were two-sided. In all analyses, P < 0.05 was taken to indicate statistical significance.
RESULTS
sall3 promoter CpG island aberrant methylation and sall3 mRNA transcription in human HCC tissues
To determine whether sall3 promoter CpG island hypermethylation changes leads to decreased sall3 mRNA transcription in human HCC tissues, the methylation levels of the sall3 promoter CpG island in 38 HCC tumors and adjacent noncancerous tissues were examined using the MassARRAY platform (Sequenom). Of the 38 tumors, 27 (71%) showed higher methylation levels at the sall3 CpG island compared with adjacent noncancerous tissue, while methylation levels were similar at the sall3 CpG island in tumor tissue and adjacent noncancerous tissue in 11 cases (29%). None of the tumors showed lower methylation levels at the sall3 CpG island compared with adjacent noncancerous tissues. Average methylation levels for each CpG unit in the 38 tumor tissues and adjacent noncancerous tissues are listed in Figure 1. Among the total of 24 CpG units (1-3 CpG sites per unit), the average methylation levels of 20 CpG units in 38 tumors were significantly higher than those in adjacent noncancerous tissues.
Figure 1.
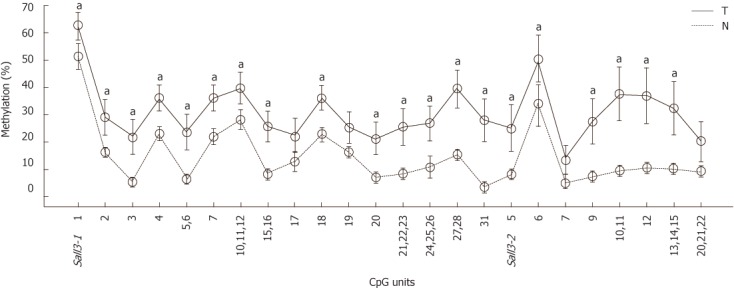
Average methylation levels were calculated from 38 tumors (T) and adjacent non-cancerous tissues (N) on 24 CpG units from sall3 CpG island respectively. The data was analyzed by Wilcox rank sum test (aP < 0.05 vs non-cancerous tissues ). Error bar, 95% confidence interval.
To determine whether aberrant CpG island DNA methylation in HCC tissues may be correlated with the decreased sall3 mRNA transcription, sall3 mRNA levels in 38 paired samples were determined by qRT-PCR (Figure 2). The relative ratio between methylation and mRNA expression levels of sall3 are negatively correlated in 26 of 38 tumor tissues vs corresponding adjacent noncancerous tissues (Figure 3). Of the 38 tumor tissues, 33 (86.8%) showed a decreased sall3 mRNA level compared to adjacent noncancerous tissues (Figure 2). Among these 33 tumor tissues with lower mRNA levels, 24 showed higher methylation levels and a negative association was found between CpG island methylation and mRNA expression (Figure 3); nine tumor tissues showed methylation levels that were not significantly different from adjacent noncancerous tissues. Of the 38 tumor tissues examined, three (7.9%) showed higher sall3 mRNA expression than adjacent noncancerous tissues (Figure 2). Methylation level was similar to the adjacent noncancerous tissue in one of these three tumor tissues, while the methylation levels in the remaining two tumor tissues were higher than those in the adjacent noncancerous tissue. Of the 38 tumor tissues, two (5.3%) showed similar sall3 mRNA expression to adjacent noncancerous tissues (Figure 2). Of these two tumor tissues, one showed no significant difference in methylation level compared with adjacent noncancerous tissues, while the methylation level in the other was higher than that the in the adjacent noncancerous tissue. Together, these results indicated a negative correlation between sall3 promoter CpG island hypermethylation and sall3 mRNA transcription in 24 tumor tissues and their adjacent noncancerous tissues (Figure 3). sall3 mRNA transcription in other tissues showed no association with sall3 promoter CpG island hypermethylation, suggesting regulatory mechanisms other than those involved in HCC.
Figure 2.
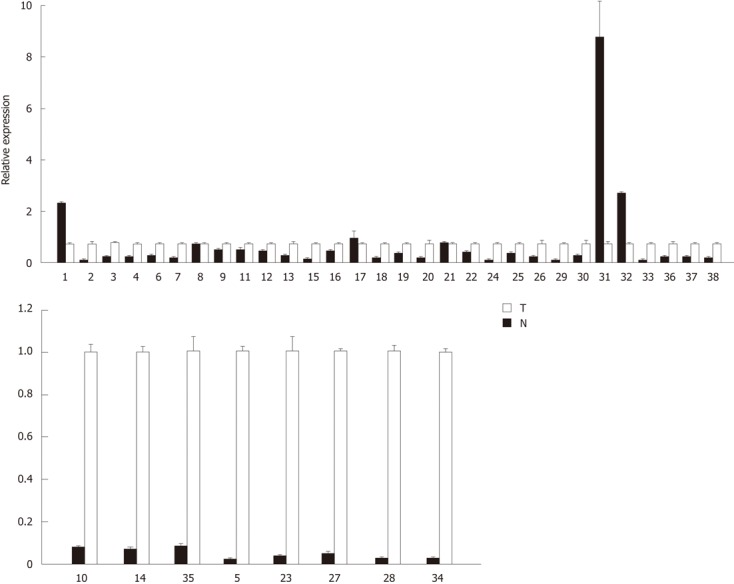
sall3 mRNA is analyzed in 38 tumor tissues (T) and adjacent non-cancerous tissues (N). Error bars, SD from triplicates.
Figure 3.
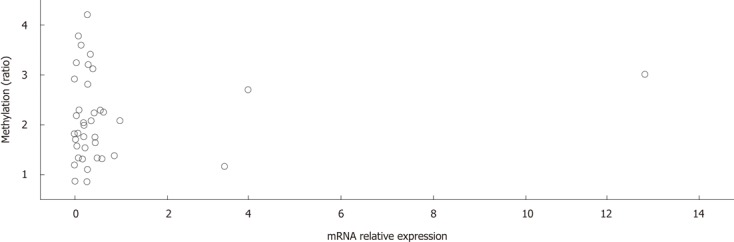
Correlation between methylation levels and mRNA expression of sall3 in 24 tumor tissues and corresponding adjacent noncancerous tissues.
sall3 promoter CpG island hypermethylation correlates its mRNA transcription in human HCC cell lines
To determine whether decreased sall3 mRNA transcription levels in human HCC tissues are due to promoter CpG island hypermethylation, six human HCC cell lines (Huh7, HepG2, SK-HEP1, SMMC7721, Bel7402 and QGY7703) were exposed to 0, 0.1, 0.5 or 2.5 μmol 5-aza-CdR, an inhibitor of DNMTs, for 72 h. As expected, after treatment with 0.1, 0.5 or 2.5 μmol 5-aza-CdR, promoter CpG island methylation levels showed dose-dependent downregulation in all six cell lines (Figure 4). sall3 mRNA levels also showed dose-dependent upregulation in Huh7, HepG2, SK-HEP1 and SMMC7721 cells, while it was irregularly upregulated in Bel7402 and QGY7703 cells (Figure 5). These data indicated that sall3 promoter CpG island hypermethylation is likely responsible for the decrease in sall3 mRNA transcription level.
Figure 4.
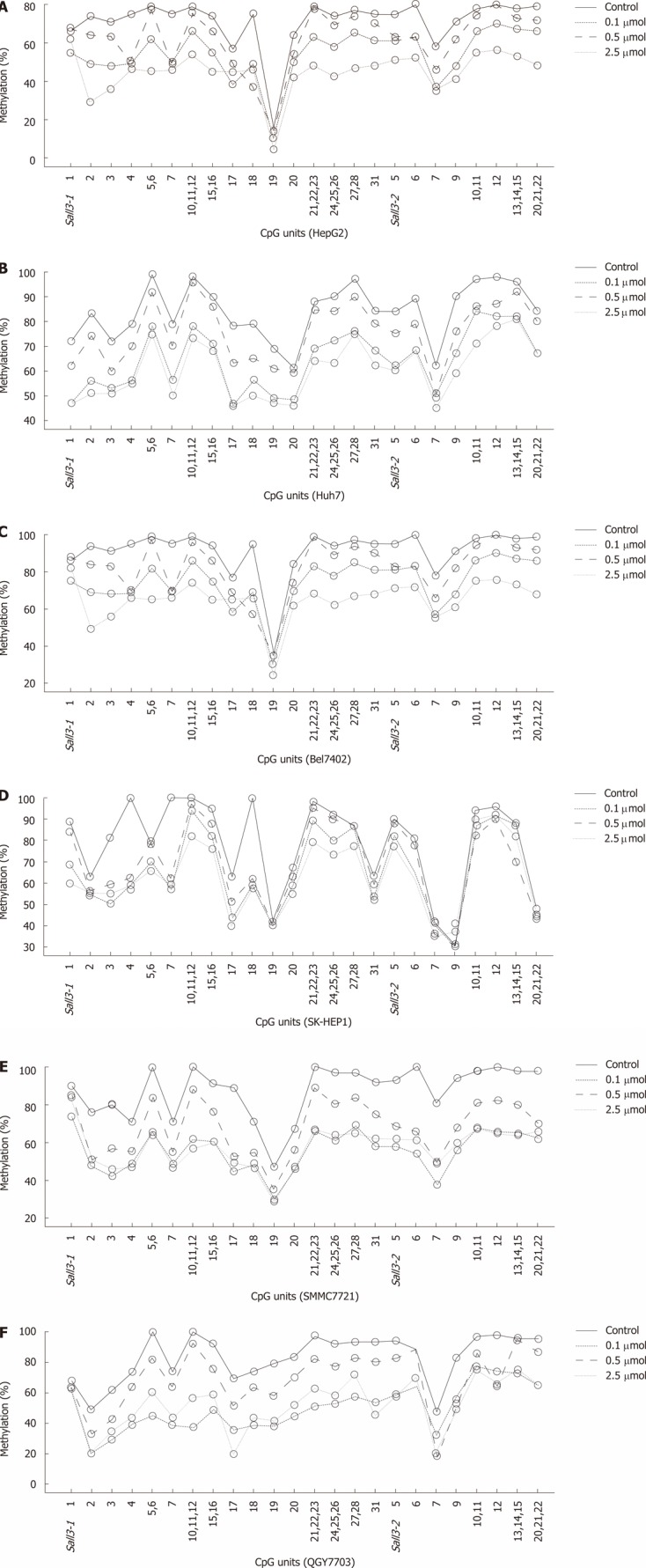
Quantitative methylation analysis on each CpG unit of sall3 CpG island in HCC cells after 5-Aza-CdR treatment or control (A-F).
Figure 5.
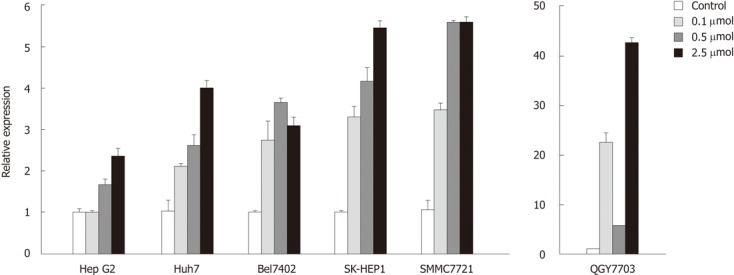
Relative sall3 expression in six hepatocellular carcinoma cells after 5-AZA-CdR treatment. Error bars, SD from triplicates.
DISCUSSION
HCC is one of the most common gastrointestinal malignancies, ranking fifth in the occurrence of common cancers, and third in common causes of cancer-related death[18]. Early-stage HCC is potentially curable with surgical resection or hepatic transplantation[19,20]. However, most patients present with advanced disease and are not amenable to surgical resection or transplantation. Therefore, they have a poor prognosis[21], and improved methods for early diagnosis are urgently required. DNA methylation is a gene expression regulatory mechanism and plays a fundamental role in carcinogenic processes[22,23]. Aberrant CpG island methylation of tumor-related genes is an early and frequent event in the process of carcinogenesis, and DNA methylation status of tumor-related genes is a potential diagnostic marker[24-26]. As it is possible to restore the function of methylated tumor suppressor genes, combinations of epigenetic modifiers and other therapeutic agents may also become a promising alternative to conventional treatments[27-29].
Although an increasing number of genes undergoing aberrant CpG island methylation have been reported in HCC[4,5,30,31], the methylated genes have not yet been fully characterized. Yu et al[14] reported that sall3 was a novel target of aberrant methylation in bladder cancer. Xia et al[15] reported that the hypermethylation frequency in tumor tissues was significantly higher than those in adjacent noncancerous tissues, but whether the decreased transcription of sall3 was caused by hypermethylation of the promoter CpG island in HCC remained unknown.
In the present study, we found that the levels of sall3 mRNA were decreased in 33 of 38 tumor tissues compared with those in adjacent noncancerous tissues. Twenty-four of these 33 tumor tissues with reduced sall3 mRNA levels showed elevated methylation levels, suggesting that the decreased sall3 mRNA transcription levels were likely caused by promoter CpG island hypermethylation. We also confirmed that six HCC cell lines (Huh7, HepG2, SK-HEP1, SMMC7721, Bel7402 and QGY7703) were hypermethylated at the sall3 promoter. Using the demethylating agent 5-aza-CdR, the transcription of sall3 could be restored in these cells when the sall3 promoter region was partially demethylated. Taken together with the results in HCC tissue samples, it is clear that promoter hypermethylation in sall3 was strongly associated with the reduced transcription of sall3 mRNA. However, in more than 25% of the HCC cases (9/33), decreased mRNA transcription was observed with similar CpG island methylation levels. In addition, there were three tumor tissues with promoter hypermethylation without decreased mRNA transcription compared with corresponding nontumorous tissues; the levels of mRNA transcription were increased in two of these cases and similar in the remaining one case. These results suggested that other regulatory mechanisms unrelated to promoter hypermethylation may also be involved. Further studies are needed to clarify this issue. However, the overall strong association between promoter hypermethylation and decreased mRNA transcription suggests a causative role of aberrant sall3 promoter methylation and decreased mRNA transcription in most cases of HCC.
In conclusion, we showed that sall3 mRNA transcription was downregulated in most (33/38) tumor tissues compared with adjacent noncancerous tissues in HCC. Most cases (24/33) with downregulated mRNA transcription were strongly associated with promoter CpG island hypermethylation. This association was further confirmed by subsequent experiments in cell lines; treatment of the cell lines Huh7, HepG2, SK-HEP1, SMMC7721, Bel7402 and QGY7703 with the DNA methyltransferase inhibitor 5-aza-2-deoxycytidine reversed promoter CpG island hypermethylation and restored sall3 mRNA transcription. These results indicated that promoter CpG island hypermethylation is the main reason for downregulation of sall3 mRNA transcription in HCC. These finding regarding sall3 mRNA transcription and DNA methylation associated with human HCC provide new insights into the pathogenesis of HCC and may serve as a powerful molecular marker for detecting HCC in biopsy tissues of HCC patients.
COMMENTS
Background
Inactivation of tumor suppressor genes by promoter CpG island hypermethylation plays a key role in cancer pathogenesis.
Research frontiers
Aberrant CpG island methylation of tumor-related genes is a early and frequent event in carcinogenetic process and DNA methylation status of tumor-related genes is a potential diagnostic marker. Using demethylating agents to restore the function of methylated tumor suppressor genes also becomes a promising alternative to conventional treatments.
Innovations and breakthroughs
sall3 mRNA transcription was downregulated in most tumor tissues compared with adjacent noncancerous tissues. Downregulation of mRNA transcription was strongly associated with hypermethylation of the promoter CpG island. This association was further confirmed by subsequent cell line experiments; treatment of the cell lines with the DNA methyltransferase inhibitor 5-aza-2-deoxycytidine reversed promoter CpG island hypermethylation and restored sall3 mRNA transcription.
Applications
The finding of sall3 mRNA transcription and DNA methylation associated with human hepatocellular carcinoma (HCC) provide new insights into pathogenesis of HCC and may serve as a powerful molecular marker for detecting HCC in biopsies tissues of HCC patients.
Terminology
To concisely and accurately describe, define or explain the specific, unique terms that are not familiar to majority of the readers, but are essential for the readers to understand the article. DNA methylation typically occurs at CpG sites (cytosine-phosphate-guanine sites, that is, where a cytosine is directly followed by a guanine in the DNA sequence). This methylation results in the conversion of the cytosine to 5-methylcytosine. Hypermethylation typically occurs at CpG islands in the promoter region and is associated with gene inactivation. Demethylation is the chemical process resulting in the removal a of methyl group (CH3) from a molecule.
Peer review
This is a nice human study conducted by a group of competent researchers. The study is nicely done. Also, the paper is well written.
Footnotes
Supported by Key Programs for Science and Technology Development of Guangzhou, No. 2008A1-E4151; and the National High Technology Research and Development Program of China, No. 2006AA02A311
Peer reviewers: Bao-Ting Zhu, MD, PhD, Professor, Department of Pharmacology, Toxicology and Therapeutics, School of Medicine, University of Kansas Medical Center, MS-1018, room-KLSIC 4061, 2146 W. 39th Ave, Kansas City, KS 66160, United States; Ralph Graeser, PhD, Group Leader, Molecular and Cellular Biology, ProQinase GmbH, Breisacher Str. 117, 79106 Freiburg, Germany
S- Editor Cheng JX L- Editor A E- Editor Xiong L
References
- 1.Lai EC, Lau WY. The continuing challenge of hepatic cancer in Asia. Surgeon. 2005;3:210–215. doi: 10.1016/s1479-666x(05)80043-5. [DOI] [PubMed] [Google Scholar]
- 2.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 3.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 4.Zhu J. DNA methylation and hepatocellular carcinoma. J Hepatobiliary Pancreat Surg. 2006;13:265–273. doi: 10.1007/s00534-005-1054-4. [DOI] [PubMed] [Google Scholar]
- 5.Tischoff I, Tannapfe A. DNA methylation in hepatocellular carcinoma. World J Gastroenterol. 2008;14:1741–1748. doi: 10.3748/wjg.14.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garinis GA, Patrinos GP, Spanakis NE, Menounos PG. DNA hypermethylation: when tumour suppressor genes go silent. Hum Genet. 2002;111:115–127. doi: 10.1007/s00439-002-0783-6. [DOI] [PubMed] [Google Scholar]
- 7.Strathdee G, Brown R. Aberrant DNA methylation in cancer: potential clinical interventions. Expert Rev Mol Med. 2002;4:1–17. doi: 10.1017/S1462399402004222. [DOI] [PubMed] [Google Scholar]
- 8.Strathdee G, Brown R. Epigenetic cancer therapies: DNA methyltransferase inhibitors. Expert Opin Investig Drugs. 2002;11:747–754. doi: 10.1517/13543784.11.6.747. [DOI] [PubMed] [Google Scholar]
- 9.Jürgens G. Head and tail development of the Drosophila embryo involves spalt, a novel homeotic gene. EMBO J. 1988;7:189–196. doi: 10.1002/j.1460-2075.1988.tb02799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mollereau B, Dominguez M, Webel R, Colley NJ, Keung B, de Celis JF, Desplan C. Two-step process for photoreceptor formation in Drosophila. Nature. 2001;412:911–913. doi: 10.1038/35091076. [DOI] [PubMed] [Google Scholar]
- 11.Dostal A, Nemeckova J, Gaillyova R. The 18q deletion syndrome and analysis of the critical region for orofacial cleft at 18q22.3. J Craniomaxillofac Surg. 2009;37:272–275. doi: 10.1016/j.jcms.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Parrish M, Ott T, Lance-Jones C, Schuetz G, Schwaeger-Nickolenko A, Monaghan AP. Loss of the Sall3 gene leads to palate deficiency, abnormalities in cranial nerves, and perinatal lethality. Mol Cell Biol. 2004;24:7102–7112. doi: 10.1128/MCB.24.16.7102-7112.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shikauchi Y, Saiura A, Kubo T, Niwa Y, Yamamoto J, Murase Y, Yoshikawa H. SALL3 interacts with DNMT3A and shows the ability to inhibit CpG island methylation in hepatocellular carcinoma. Mol Cell Biol. 2009;29:1944–1958. doi: 10.1128/MCB.00840-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu J, Zhu T, Wang Z, Zhang H, Qian Z, Xu H, Gao B, Wang W, Gu L, Meng J, et al. A novel set of DNA methylation markers in urine sediments for sensitive/specific detection of bladder cancer. Clin Cancer Res. 2007;13:7296–7304. doi: 10.1158/1078-0432.CCR-07-0861. [DOI] [PubMed] [Google Scholar]
- 15.Xia W, Ni W, Fei Q, Sun J, Zhao Y, Zhang H, Gu J, He Y, Yu J. Methylation of Sall3 gene in hepatocellular carcinoma. Zhenduanxue Lilun Yu Shijian. 2010;9:491–494. [Google Scholar]
- 16.Ehrich M, Zoll S, Sur S, van den Boom D. A new method for accurate assessment of DNA quality after bisulfite treatment. Nucleic Acids Res. 2007;35:e29. doi: 10.1093/nar/gkl1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Villanueva A, Minguez B, Forner A, Reig M, Llovet JM. Hepatocellular carcinoma: novel molecular approaches for diagnosis, prognosis, and therapy. Annu Rev Med. 2010;61:317–328. doi: 10.1146/annurev.med.080608.100623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bergsland EK, Venook AP. Hepatocellular carcinoma. Curr Opin Oncol. 2000;12:357–361. doi: 10.1097/00001622-200007000-00013. [DOI] [PubMed] [Google Scholar]
- 20.Llovet JM. Updated treatment approach to hepatocellular carcinoma. J Gastroenterol. 2005;40:225–235. doi: 10.1007/s00535-005-1566-3. [DOI] [PubMed] [Google Scholar]
- 21.Thomas MB, Abbruzzese JL. Opportunities for targeted therapies in hepatocellular carcinoma. J Clin Oncol. 2005;23:8093–8108. doi: 10.1200/JCO.2004.00.1537. [DOI] [PubMed] [Google Scholar]
- 22.Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999;59:793–797. [PubMed] [Google Scholar]
- 23.Tessema M, Länger F, Dingemann J, Ganser A, Kreipe H, Lehmann U. Aberrant methylation and impaired expression of the p15(INK4b) cell cycle regulatory gene in chronic myelomonocytic leukemia (CMML) Leukemia. 2003;17:910–918. doi: 10.1038/sj.leu.2402891. [DOI] [PubMed] [Google Scholar]
- 24.Hua D, Hu Y, Wu YY, Cheng ZH, Yu J, Du X, Huang ZH. Quantitative methylation analysis of multiple genes using methylation-sensitive restriction enzyme-based quantitative PCR for the detection of hepatocellular carcinoma. Exp Mol Pathol. 2011;91:455–460. doi: 10.1016/j.yexmp.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 25.Lambert MP, Paliwal A, Vaissière T, Chemin I, Zoulim F, Tommasino M, Hainaut P, Sylla B, Scoazec JY, Tost J, et al. Aberrant DNA methylation distinguishes hepatocellular carcinoma associated with HBV and HCV infection and alcohol intake. J Hepatol. 2011;54:705–715. doi: 10.1016/j.jhep.2010.07.027. [DOI] [PubMed] [Google Scholar]
- 26.Jacinto FV, Esteller M. MGMT hypermethylation: a prognostic foe, a predictive friend. DNA Repair (Amst) 2007;6:1155–1160. doi: 10.1016/j.dnarep.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther. 2009;8:1409–1420. doi: 10.1158/1535-7163.MCT-08-0860. [DOI] [PubMed] [Google Scholar]
- 28.Ganesan A, Nolan L, Crabb SJ, Packham G. Epigenetic therapy: histone acetylation, DNA methylation and anti-cancer drug discovery. Curr Cancer Drug Targets. 2009;9:963–981. doi: 10.2174/156800909790192428. [DOI] [PubMed] [Google Scholar]
- 29.Cortez CC, Jones PA. Chromatin, cancer and drug therapies. Mutat Res. 2008;647:44–51. doi: 10.1016/j.mrfmmm.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee S, Lee HJ, Kim JH, Lee HS, Jang JJ, Kang GH. Aberrant CpG island hypermethylation along multistep hepatocarcinogenesis. Am J Pathol. 2003;163:1371–1378. doi: 10.1016/S0002-9440(10)63495-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moribe T, Iizuka N, Miura T, Kimura N, Tamatsukuri S, Ishitsuka H, Hamamoto Y, Sakamoto K, Tamesa T, Oka M. Methylation of multiple genes as molecular markers for diagnosis of a small, well-differentiated hepatocellular carcinoma. Int J Cancer. 2009;125:388–397. doi: 10.1002/ijc.24394. [DOI] [PubMed] [Google Scholar]