

Abstract
Intracellular (clade B) ovalbumin (ov)-serpin protease inhibitors play an important role in tissue homeostasis by protecting cells from death in response to hypoosmotic stress, heat shock and other stimuli. Whether these serpins influence immunological tolerance and the risk for autoimmune diseases is not known. We found that a fraction of young autoimmune diabetes-prone non-obese diabetic (NOD) mice had elevated levels of autoantibodies against a member of clade B family known as serpinB13. High levels of anti-serpinB13 antibodies were accompanied by low levels of anti-insulin autoantibodies, reduced numbers of islet-associated T-cells and delayed onset of diabetes. Exposure to anti-serpinB13 monoclonal antibody (mAb) alone also decreased islet inflammation and co-administration of this reagent and a suboptimal dose of anti-CD3 mAb accelerated recovery from diabetes. In a fashion similar to that discovered in the NOD model, a deficiency in humoral activity against serpinB13 was associated with early onset of human type 1 diabetes. These findings suggest that in addition to limiting exposure to proteases within the cell, clade B serpins help to maintain homeostasis by inducing protective humoral immunity.
INTRODUCTION
Type 1 diabetes mellitus (T1D) is thought to be primarily a T-cell mediated disease that results from destruction of the insulin producing β-cells in the pancreatic islets (1-3). The incidence of this condition has significantly increased in developed countries over the last decade (4, 5) and hygiene has been added to the growing list of potential contributors to this worrying trend. One hypothesis for the role of hygiene in the risk for T1D is based on the assumption that adequate hygiene causes a change in exposure to certain pathogens and leads to reduced innate immunity and the output of regulatory T (Treg) cells with anti-inflammatory properties (6-8). According to an alternative model, hygiene may contribute to exacerbation of destructive autoimmunity by decreasing the total amount of tissue damage and impeding the development of protective autoimmune response.
We examined the role of protective autoimmunity in the risk for T1D by focusing on intracellular molecules of the B-clade family, also known as ovalbumin (ov)-serine proteinase inhibitors (serpins) (9, 10). We hypothesized that serpins can stimulate an immune response that could influence the severity of autoimmune inflammation. To investigate this possibility, we studied the immune response against clade B serpins during the immune-mediated destruction of pancreatic islets in nonobese diabetic (NOD) mice (1, 2). We chose this model because the cathepsin proteases have been implicated in the pathogenesis of autoimmune diabetes (11-15) and clade B serpins are potent inhibitors of these proteases (16, 17).
In this study we focused on a novel autoantibody against serpinB13. We found that in contrast to the autoantibodies that are associated with an elevated risk for T1D, anti-serpinB13 autoantibody supports protective outcomes, including a diminished inflammatory response in the pancreatic islets. The identification of this autoantibody provides new information regarding the etiology of T1D and contributes to our understanding of interrelationships between the immune system and other biological pathways.
MATERIALS and METHODS
Human subjects
Patients with recent-onset T1D (n=55) and healthy controls (n=53) aged 3 to 20 years were recruited consecutively by the Belgian Diabetes Registry (BDR). After obtaining written informed consent from each subject or the subject’s parents, the investigators collected blood samples and stored them at -80°C until they could be analyzed for serpinB13 serum binding activity. The study was approved by Institutional Review Board (IRB) at the BDR and Yale University.
Mice
NOD, NOR, NOD SCID, Balb/c and C57BL/6J mice were used as donors of T cells, serum and pancreatic islets. The NOD/Caj mice were kindly provided by Dr. L. Wen (Yale University). The NOD/LtJ mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and used to study the effects of treatment with anti-serpinB13 mAb on blood glucose levels. Mice were considered diabetic after 2 consecutive blood or urine glucose levels exceeding 200 mg/dL and 250 mg/dL, respectively. The University Animal Care and Use Committee approved all mouse experiments.
Peptides
Two peptide libraries were purchased from Proimmune, each consisting of 38 overlapping peptides representing (1) the first 200 AAs of OVA (peptides 1-19), moth cytochrome c (peptides 20-38), and (2) the entire sequence of serpinB13. The overlap between peptides was 10 AA in length. The pMOG sequence (MEVGWYRSPFSRVVHLYRNGK) was synthesized in the Keck Facility at Yale University.
Serpins
Purified mouse serpinB13 and serpinB8 expressed either in baculovirus were obtained from GeneScript.
Antibodies
The 2C11 anti-CD3 mAb was used to stimulate CD4 T cells isolated from various mouse strains and treat diabetic mice. An anti-His(6) epitope tag (Rockland) and anti-alpha tubulin antibodies (Cell Signaling; Millipore, Billerica, Mass) were used to coat the Luminex beads and stain the Western blots. Phycoerythrin (PE)- and APC-conjugated antibodies were used against CD4 (RM4-5), CD11b (M1/70), CD11c (HL3), CD45 (30-F11), B220 (RA3-6B2) and T-cell receptor (H57-597) (BD Biosciences, Billerica, Mass).
Production of anti-serpinB13 mAb
A single dose of 20 μg of purified mouse serpinb13 resuspended in 200 μL of complete Freund’s adjuvant was injected subcutaneously into a 6-week-old female NOD mouse. Two weeks later, the same animal was injected with the same amount of antigen resuspended in incomplete Freund’s adjuvant. The spleen was harvested 3 days after the second injection and was used to generate hybridoma fusions. After 2 weeks of selection with HAT media supplement (Sigma; St. Louis, MO), colonies were visible in 30 wells. These wells were screened for evidence of anti-serpinb13 antibody secretion using a Luminex assay. The supernatant obtained from the culture in one well demonstrated strong anti-serpinB13 binding activity. Hybridoma cells derived from this well were subcloned by serial dilution and retested for their ability to produce the antibody and its immunoglobulin G (IgG) isotype. In addition to the anti-serpinB13, we produced an IgG mAb with unknown specificity. This antibody was generated in NOD mouse and used as an IgG control in studies described in Figure 8.
Figure 8. Protective properties of the anti-serpinB13 mAb antibody.
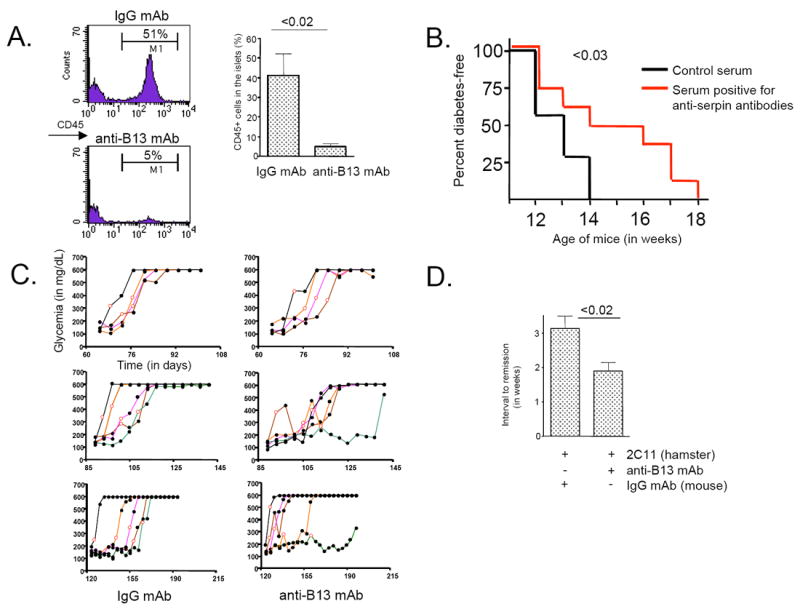
(A) FACS analysis of CD45+ cells (stained with 30-F11 mAb) in islets from mice treated with anti-serpinB13 mAb. Four-week-old female NOD mice received 4 injections of mAb (n=4) or the IgG mAb control (n=4) (100 μg/injection) over 10 days. The animals were sacrificed at 5-to-6 weeks of age. Right: The average of 3 independent experiments is shown. (B) Diabetes-free survival in NOD mice treated with serum containing anti-serpinB13 autoantibodies. The recipient mice were prescreened for low anti-serpinB13 autoantibody levels and were injected every 3 days with a 100-μL sample of serum containing either a high (n=7) or low (n=7) levels of anti-serpin autoantibodies. A total of 10 injections were made, the first at 3.5 weeks of age. After the last injection, the animals were followed weekly for evidence of glucosuria. (C) Blood glucose levels in NOD mice following treatment with anti-serpinB13 mAb. Animals were injected 4 times with anti-serpinB13 mAb (right) or IgG mAb control (left) (100 μg/injection) starting on the day of diagnosis (open circles), and their glucose levels were measured every 5 days thereafter. Data are displayed according to age at onset of diabetes: early (<12 weeks [upper]), intermediate (12-16 weeks [middle]) and late (>16 weeks [upper]) onset. The trajectory of each mouse towards the development of disease is shown in a different color. In groups treated with anti-serpinB13 mAb, responders are depicted in green, orange and brown and nonresponders are shown in black and purple. To determine the difference between control and treatment groups that develop diabetes after 12 weeks, animals from intermediate- and late-onset groups were combined. The Fisher’s exact test was used for the statistical analysis. (D) Timing of recovery from diabetes. Animals were treated with 2C11 mAb (10 μg/injection) and IgG mAb control (n=10) or 2C11 and anti-serpinB13 mAb (n=14). This injection regimen was similar to that described in C. The data are from one experiment and are representative of two to three independent experiments.
Luminex assays
Luminex-based technology was used to measure the serum-binding activity of ovserpins, green fluorescent protein (GFP), and secretagogin. Beads labeled with distinct dyes and blocked with a 1% bovine serum albumin (BSA) solution were precoated with anti-His(6) rabbit polyclonal antibody overnight at 4°C using a BioRad (Hercules, Calif) labeling kit. The beads were then washed 2 times and incubated for 24 to 48 hours at 4°C with lysates of 293T cells overexpressing individual His(6)-tagged proteins. The beads then underwent extensive washing and were pooled, incubated for 2 hours at room temperature with mouse serum samples (final dilution: 1:12), and then stained with biotinylated goat anti-mouse IgG polyclonal antibody (Invitrogen/Molecular Probes, Grand Island, NY) (dilution 1:100). To determine the IgG subclass of anti-serpinB13 autoantibodies, the beads were incubated with mouse serum samples and stained separately with biotinylated anti-mouse IgG1 (clone A85-1), IgG2a (clone R19-15), IgG2b (clone R12-3) or IgG3 (clone R40-82). These mAbs were purchased from BD Pharmingen (San Diego, Calif) and used at a dilution of 1:500. The secondary reagents were preabsorbed with rabbit IgG (Invitrogen/Molecular Probes) (dilution: 1:100) in a 1% BSA solution before use. The final step was to incubate the beads with streptavidin (Caltag catalog # SA1004-4; Invitrogen) (dilution: 1:200) for 10 min. at room temperature. Fluorescence intensity (FI) was measured using Luminex 100 and BioRad-Bioplex software. Specific binding to serpinB13 was calculated as FI units after subtracting FI due to serum binding activity in the presence of beads precoated with a control lysate (293T cells transfected with GFP [mouse] or GFP + secretagogin [human]). To demonstrate the specificity of antibody binding to serpinB13, representative mouse serum samples producing high (n=15), intermediate (n-14) or low (n=13) FI were subjected to competitive inhibition with soluble serpinB13 (1 μg/mL) in a fashion similar to that described for human serum samples (as described in the following paragraph).
To detect anti-serpinB13 antibodies in the human serum, these samples were divided into 2 halves, then incubated for 12 hours at 4°C with either soluble serpinB13 or serpinB8 as specific and nonspecific competitor, respectively. After incubation, the beads were precoated with individual antigens, then added to the samples, which were incubated for 2 hours at room temperature on a shaker (final serum dilution of serum: 1:10), and stained using a mix of biotinylated mouse anti-human kappa chain and lambda chain mAbs (BD Biosciences) (dilution: 1:300). The final steps of the assay were performed exactly as for the detection of mouse antibodies. The samples were evaluated based on the degree of inhibition of binding to serpinB13-coated beads by soluble serpinB13 compared with soluble serpinB8. Serum samples, in which the degree of inhibition was 25% to 49% or 50% to 100% were considered positive (+) and strongly positive (++), respectively.
IAA levels were measured using color-coated Luminex beads covalently conjugated with human insulin (Roche Molecular Biochemicals, Mannheim, Germany). The coupling of insulin to these beads was carried out according to a protocol provided by the vendor (BioPlex Amine Coupling Kit 171-406001; BioRad). To demonstrate antigen specificity, we divided each serum sample into 2 smaller aliquots that were preincubated with either free insulin (final concentration: 30μg/mL), to serve as a binding competitor, or without free insulin. The rest of the assay was similar to that described above.
Western blots
To verify the expression of His-tagged proteins, including clade B serpins, 293T-cell transfectants were lysed in a buffer containing 1% NP40, 150 mM NaCl, 50 mM Tris, (pH 7.4), 1 mM Na3VO4, 1mM phenylmethylsulfonyl fluoride and a commercial protease inhibitor (Roche Diagnostics, Indianapolis, Ind). Protein samples from precleared cell lysates were fractionated under reducing conditions on a 9% sodium dodecyl sulfate-polyacrylamide gel. After electrophoresis, the proteins were electroblotted onto nitrocellulose membranes (Bio-Rad), blocked with 5% nonfat dry milk, and probed with anti-His(6) rabbit antibody (1 μg/mL) or anti-serpinB13 mAb (1 μg/mL) followed by horseradish peroxidase (HRP)-linked protein A (Sigma) (1:2000) and goat anti-mouse secondary antibody (GE Healthcare, Uppsala, Sweden) (1:10000), respectively. The immunoblots were developed using an enhanced chemiluminescence detection system (GE Healthcare-Amersham Biosciences).
Molecular constructs
cDNAs samples encoding mouse plasminogen activator inhibitor 2 (also known as serpinB2), serpinB3a, serpinB3b, serpinB13 (both mouse and human), cathepsin L, secretagogin, and green fluorescent protein (GFP) were obtained from Open Biosystems (Thermoscientific, Lafayette, Colorado) as IMAGE clones and subcloned into a pcDNA™3.1 Directional V5-His-TOPO vector. The sequence of every construct was verified at the Keck Facility at Yale University.
Transfections
To obtain 293T cells expressing ovserpins, secretagogin, or GFP, we transfected individual cDNAs using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. The cells were harvested 48 hours after transfection and used to make the lysates.
Preparation of CD4 T cells and antigen presenting cells
CD4+ cells were isolated by negative selection using mAb to CD8 (TIB-210), MHC class II (10.2.16), FcR (2.4.G2), NK cells (HB191), and B cells (TIB164) then incubated with anti-mouse and rat Ig-coated magnetic beads (Qiagen; Valencia, Calif). The purity of the recovered CD4+ cells was 85% to 90% as determined by staining with anti-CD4 mAb (GK1.5). T-cell depleted antigen presenting cells (APCs) were prepared by Ab-mediated complement lysis of NOD splenocytes. Briefly, spleen cells were depleted of erythrocytes by centrifugation on a lymphocyte separation medium (MP Biomedicals; Solon, Ohio) then incubated, first with a mixture of anti-Thy 1 (Y-19), anti-CD8 (TIB-105), and anti-CD4 (GK1.5) mAbs and then with low-toxicity rabbit complement and 50 μg/ml mitomycin C (Sigma-Aldrich). Purity of the APC was 90% to 95%, as determined by staining with anti-MHC class II mAb.
T-cell culture and activation
All CD4+ T-cell cultures were maintained in Click’s medium containing 2-mercaptoethanol supplemented with 10% fetal calf serum (FCS), 10 mM HEPES, and antibiotics. Purified CD4+ cells were incubated with APCs and stimulated with either peptide pools (final concentration: 10 μg/mL each), single peptides, or 2C11 mAb (1 μg/mL). In some experiments (Fig. 1), T cells were subjected to several rounds of stimulation at 2-week intervals.
Figure 1. Early immune response to clade B serpins precedes the clinical onset of autoimmune diabetes.
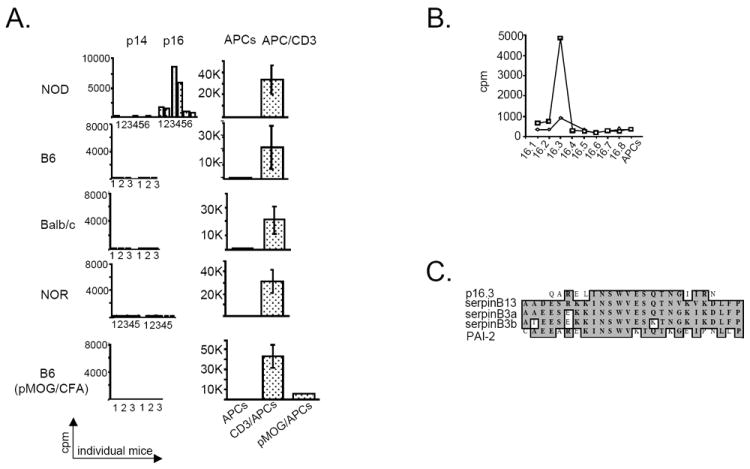
(A) CD4+ T-cell proliferation in response to OVA peptides (pool 16 [p16]) and control cytochrome c peptides (p14) in various mouse strains. Splenic CD4+ T cells isolated from NOD (n=6), B6 (n=3), Balb/c (n=3), NOR (n=5), and B6 mice immunized with pMOG (n=3), were stimulated 3 times at 2-week intervals with either p14 or p16 (left column). In addition, CD4+ T cells from each strain were stimulated once with APCs and with anti-CD3 mAb (right column). T cells from pMOG-exposed mice were also stimulated with APCs and pMOG. (B) CD4+ T-cell proliferation in response to individual p16 peptides. The cells isolated from 8-week-old NOD mice (n=2) were stimulated with 10 μg/mL of individual p16 peptides (p16.1 - p16.8). Cpm, counts per minute. (C) Blast alignment of AA sequences in p16.3 and several clade B serpins.
Proliferation assay
To measure proliferative activity, T cells were stimulated with APCs and the peptide in round 96-well plates for 56 hours, pulsed for the next 16 hours with [3H] thymidine, and then harvested to count cells that had incorporated the isotope.
Isolation of islets
Pancreatic islets were isolated using the collagenase/DNAase I digestion method and handpicked under a stereomicroscope. Islet cell suspensions were obtained by treating the islets with Cellstripper buffer (Invitrogen; cat. # 25-056-Cl) for 5 minutes at 37°C. We used 100 μL of tissue digest devoid of the islets to analyze proteases activity in the exocrine pancreas.
Statistics
Statistical analyses were carried out using the t test (Fig. 2, 3, 6, 8A, 8D and Supplemental Fig. S2-C), Kaplan-Meier Test (Fig. 8B) and Fisher’s exact test (Fig. 8C). Generalized estimating equations (GEE) were used to model the relationship for (i) anti-serpinB13 autoantibody responses and the 3 groups indicated in Fig. 4, (ii) antibody type and age of mice (Fig. 5A), and (iii) anti-serpinB13 autoantibody response in young mice and age at diabetes onset (Fig. 5B). To determine the relationship between age and the prevalence of anti-serpin antibody in diabetic patients with diabetes versus controls (Table 1), we used a generalized linear model with a logit link and Bernoulli distribution of the outcome estimated by maximizing the likelihood function. A P value less than .05 was used to indicate significance. Data are presented as mean ± standard deviation.
Figure 2. The specificity of assay detecting anti-serpinB13 autoantibodies.

Serum binding activity to serpinB13 was measured in the sera of NOD mice that were considered as negative (A, [n = 13]), weakly positive (B, [n = 14]) or positive (C, [n-15]) for anti-serpinB13 autoantibodies. Each sample was divided and preincubated at 4°C overnight with BSA (left) or with serpinB13 (right) at 1.0 μg/mL.
Figure 3. Immune response to clade B serpins in T1D is confined to serpinB13.
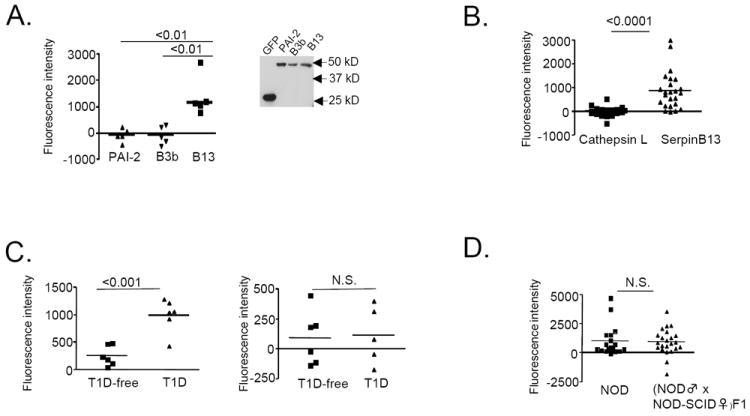
(A) Serum binding activity for serpin PAI-2 (n=5), serpinB3b (n=5) and serpinB13 (n=5) in NOD mice. Data are expressed as fluorescent intensity (FI) units and represent the total FI minus the FI due to serum binding activity in the presence of beads precoated with a control lysate (293 T cells transfected with GFP). Insert: Western blot analysis of cell lysates expressing individual proteins and used to coat the beads. (B) Serum-binding activity for cathepsin L and serpinB13 in NOD mice. A mixture of 3 sets of fluorescent beads precoated with cell lysates expressing cathepsin L, serpinb13, or GFP (control) was incubated with serum samples from 4-to 5-week-old NOD mice. (C) Serum binding to serpinB13 (left) and secretagogin (right) in 12-week-old NOD SCID mice at 6 weeks after receiving an adoptive transfer of splenocytes (5 × 106 cells/mouse) isolated from normal (T1D-free, n=6) or diabetic (T1D) NOD mice (n=6). (D) Serum-binding activity for serpinB13 in the offspring of wild type and immunodeficient NOD mice. Blood samples were obtained for testing from all mice at 4.5 weeks of age. The number of animals examined was 19 (wild type) and 23 (immunodeficient). Serum binding assay and verification of protein expression (data not shown) in B, C, and D was performed exactly as described in A. Data are from one experiment and are representative of at least two independent experiments.
Figure 6. Immunological response to serpinB13 is associated with diminished diabetogenic response.
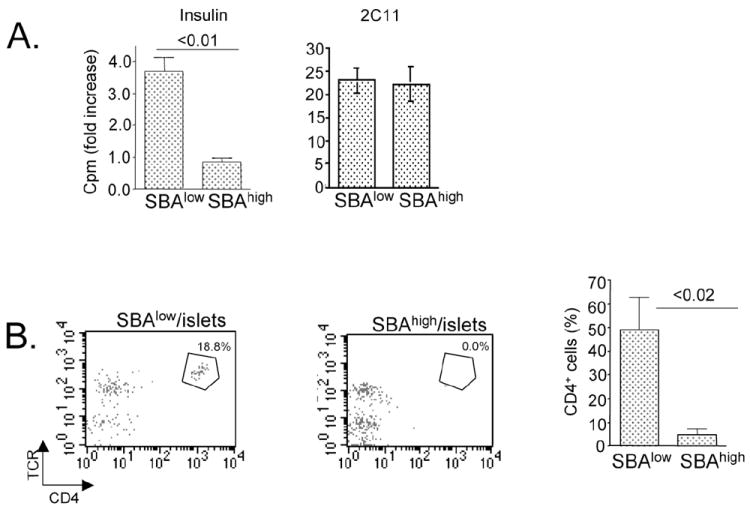
(A) T-cell proliferation in NOD mice with high (SBAhigh, n=4) and low (SBAlow, n=4) levels of anti-serpinB13 autoantibodies. T cells were isolated from the pancreatic lymph nodes of 5-week-old NOD mice and stimulated with insulin (10 μg/mL) or 2C11 mAb (1.0 μg/mL) in the presence of APCs (T-cell depleted splenocytes). Proliferation was measured using a thymidine incorporation assay. Data are expressed as fold induction over stimulation with APCs alone. Error bars indicate standard deviation for assay performed in triplicate. (B) Analysis of CD4+ T cells in the pancreatic islets of 5-week-old NOD mice that were negative or positive for anti-serpin autoantibodies. The cell suspensions were obtained by exposing pancreatic islets to cellstripper buffer. The cells were stained with anti-CD4 and anti-TCR mAbs and analyzed by FACS. Right: The average of 4 independent experiments is shown.
Figure 4. Immune response to serpinB13 in various mouse strains.
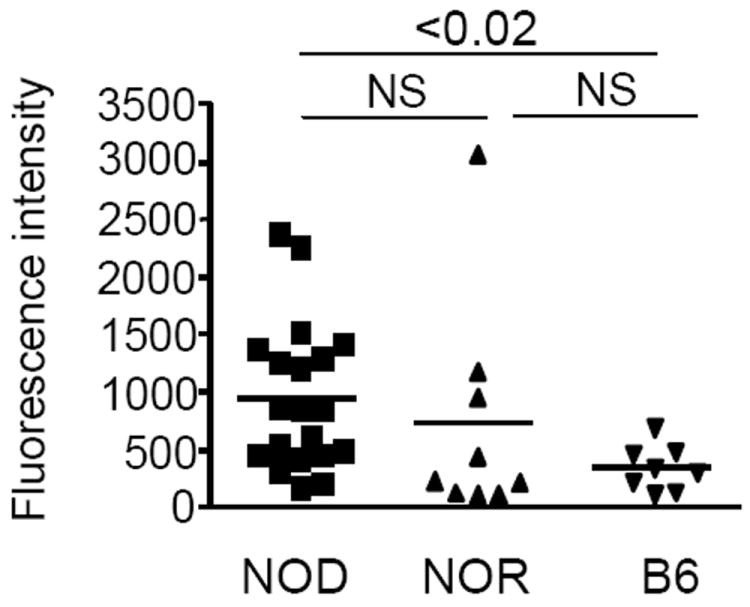
Serum-binding activity of serpinB13 in NOD (n = 20), NOR (n = 9), and B6 (n = 8) mice (A). The assay was performed exactly as described in Figure 3. Generalized estimating equations were used to model the relationship of antibody secretion among the three groups indicated. The overall chi-square test was significant (p=0.0354) as was the post-hoc comparison of NOD vs. B6 (p=0.0001).
Figure 5. Immunological response to serpinB13 is associated with prevention of early-onset autoimmune diabetes.
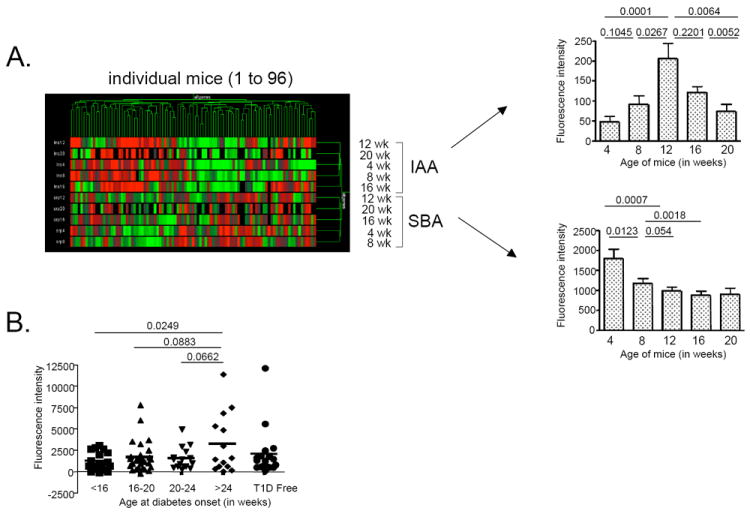
(A) Left: Hierarchical clustering of anti-insulin autoantibody (IAA; 5 upper rows) and anti-serpinB13 autoantibody (SBA; 5 lower rows) in a cohort of NOD mice (n=96) from whom blood samples were taken at age 4 weeks then every 4 weeks thereafter until the onset of diabetes. Both antibodies were measured using Luminex technology. Right: The kinetics of IAA and SBA responses based on the data displayed in left panel. Generalized estimating equations were used to model the response variable in association with the relationship of antibody type and mouse age, using the robust sandwich estimator to accommodate correlations introduced by the longitudinal data. Pairwise comparisons of interest were carried out as indicated. (B) Distribution of anti-serpinB13 serum binding activity in 4-week-old NOD mice (n=96) by age of disease onset. Generalized estimating equations were used to model the relationship of antibody response and age at diabetes onset.
Table 1.
Analysis of anti-serpinB13 antibodies in patients with recent-onset T1D.
| Controls | Diabetic subjects | |||
|---|---|---|---|---|
| Age at onset (yrs.) | N | SBA Pos (%) | N | SBA Pos (%) |
| 0-5 | 7 | 71% | 6 | 17% |
| 6-10 | 19 | 37% | 21 | 24% |
| 11-15 | 9 | 33% | 16 | 44% |
| 16-20 | 18 | 22% | 12 | 50% |
| Trend P* | 0.07 | 0.02 | ||
The data are expressed as the percentages of subjects in each age group that were positive for anti-serpin antibodies (SBA). Computed using logistic regression model with age as a continuous covariate; age-by-disease status interaction P=0.003
RESULTS
Immunological response against clade B serpins is induced in NOD mice at a young age
In the first attempt to determine whether NOD mice develop immunity to clade B serpins we studied their immunological response to chicken ovalbumin (OVA), a founding member of this protein family (18). Following in vitro stimulation, the proliferation of CD4+ T cells isolated from 2-month-old NOD mice was observed in all of the examined animals in response to pool 16 (p16) OVA peptides (amino acids [AAs] 121-200]. This response was not observed with CD4+ T cells isolated from NOR, Balb/c, or B6 mouse strains including B6 mice immunized with myelin oligodendrocyte glycoprotein peptide (pMOG) 36-50 (Fig. 1A). These results suggest that NOD mice have a relatively high frequency of OVA-specific CD4+ T cells and that stimulation of immune system with a random antigen (eg, pMOG) without coexisting pathologic changes in the peripheral tissues is not likely to induce anti-clade B serpin immunity.
To identify the specific OVA peptide responsible for the CD4+ T-cell response in NOD mice, we stimulated NOD T cells with individual peptides from p16 (p16.1 - p16.8). We found that the AA sequence QARELINSWVESQTNGIIRN (p16.3) was the best candidate (Fig. 1B). This sequence shares strong similarity with other members of clade B serpin family, including serpinB3a, serpinB3b, plasminogen-activated inhibitor (PAI)-2, and serpinB13 (Fig. 1C) (19). Thus, CD4+ T-cell response to p16.3 may represent the ability of NOD T cells to recognize several distinct clade B serpins.
To determine whether NOD mice develop a humoral response against ovserpins, we used Luminex-based technology to assay the serum-binding activity of PAI-2, serpinB3b, and serpinB13 simultaneously in serum samples taken from 5-week-old NOD mice. The specificity of this assay was verified by competitive inhibition with soluble serpinB13 (Fig. 2). The binding activity of serum samples (which were considered positive for anti-serpinB13 autoantibodies) to the beads precoated with serpinB13 was effectively blocked by preincubation of these serum samples with soluble serpinB13 (Fig. 2C, p=0.0103). By contrast, the binding activity in sera that were considered weak-positive was less affected by preincubation with soluble serpinB13 (Fig. 2B, p=0.0724) and completely unaffected by this treatment in negative serum samples (Fig. 2A, p=0.5007).
We found that the binding activity was significantly higher for serpinB13 (which is expressed in the pancreas [Supplemental Fig. S1]) compared with PAI-2, serpinB3b (Fig. 3A) or cathepsin L (which is inhibited by serpinB13 [16, 17]) (Fig. 3B). Importantly, the antibody response to serpinB13 in NOD mice was associated with pancreatic tissue damaged by diabetogenic splenocytes (Fig. 3C) rather than passive acquisition from NOD mothers during feeding period (Fig. 3D). These findings demonstrate that both T- and B-cell-mediated immunity against clade B serpins is active during the prediabetes stage in NOD mice and that immune-mediated injury of pancreatic islets plays a role in promoting this immunological response.
Additional characterization of anti-serpin humoral responses revealed that the serum-binding activity to serpinB13 usually occurred at a titer of less than 1:100 (Supplemental Fig. S2-A), was predominantly IgG2b (Supplemental Fig. S2-B), and at a lower level in NOR mice compared with NOD mice, although this difference between these animals was not statistically significant (Fig. 4). By contrast, the autoantibody response to serpinB13 was significantly lower in B6 mice compared with NOD mice (p=0.0354), but not with NOR mice. Although these observations suggest that an inflammatory lesion in pancreatic tissue helps induce the anti-serpin response, male NOD mice - which usually develop diabetes at a much lower rate - had similar levels of anti-serpinB13 autoantibodies compared with age-matched female NODs (Supplemental Fig. S2-C).
Secretion of anti-clade B serpin autoantibodies is associated with protection from early-onset T1D
To determine whether anti-clade B serpin immunity plays a role in the pathogenesis of autoimmune diabetes, we compared the expression of anti-serpinB13 autoantibodies with that of anti-insulin autoantibodies (anti-IAAs), a known marker of an elevated risk for autoimmune diabetes (20, 21). We found that a strong IAA response was associated with relatively weak anti-clade B serpin autoantibody response, whereas a strong anti-clade B serpin autoantibody response was associated with a relatively weak expression of IAAs (Fig. 5A, left). This inverse relationship could be explained by different kinetics for anti-insulin versus anti-serpinB13 autoantibody responses. We found that the secretion of anti-serpinB13 autoantibodies was strongest early in life (eg at age 4 weeks) and declined over the time, whereas IAAs levels peaked at age 12 weeks (Fig. 5A, right). We also found that a small subset of mice (approximately 16% [Supplemental Table S1]) generated high levels of anti-serpinB13 antibodies and that these mice showed a tendency toward a later onset of disease. Specifically, almost half of the animals with late-onset diabetes (age >24 weeks) had a strong anti-serpinB13 autoantibody response when they were 4 weeks old. By contrast, none of the NOD mice that developed early-onset diabetes (age <16 weeks) produced high levels of the anti-serpinB13 autoantibody at a young age (<16 weeks vs. >24 weeks; p= 0.0249) (Fig. 5B and Supplemental Table S1). This observation suggests the existence of a protective mechanism mediated through anti-clade B serpin immunity. We found additional support for this role when we compared T cells responses in NOD mice with different levels of anti-serpinB13 autoantibodies. T cells isolated from pancreatic lymph nodes in NOD mice with high levels of anti-serpin autoantibodies proliferated defectively in response to stimulation with diabetogenic antigens, including insulin (22) (Fig. 6A) and BDC2.5 mimotope (data not shown) (23). The number of islet-associated CD4 T cells was also reduced in these animals (Fig. 6B).
Antibodies against serpinB13 demonstrate anti-inflammatory and therapeutic properties
To determine whether antibody contributes directly to the protective process in autoimmune diabetes, we injected anti-serpinB13 monoclonal antibody (mAb) that was produced in our laboratory mAb (Fig. 7) into NOD mice that secreted relatively low levels of autoantibodies against this molecule. The response was similar to that observed in mice with high levels of endogenous anti-serpinB13 autoantibodies, ie, the numbers of islet CD45+ cells decreased in the islets considerably when this mAb was administrated soon after weaning (Fig. 8A). In addition, and in support of another report that the decrease in β-cell replication is reduced during diabetes remission (24), we found that the mAb-mediated reduction in lymphocyte levels in the peri-islet region was associated with a reduction in the proliferation of insulin-producing cells (data not shown). In light of these results, we decided to examine the possibility that enhanced anti-serpinB13 humoral immunity slows down the progression of autoimmune diabetes. NOD mice that received serum containing high levels of anti-serpinB13 autoantibodies exhibited a delayed onset of diabetes compared with animals that received serum containing low levels of these autoantibodies (Fig. 8B). Moreover, although the anti-serpinB13 mAb injection did not appear to influence the rate of diabetes reversal in animals that developed the disease at an early age; up to 60% of mice with later-onset diabetes (age >12 weeks) responded to this autoantibody with a transient or sustained reduction in blood glucose levels (p=0.0108) (Fig. 8C, middle and lower combined). Finally, to determine whether normoglycemia could be achieved sooner through the co-administration of anti-serpinB13 mAb and another agent with a proven anti-diabetic effect, we added an anti-CD3 mAb (25-27) to the treatment regimen in these animals. We found that co-administration of anti-serpinB13 mAb and a suboptimal dose of anti-CD3 mAb (28) in NOD mice with severe diabetes at onset (blood glucose > 300 mg/dL) was followed by the accelerated restoration of a normoglycemic state (Fig. 8D). These data together show that an enhancement of anti-serpinB13 humoral immunity can be therapeutically beneficial.
Figure 7. Epitope mapping of anti-serpinB13 mAb.
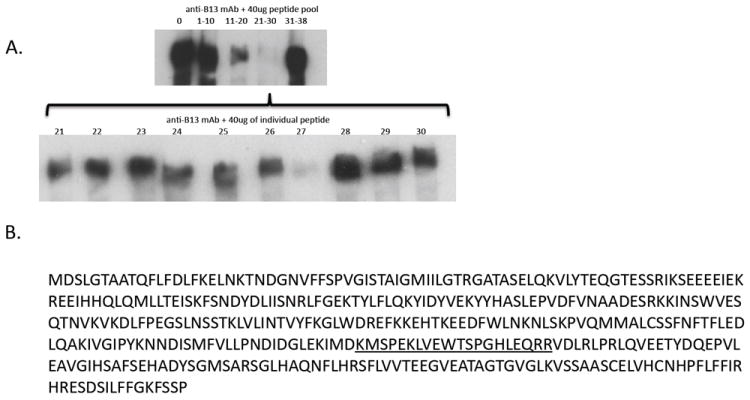
(A) Western blot analysis of serpinB13 staining with a mAb raised against this protein and preincubated with sequential pools of peptides (upper) or individual peptides (lower) corresponding to serpinB13. A 40 μg pool of peptides or individual peptides were incubated with anti-serpinB13 mAb in a volume of 100 μL at 4°C overnight and used for staining. (B) Full AA sequence of serpinB13. The sequence corresponding to peptide 27, which blocks staining with anti-serpinB13 mAb, is underlined.
Expression of anti-serpinB13 antibodies in humans with T1D
To determine whether an immunological response to serpinB13 is associated with the pathogenesis of autoimmune diabetes in humans, we measured anti-serpin autoantibody levels in patients with recent-onset T1D (Table 1 and Supplemental Table S2). We found that the anti-serpin activity increased significantly with age in these patients (p=0.02) but decreased with age in healthy controls, although not to a statistically significant degree (p=0.07). The test of age-by-disease status interaction in the logistic model was highly significant (p=0.003), suggesting that the relationship between age and anti-serpin humoral activity differs between patients with T1D and healthy controls. These data, together with our observations in the NOD mouse demonstrate that early-onset autoimmune diabetes is associated with a relatively low level of anti-serpin immunity.
DISCUSSION
Type 1 diabetes (T1D) is thought to be primarily a T-cell mediated disease (1-3); however, B cells (29-32) and antibodies (33-35) have also been implicated in its pathogenesis. Remarkably, certain autoantibodies have been found to be associated with protection from T1D (36-38). For example, vertical autoantibody transmission from the mothers with T1D to their progeny significantly lowers risk for the subsequent development of multiple islet autoantibodies and diabetes (38, 39). In this study we have demonstrated the existence of a novel autoantibody that recognizes serpinB13 and is associated with the protection against T1D. Experiments with a monoclonal antibody against serpinB13 that was produced in our laboratory suggest active defense mechanism that slows down progression to clinical diabetes. However, exact molecular events following enhancement of anti-serpin immunity remain to be determined. One possibility is that the binding serpinB13 to its antibody impairs its inhibitory effect and consequently allows the activity of its protease targets to increase. In turn, upregulated proteases may cause the damage of inflammatory cells that accumulate in the pancreatic islets; alternatively they may increase the rate of conversion of molecules with a potential to regulate β-cell survival and islet regeneration. These hypothetical scenarios are currently being tested in our laboratory.
It is interesting to know whether anti-serpin B-cell immunity occurs in disease models other than NOD mice. Based on the preliminary work we found that B6 mice produce anti-serpin autoantibodies following subcutaneous injection of myelin oligodendrocyte glycoprotein (pMOG) autoantigen coupled with intravenous injection of pertussis toxin (this treatment leads to experimental autoimmune encepohalomyelitis). On the other hand, exposure to pMOG alone does not result in EAE and fails to induce the secretion of these autoantibodies. These observations, together with other data described in our manuscript, suggest a two-step model. Step 1 consists of the formation of an inflammatory lesion, which helps to induce the anti-serpin response; and Step 2, which follows the generation of these antibodies, helps confer protection against further progression of inflammation. Of note, our observations regarding the protective role of anti-serpin humoral activity concerns mainly autoimmune diabetes. We do not know whether anti-serpin antibodies secreted during the weeks preceding the onset of EAE are protective or harmful.
In addition to their induction following an inflammatory tissue injury, anti-serpin autoantibodies, may be derived from B1 B cells. These cells are responsible for the very early antibody response and are known to be active in NOD mice (40). In our study of CD5+ B1 B cells isolated from the peritoneal cavity and spleen in NOD mice, we found no detectable levels of anti-serpinB13 immunoglobulins in the culture supernatants after stimulating these cells with LPS (data not shown). Thus, it is not very likely that B1 B cells are the main source of anti-serpin activity. Consistent with this conclusion is a recent report that B1 B cells promote, rather than inhibit, the inflammatory response in the pancreas (41).
The reason why treatment with anti-serpinB13 mAb inhibited late-onset diabetes and endogenous anti-serpinB13 autoantibodies were associated with protection from the early-onset diabetes in our study remains unclear. The existence of distinct mechanisms of protection that are regulated differently by anti-serpin antibodies in young versus old mice may be responsible for this observation. For example, one may argue that anti-serpin antibodies are more effective in mice with late-onset disease because islet inflammation progresses less aggressively in these animals compared with mice that develop early-onset T1D. By contrast, during the first weeks of life, anti-serpin antibodies may - in addition to dampening the inflammatory response in islets - enhance the regeneration of insulin-producing cells. Consistent with this model, and with the notion that islet regeneration is more active early in life, the presence of anti-serpin activity during that early period would mainly benefit animals that are destined to develop early-onset (rather than late-onset) T1D.
Our observations in the NOD mouse model are relevant to human disease, because we have found that children with T1D onset during the first decade of life are deficient in anti-clade B serpin antibodies compared with age-matched healthy counterparts. Interestingly, the opposite is true for individuals who developed T1D during the second decade of life. Specifically, anti-serpin activity is seen more frequently in healthy young children (0-10 y; 46%) compared with young children with recent-onset diabetes (21%) or healthy adolescents (11-20 y; 26%). The reason of this finding is not clear. Perhaps, the secretion of anti-serpin antibodies is a normal response to tissue damage caused by environmental factors. Indeed, certain members of the ovalbumin-serpin family are up-regulated by Th2-type cytokines in bronchial epithelial cells and their expression is augmented in the bronchial lesions of patients with asthma (42). Exposure to air pollutants, seasonal infections, or physical injuries that frequently occur at a young age may all contribute to the release of intracellular serpins from damaged tissue. By contrast, highly hygienic conditions may reduce the amount of tissue damage, thereby reducing the production of protective anti-serpin antibodies and promoting a more rapid progression toward T1D in individuals at risk for the disease. Of note, the incidence of T1D is on the rise in the developed countries (4, 5) and a reduced output of anti-serpin antibodies, especially in the young children, may contribute to this trend.
The findings regarding anti-serpin antibodies described here are important for several reasons. First, if these antibodies induce responses in humans that are analogous to those observed in NOD mice, we may be able to improve our ability to predict the rate of progression toward the onset of disease in individuals at risk for T1D by monitoring their anti-ovserpin antibody levels over time. Second, information about prediabetes levels of anti-serpin autoantibodies may allow us to improve the design of T1D treatment and prevention trials. Finally, anti-clade B serpin mAbs could be used in individuals who lack immunity against these molecules to impede the accumulation of inflammatory cells in affected organs and, thus, could be therapeutically beneficial.
Supplementary Material
Acknowledgments
The authors are thankful to Drs. R. Sherwin (Yale University) and L. Wen (Yale University) for their helpful discussions and for the NOD mice; Drs. E. Scosyrev, S. Messing, and X. Tu (University of Rochester) for the statistical analysis; and the Belgian Diabetes Registry for collecting serum samples.
This work was supported by a Diabetes Endocrinology Research Center Grant awarded to J.C. and a pilot grant in Translational and Interdisciplinary Research awarded to J.C. and O.H. R.A.F. is an Investigator of the Howard Hughes Medical Institute.
References
- 1.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Ann Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 2.Solomon M, Sarvetnick N. The pathogenesis of diabetes in the NOD mouse. Adv Immunol. 2004;84:239–264. doi: 10.1016/S0065-2776(04)84007-0. [DOI] [PubMed] [Google Scholar]
- 3.Martin S, Wolf-Eichbaum D, Duinkerken G, Scherbaum WA, Kolb H, Noordzji JG, Roep BO. Development of type 1 diabetes despite severe hereditary B-lymphocyte deficiency. N Eng J Med. 2001;345:1036–1040. doi: 10.1056/NEJMoa010465. [DOI] [PubMed] [Google Scholar]
- 4.Harjutsalo V, Sjoberg L, Tuomilehto J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. Lancet. 2008;371:1777–1782. doi: 10.1016/S0140-6736(08)60765-5. [DOI] [PubMed] [Google Scholar]
- 5.Patterson CC, Dahlquist GG, Gyurus A, Green A, Soltesz H EURODIAB Study Group. Incidence trends for childhood type 1 diabetes in Europe during 1989-2003 and predicted new cases 2005-2020: a multicentre prospective registration study. Lancet. 2009;373:2027–2033. doi: 10.1016/S0140-6736(09)60568-7. [DOI] [PubMed] [Google Scholar]
- 6.Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, Chervonsky AV. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aumeunier A, Grela F, Ramadan A, Van LP, Bardel E, Gomez Alcala A, Jeannin P, Akira S, Bach J-F, Thieblemont N. Systemic Toll-like receptor stimulation suppresses experimental allergic asthma and autoimmune diabetes in NOD mice. PloS ONE. 2010;5:e11484. doi: 10.1371/journal.pone.0011484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.King C, Sarvetnick N. The incidence of type-1 diabetes in NOD mice is modulated by restricted flora not germ-free conditions. PloS ONE. 2011;6:e17049. doi: 10.1371/journal.pone.0017049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irving JA, Pike RN, Lesk AM, Whisstock JC. Phylogeny of the serpin superfamily: implications of patterns of amino acid sequences for structure and function. Genome Res. 2000;10:1845–1864. doi: 10.1101/gr.gr-1478r. [DOI] [PubMed] [Google Scholar]
- 10.Silverman GA, Whisstock JC, Askew DJ, Pak SC, Luke CJ, Cataltepe S, Irving JA, Bird PI. Human clade B serpins (ov-serpins) belong to a cohort of evolutionary dispersed intracellular proteinase inhibitor clades that protect cells from promiscuous proteolysis. Cell Mol Life Sci. 2004;61:301–325. doi: 10.1007/s00018-003-3240-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maehr R, Mintern JD, Herman AE, Lennon-Dumenil A-M, Mathis D, Benoist C, Ploegh HL. Cathepsin L is essential for onset of autoimmune diabetes in NOD mice. J Clin Invest. 2005;115:2934–2943. doi: 10.1172/JCI25485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Viken MK, Sollid HD, Joner G, Dahl-Jorgensen K, Ronningen kS, Undlien DE, Flato B, Selvaag AM, Forre O, Kvien TK, Thorsby E, Melms A, Tolosa E, Lie BA. Polymorphisms in the cathepsin L2 (CTSL2) gene show association with type 1 diabetes and early-onset myasthenia gravis. Hum Immunol. 2007;68:748–755. doi: 10.1016/j.humimm.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 13.Hsing LC, Kirk EA, McMillen TS, Hsiao SH, Caldwell M, Houston B, Rudensky AY, LeBoeuf RC. Roles of cathepsin S, L, and B in insulitis and diabetes in the NOD mouse. J Autoimmunity. 2010;34:96–104. doi: 10.1016/j.jaut.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishimaru N, Arakaki R, Katunuma N, Hayashi Y. Critical role of cathepsin-inhibitors for autoantigen processing and autoimmunity. Adv Enzyme Reg. 2004;44:309–320. doi: 10.1016/j.advenzreg.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Yamada A, Ishimaru N, Arakaki R, Katunuma N, Hayashi Y. Cathepsin L inhibition prevents murine autoimmune diabetes via suppression of CD8+ T cell activity. PloS One. 2010;5:e12894. doi: 10.1371/journal.pone.0012894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welss T, Sun J, Irving JA, Blum R, Smith AI, Whisstock JC, Pike RN, von Mikecz A, Ruzicka T, Bird PI, Abts HF. Hurpin is a selective inhibitor of lysosomal cathepsin L and protects keratinocytes from ultraviolet-induced apoptosis. Biochemistry. 2003;42:7381–7389. doi: 10.1021/bi027307q. [DOI] [PubMed] [Google Scholar]
- 17.Jayakumar A, Kang Y, Frederick MJ, Pak SC, Henderson Y, Holton PR, Mitsudo K, Silverman GA, El-Naggar AK, Bromme D, Clayman GL. Inhibition of the cysteine proteinases cathepsins K and L by the serpin headpin (SERPINB13): a kinetic analysis. Arch Biochem Biophys. 2003;409:367–374. doi: 10.1016/s0003-9861(02)00635-5. [DOI] [PubMed] [Google Scholar]
- 18.Remold-O’Donnell E. The ovalbumin family of serpin proteins. FEBS Lett. 1993;315:105–108. doi: 10.1016/0014-5793(93)81143-n. [DOI] [PubMed] [Google Scholar]
- 19.Nakashima T, Pak SC, Silverman GA, Spring PM, Frederick MJ, Clayman GL. Genomic cloning, mapping, structure and promoter analysis of HEADPIN, a serpin which is down-regulated in head and neck cancer cells. Biochim Biophys Acta. 2000;1492:441–446. doi: 10.1016/s0167-4781(00)00100-7. [DOI] [PubMed] [Google Scholar]
- 20.Palmer JP, Asplin CM, Clemons P, Lyen K, Tatpati O, Raghu PK, Paquette TL. Insulin antibodies in insulin dependent diabetics before insulin treatment. Science. 1983;222:1337–1339. doi: 10.1126/science.6362005. [DOI] [PubMed] [Google Scholar]
- 21.Atkinson MA, Eisenbarth GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;358:221–229. doi: 10.1016/S0140-6736(01)05415-0. [DOI] [PubMed] [Google Scholar]
- 22.Nakayama M, Abira N, Moriyama H, Babaya N, Liu E, Miao D, Yu L, Wegmann DR, Hutton JC, Elliott JF, Eisenbarth GS. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 2005;435:220–223. doi: 10.1038/nature03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell. 1983;74:1089–1100. doi: 10.1016/0092-8674(93)90730-e. [DOI] [PubMed] [Google Scholar]
- 24.Sherry NA, Kushner JA, Glandt M, Kitamura T, Brillantes A-MB, Herold KC. Effects of autoimmunity and immune therapy on beta-cell turnover in type 1 diabetes. Diabetes. 2006;55:3238–3245. doi: 10.2337/db05-1034. [DOI] [PubMed] [Google Scholar]
- 25.Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci USA. 1994;91:123–127. doi: 10.1073/pnas.91.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keymeulen B, Vandemeulebroucke E, Ziegler AG, Matheu C, Kaufman L, Hale G, Gorus F, Goldman M, Walter M, Candon S, Schandene L, Crenier L, De Block C, Seigneurin J-M, De Pauw P, Pierard D, Weets I, Rebello P, Bird P, Berrie E, Frewin M, Waldmann H, Bach J-F. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352:2598–2608. doi: 10.1056/NEJMoa043980. [DOI] [PubMed] [Google Scholar]
- 27.Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman S, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- 28.Mehta DS, Christmas RA, Waldmann H, Rosenzweig M. Partial and transient modulation of the CD3-T-cell receptor complex, elicited by low-dose regimens of monoclonal anti-CD3, is sufficient to induce disease remission in non-obese diabetic mice. Immunology. 2010;130:103–113. doi: 10.1111/j.1365-2567.2009.03217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NC. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2009;155:173–181. doi: 10.1111/j.1365-2249.2008.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu C-Y, Rodriguez-Pinto D, Du W, Ahuja A, Henegariu O, Wong FS, Shlomchik MJ, Wen L. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest. 2007;117:3857–3867. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Type 1 Diabetes TrialNet Anti-CD20 Study Group. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Eng J Med. 2009;361:2143–2152. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fiorina P, Vergani A, Dada S, Jurewicz M, Wong M, Law K, W E, Tian Z, Abdi R, Guleria I, Rodig S, Dunussi-Joannpoulos K, Bluestone J, Sayegh MH. Targeting CD22 reprograms B-cell and reverses autoimmune diabetes. Diabetes. 2008;57:3013–3024. doi: 10.2337/db08-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greeley SAW, Katsumata M, Yu L, Eisenbarth GS, Moore DJ, Goodarzi H, Barker CF, Naji A, Noorchashm H. Elimination of maternally transmitted autoantibodies prevents diabetes in nonobese diabetic mice. Nature Med. 2002;8:399–402. doi: 10.1038/nm0402-399. [DOI] [PubMed] [Google Scholar]
- 34.Harbers SO, Crocker A, Catalano H, D’Agati V, Jung S, Desai DD, Clynes R. Antibody-enhanced cross-presentation of self antigen breaks T cell tolerance. J Clin Invest. 2007;117:1361–1369. doi: 10.1172/JCI29470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silva DG, Daley SR, Hogan J, Lee SK, Teh CE, Hu DY, Lam K-P, Goodnow CC, Vinuesa CG. Anti-islet autoantibodies trigger autoimmune diabetes in the presence of an increased frequency of islet-reactive CD4 T cells. Diabetes. 2011;60:2102–2111. doi: 10.2337/db10-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.She J-X, Ellis TM, Wilson SB, Wasserfall CH, Marron M, Reimsneider S, Kent SC, Hafler DA, Neuberg DS, Muir A, Strominger JL, Atkinson MA. Heterofile antibodies segregate in families and are associated with protection from type 1 diabetes. Proc Natl Acad Sci USA. 1999;96:8116–8119. doi: 10.1073/pnas.96.14.8116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menard V, Jacobs H, Jun F-S, Yoon J-W, Kim SW. Anti-GAD monoclonal antibody delays the onset of diabetes mellitus in NOD mice. Pharmac Res. 1999;16:1059–1066. doi: 10.1023/a:1018939900961. [DOI] [PubMed] [Google Scholar]
- 38.Koczwara K, Bonifacio E, Ziegler A-G. Transmission of maternal islet antibodies and risk of autoimmune diabetes in offspring of mothers with type 1 diabetes. Diabetes. 2004;53:1–4. doi: 10.2337/diabetes.53.1.1. [DOI] [PubMed] [Google Scholar]
- 39.Warram JH, Krolewski AS, Gottlieb MS, Kahn CR. Differences in risk of insulin-dependent diabetes in offspring of diabetic mothers and diabetic fathers. N Eng J Med. 1985;311:149–152. doi: 10.1056/NEJM198407193110304. [DOI] [PubMed] [Google Scholar]
- 40.Corte-Real J, Duarte N, Tavares L, Penha-Goncalves C. Autoimmunity triggers in the NOD mouse: a role for natural auto-antibody reactivities in type 1 diabetes. Ann New York Acad Sci. 2009;1173:442–448. doi: 10.1111/j.1749-6632.2009.04661.x. [DOI] [PubMed] [Google Scholar]
- 41.Ryan GA, Wang CJ, Chamberlain JL, Attridge K, Schmidt EM, Kenefeck R, Clough LE, Dunussi-Joannopoulos K, Toellner K-M, Walker LSK. B1 cells promote pancreas infiltration by autoreactive T cells. J Immunol. 2010;185:2800–2807. doi: 10.4049/jimmunol.1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuyama N, Davies DE, Akaiwa M, Matsui K, Hamasaki Y, Suminami Y, Yoshida NL, Maeda M, Pandit A, Lordan JL, Kamogawa Y, Arima K, Nagumo F, Sugimachi M, Berger A, Richards I, Roberds SL, Yamashita T, Kishi F, Kato H, Arai K-I, Ohshima K, Tadano J, Hamasaki N, Miyatake S, Sugita Y, Holgate ST, Izuhara K. Analysis of novel disease-related genes in bronchial asthma. Cytokine. 2002;19:287–296. doi: 10.1006/cyto.2002.1972. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.