

Abstract
Deregulated expression of most members of the E2F family has been detected in many human cancers. We examined the association of common single nucleotide polymorphisms (SNPs) of E2F1 and E2F2 with risk of squamous cell carcinoma of the head and neck (SCCHN) in 1,096 SCCHN patients and 1,090 cancer-free controls. We genotyped ten selected SNPs in E2F1 and E2F2, including those at the near 5′ UTR, miRNA binding sites at the near 3′ UTR and tagSNPs according to bioinfotmatics analysis. Although none of the selected SNPs alone was significantly associated with risk of SCCHN, there was a statistically significantly increased risk of SCCHN associated with the combined risk genotypes (i.e. rs3213182 AA, rs3213183 GG, rs3213180 GG, rs321318121 GG, rs2742976 GT+TT, rs6667575 GA+AA, rs3218203 CC, rs3218148 AA, rs3218211 CC, rs3218123 GT+TT). Compared with those with 0–4 risk genotypes, an increased risk was observed for those who carried 5–8 risk genotypes (adjusted OR = 1.04; 95% CI = 0.86–1.26) and 9–10 risk genotypes (adjusted OR = 1.62; 95% CI = 1.14–2.30) in a dose-response manner (P = 0.045). Furthermore, the joint effect was more pronounced among patients with oropharyngeal cancer, younger adults (≤57 years old), men, non-smokers, non-drinkers, and individuals with family history of cancer first-degree relatives. Additionally, we also observed that those with 5–10 risk genotypes had an earlier SCCHN onset than those with 0–4 risk genotypes, particularly for non-smokers and/or non-drinkers. We concluded that E2F1 and E2F2 genetic variants may jointly play important roles in head and neck carcinogenesis.
Keywords: E2F1, E2F2, head and neck cancer, polymorphisms, age at onset
INTRODUCTION
Squamous cell carcinoma of the head and neck (SCCHN) mainly comprises malignant tumors at the oral cavity, oropharynx, hypopharynx and larynx. Approximately 52,140 new SCCHN cases occurred, accounting for above 3.2% of all newly diagnosed non-cutaneous malignancies, with 11,460 deaths in 2011 in the United States [1]. Tobacco and alcohol use, as well as infection of human papillomavirus, are the known risk factors for SCCHN [2–5]. However, only a small part of exposed individuals develops SCCHN, suggesting that genetic susceptibility factors also play critical roles in carcinogenesis of the head and neck. Increasing evidence has showed that inherited differences in efficiencies of carcinogen metabolism, DNA repair, cell cycle control, apoptosis, or a combination of these factors may modulate individual risk of developing SCCHN [6–11]. However, the roles of genetic variants in the E2F family members in the etiology of SCCHN have not been elucidated.
The E2F family are transcriptional factors that affect cell fate in general, and cancer development in particular [12], because they play pivotal roles in regulating both cell proliferation and antiproliferative processes [13]. Deregulated expression or activity of most members of the E2F family has been detected in many human cancers [14]. For example, E2F1-E2F8 are downstream effectors of the Rb tumor suppressor, which contain one or more conserved DNA binding domains (DBDs) that bind to the target promoters and regulate their expression [15,16]. E2F1, E2F2 and E2F3a, which interact only with pRB, have been classified as the ‘activator E2Fs’ that functionally activate gene expression, and E2F1 and E2F3 can influence the p53 activity by binding directly to p53 [13]. Furthermore, the E2F1 or E2F3 acting as oncogenes are also found to be amplified in many malignancies, and large chromosomal deletions of the regions of E2F1, E2F2 and E2F3 genes have been detected in some patients with neuroblastoma, thyroid cancer, and pancreatic cancer [17].
The E2F1 gene is located on chromosome 20q, spanning approximately 10.71 kb, and it contains 7 exons (National Center for Biotechnology Information reference sequences, NCBI RefSeq). The E2F2 gene is located on chromosome 1p, spanning approximately 24.79 kb, and it also contains 7 exons (National Center for Biotechnology Information reference sequences, NCBI RefSeq). Generally, E2F1−/− may increase susceptibility to tumorigenesis in different tissues [12,13], because E2F1 can induce apoptosis that is mediated by both p53-dependent and p53-independent pathways [14]. One study showed that the absence of E2F2 cooperates with Myc to accelerate tumorigenesis in the oral cavity [18].
It is well known that genetic variants in the coding, promoter, and miRNA binding regions can result in altered gene functions. However, some studies have shown inconsistent results of E2F1 and E2F2 variants in association with risk of cancer, including SCCHN [19,20]. Considering that E2F3, including E2F3a and E2F3b, is more complicated to study, because they are generated by alternative promoters [15]. Here, we tested our hypothese that E2F1 and E2F2 variants or their combination are associated with risk of SCCHN by performing a case-control study to examine the association of common (with minor allele frequency ≥5%) single nucleotide polymorphisms (SNPs) of E2F1 and E2F2 with SCCHN susceptibility.
MATERIALS AND METHODS
Study Subjects and Sample Collection
Study subjects were recruited from our ongoing SCCHN study as described previously [9]. Briefly, this study included 1,096 patients with newly diagnosed and histopathologically confirmed SCCHN and 1,090 self-reported cancer-free controls recruited between October 1999 and October 2009 at The University of Texas M. D. Anderson Cancer Center. Patients were excluded, if they had received prior surgery (other than diagnostic biopsies), chemotherapy or radiation therapy before recruitment, any blood transfusion during the preceding 6 months, any malignancies other than SCCHN, second SCCHN primary tumors, primary tumors of nasopharynx or sinonasal tract, or primary tumors outside the upper aerodigestive tract. Of the 1096 SCCHN patients included in the analysis, 323 (29.55%) had cancers of the oral cavity, 557 (50.8%) of the oropharynx, and 216 (19.7%) of the larynx (including 44 of the hypopharynx). Controls were enrolled from those genetically unrelated hospital visitors who were not seeking health care but accompanying other cancer patients to visit clinics. Controls were frequency-matched to the cases by age (±5 years), sex and ethnicity. Approximately 92% of eligible patients and 85% of controls contacted agreed to participate in this study.
Because of the expected differences in genotype frequencies between ethnic groups, only non-Hispanic whites were included in the analysis. All recruited subjects who were willing to participate in studies were interviewed by trained interviewers. Demographic (e.g., age, sex, and ethnicity) and risk factor (e.g., the history of tobacco smoking and alcohol consumption) information were collected using a standardized, structured questionnaire [21]. Each subject signed for an informed consent and donated a one-time 30-ml venous blood sample (after the diagnosis and before the initiation of treatment for the cases), and the research protocol was approved by the M. D. Anderson institutional review board.
Bioinformatics Analysis and Selection of Polymorphisms
We selected SNPs according to the following strategies: firstly, we identified tagging SNPs of E2F1 and E2F2 genes extending 2000 bps at each end of the gene by Hapmapview4.2 (http://hapmap.ncbi.nlm.nih.gov/cgi-perl/gbrowse/hapmap24_B36/#search). Secondly, we used the bioinformatics tool of SNP Function Prediction (FuncPred, http://snpinfo.niehs.nih.gov/snpfunc.htm) to identify their potential functional relevance as followed: (1) affecting transcription factor binding sites (TFBS) activity in the putative promoter region (here we defined as 2-kb upstream from the first exon) ( http://molsun1.cbrc.aist.go.jp/research/db/TFSEARCH.html), (2) affecting the microRNA (miRNA) binding site activity. We limited the SNPs to those with minor allele frequency ≥5% in the Hapmap CEU populations. We also calculated pairwise linkage disequilibrium (LD) of all SNPs in the same gene and chose those SNPs that were not in LD (r2 < 0.8), and plotted LD maps of those SNPs in E2F1 and E2F2 genes with the program Haploview (Vision 4.2) (Figure 1)
Figure 1.
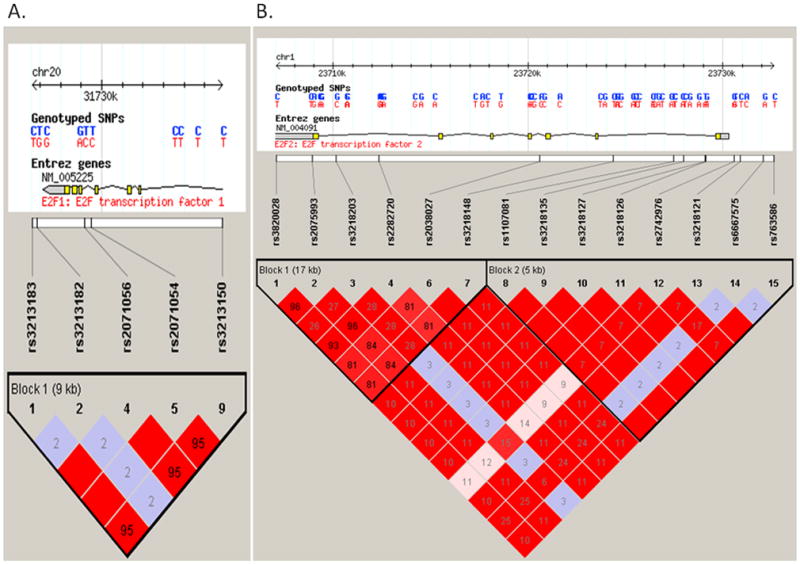
The top panel shows the E2F1 (Figure 1A) and E2F2 (Figure 1B) gene region. The lower panel shows the linkage disequilibrium plot of the E2F1 and E2F2 region, using the CEU population from the International HapMap project. Each square number represents the pairwise r2 relationships between the relevant two SNPs.The color of each SNP spot reflects its D′ value, which changes from red to white as the D′ value decreases. The haplotype blocks were estimated with the program Haploview.The minor allele frequency of all above alleles are more than 0.05. Other selected functional alleles predicted in our study rs3213180 in E2F1 and rs3218123, rs3218211 in E2F2 were not included in the International HapMap data.
As a result, we identified two common tagSNPs for the E2F1 gene: rs3213182 (in the 3′ near gene) and rs2071054 (in intron 4). We found another SNP rs3213183 in the 3′ near gene in complete LD with rs2071054 (r2 = 1). We also selected an additional SNP rs3213180 (in the 3′ UTR) that was predicted to affect the microRNA (miRNA) binding site activity. Thus, three potentially functional SNPs were identified: rs3213180 (E2F1 c.*914 C>G), rs3213182 (E2F1 3′ 256 T>G) and rs3213183 (E2F1 3′ 527 C>T) (Table 1).
Table 1.
Information of Selected SNPs of E2F1 and E2F2 Genes
| Gene | Selected SNPs | Location | TagSNPs | Function Predictedb | Position | Allele | MAFc |
|---|---|---|---|---|---|---|---|
| E2F1 | rs3213180 | 3′ UTR | NA | miRNA bing site | c.*914 | C>G | 0.056 |
| rs3213182 | 3′ near gene | Yes | NA | 3′ 256 | T>G | 0.067 | |
| rs3213183 | 3′ near gene | Yesa | NA | 3′ 527 | C>T | 0.267 | |
| E2F2 | rs6667575 | 5′ near gene | Yes | TFBS: G:GATA-2/A:GATA-1,2 | −1808 | G >A | 0.200 |
| rs3218121 | 5′ near gene | Yesa | TFBS | −604 | C >T | 0.100 | |
| rs2742976 | 5′ near gene | Yes | TFBS:G: STATx/T: STATx | −289 | G >T | 0.408 | |
| rs3218123 | 5′ near gene | NA | TFBS:G:SRY /T: Elk-1 | −143 | G >T | 0.140 | |
| rs3218148 | Intron 1 | Yesa | NA | 5926 | C >T | 0.492 | |
| rs3218203 | Intron 6 | Yes | NA | 20153 | G >C | 0.217 | |
| rs3218211 | 3′ UTR | NA | miRNA bing site | c.*578 | C >T | 0.500 |
complete LD with TagSNP: SNP rs3213183 in the 3′ near gene in complete LD (r2=1) with rs2071054; rs3218121 (in the 5′ near gene) is in complete LD (r2=1) with rs3218127 and that rs3218148 (in intron 1, MAF > 5%) is in complete LD (r2=1) with rs2038027 (in intron 3, MAF < 5%)
Function Predicted were used the bioinformatics tool of SNP: FuncPred, http://snpinfo.niehs.nih.gov/snpfunc.htm) and http://molsun1.cbrc.aist.go.jp/research/db/TFSEARCH.html
MAF of rs3213180, rs3218123, and rs3218211 were cited from http://www.ncbi.nlm.nih.gov/SNP, others were form Hapmap-CEU data
For the E2F2 gene, five common tagSNPs were identified, including rs3218127 (in intron 1), rs2038027 (in intron 3), rs2742976 (in the 5′ near gene), rs3218203 (in intron 6), and rs6667575 (in the 5′ near gene). We found that rs3218121 (in the 5′ near gene) is in complete LD (r2 = 1) with rs3218127 and that rs3218148 (in intron 1, minor allele frequency, MAF > 5%) is in complete LD (r2 = 1) with rs2038027 (in intron 3, MAF < 5%); furthermore, we also identified two additional functional SNPs: rs3218211 (in the 3′UTR, miRNA bing site) and rs3218123 (TFBS) in the E2F2 gene. After we limited the SNPs to those with minor allele frequency ≥5% in the Hapmap CEU population, seven SNPs were selected for genotyping: rs6667575 (E2F2 −1808 G >A), rs3218121 (E2F2 −604 C >T), rs2742976 (E2F2 −289 G >T), rs3218123 (E2F2 −143 G >T), rs3218148 (E2F2 intron 1 C >T) and rs3218203 (E2F2 intron 6 G >C), and rs3218211 (E2F2 c.*578 C >T). Of these, E2F2 rs2742976, rs3218121, rs3218123, and rs6667575 may be at TFBS, according to bioinformatics analysis (Table 1).
Genotyping of E2F1 and E2F2 Polymorphisms
The DNA was extracted from peripheral blood leukocytes by the DNA Blood Mini Kit (Quagen, Valencia, CA), according to the protocol of the manufacturer. The quantification of DNA was determined by a Nanodrop analyzer (ND-1000 spectrophotometer [Nano Drop Technologies, Inc., Wilmington, DE]). All SNPs were genotyped using the TaqMan assay. The PCR amplification was run and the plate was read with the built-in Sequence Detection Software on an ABI-Prism 7900 instrument (Applied Biosystems, Foster City, CA). Each plate included four negative controls, duplicated positive controls and eight repeated samples. The conditions of amplification were as follows: 50°C for 2 min, 95°C for 10 min followed by 40 cycles of 95°C for 15 sec, and 60°C for 1 min.
Statistical Analysis
The differences in the distribution of categorical variables, including demographic characteristics, smoking and drinking status and E2F1and E2F2 genotype/allele frequencies between cases and controls, were tested by Chi-square or Fisher’s exact test (when an expected cell count was less than 5). The deviation from Hardy-Weinberg equilibrium in genotype distribution among controls was tested by Chi-square goodness-of-fit test, and the LD coefficient r2 for each of the SNP pairs was also examined. The crude and adjusted odds ratios (ORs) with the 95% confidence intervals (CI) were estimated by unconditional univariate and multivariate logistic regression analyses to assess the association between E2F1 and E2F2 variant genotypes and risk of SCCHN, combined effects of the ten variants, which were further stratified by the primary tumor site, age, sex, smoking, drinking status, and family history of cancer in first-degree relatives, and which were evaluated by the P value in multivariate logistic regression models with adjustment for age, sex, smoking, drinking status, and family history of cancer in first-degree relatives. The P values of trend test was also estimated by unconditional logistic regression analyses, which were carried out to examine the homogeneity and trend of ORs, after controlling for possible confounding of age, sex, smoking status, alcohol use, and family history of cancer first-degree relatives. To adjust multiple tests, we performed permutations by simulating an additional 1000 same logistic models for each SNP and combined ten SNPs by randomly permuting case-control status of all subjects to generate a null distribution with correction for the overfitting caused by selection of a model including ten SNPs [22]. We used Kaplan-Meier curves to describe the cumulative hazard of combined genotypes for age at onset of SCCHN. Statistical significance was tested using Log rank test. All statistics tests were two-sided with a 0.05 significance level. Data were analyzed using SAS statistical software (SAS version 9.2; SAS Institute Inc., Cary, NC) and Stata software (version 8.2; StataCorp LP, College Station, TX)..
RESULTS
Characteristics of the Study Population
The frequency distributions of selected variables between SCCHN cases and controls are described in Table 2. There was no significant difference in the distributions of age between the cases and the controls (P = 0.510) with a mean age of 57.1 (±11.1 years) for the cases and 56.6 (±11.0 years) for the controls. Of all the subjects, 75.4% of the cases and 76.3 % of the controls were men (P = 0.598). The cases were more likely to be current smokers (37.9%) and current drinkers (50.9%) or former drinkers (22.0%), compared with the controls (14.5%, 40.5% and 16.1%, respectively) (P < 0.001 for all). Approximately 60.4% of the SCCHN cases had a family history of cancer in their first-degree relatives, compared with 62.2% of the controls (P = 0.388), which may reflect the nature of controls selected from visitors to a cancer hospital.
Table 2.
Frequency Distribution of Selected Variables among SCCHN Patients and Cancer-free Controls
| Variable | Cases n (%) | Controls n (%) | P-valuea |
|---|---|---|---|
| All subjects | 1096 (100) | 1090 (100) | |
| Age (mean ± standard deviation in years) | 57.14±11.14 | 56.61±11.01 | 0.258 |
| Age (years) | |||
| ≤ 57 | 585 (53.4) | 572 (52.5) | 0.674 |
| > 57 | 511 (46.6) | 518 (47.5) | |
| Sex | |||
| Male | 826 (75.4) | 832 (76.3) | 0.598 |
| Female | 270 (24.6) | 258 (23.7) | |
| Smoking status | |||
| Current | 415 (37.9) | 158 (14.5) | <0.001 |
| Former | 377 (34.4) | 397 (36.4) | |
| Never | 304 (27.7) | 535 (49.1) | |
| Drinking status | |||
| Current | 558 (50.9) | 441 (40.5) | <0.001 |
| Former | 241 (22.0) | 176 (16.1) | |
| Never | 297 (27.1) | 473 (43.4) | |
| Family history of cancer in first-degree relatives | 0.388 | ||
| Yes | 662 (60.4) | 678 (62.2) | |
| No | 434 (39.6) | 412 (37.8) | |
| Tumor site | |||
| Oropharynx | 557 (50.8) | ||
| Non-oropharynx b | 539 (49.2) | ||
The differences between cases and controls were evaluated by two-sided χ2 – test for discontinuous variables and Student’s t-tests for continuous variables.
Included patients with oral cavity (n = 323), patients with laryngeal cancer (n = 172) and patients with hypopharyngeal cancer (n = 44).
E2F1 and E2F2 Genotypes Distribution between Cases and Controls
The frequency distribution of genotypes and alleles of all SNPs are showed in Table 3. The observed genotype frequencies among the controls were all consistent with Hardy-Weinberg equilibrium (all P > 0.05). Although there was no altered risk associated with any of the selected SNPs alone, the frequencies of rs2742976 T, rs6667575 A, and rs3218123 T alleles (0.378, 0.281, and 0.134, respectively) among the cases were slightly higher than those among the controls (0.371, 0.274, and 0.133, respectively), whereas the frequencies of rs3213182 C, rs3213183 A, rs3213180 C, rs3218121 A, rs3218203 G, rs3218148 G, and rs3218211 T alleles (0.102, 0.293, 0.090, 0.760, 0.186, 0.448, and 0.472, respectively) among the cases were slightly lower than those among the controls (0.106, 0.301, 0.100, 0.770, 0.188, 0.449, and 0.485, respectively). As a result, we assumed in further combined analyses that rs2742976 T, rs6667575 A, rs3218123 T, rs3213182 C, rs3213183 A, rs3213180 C, rs3218121 A, rs3218203 G, rs3218148 G, and rs3218211 T alleles may be the putative risk alleles. To further adjust for multiple tests, we performed the permutations but did not find any directional change in the estimates for each of the SNPs (Supplement Table 1).
Table 3.
E2F1 and E2F2 Genotype and Allele Frequencies and their Association with Risk of SCCHN
| Genotypes | Cases, n (%) | Controls, n (%)b | Adjusted OR (95% CI)c | Ptrend-testc |
|---|---|---|---|---|
| E2F1 rs3213182 | ||||
| AAa | 880 (80.3) | 867 (79.6) | 1.00 (reference) | |
| AC | 208 (19.0) | 214 (19.6) | 0.96 (0.77–1.20) | |
| CC | 8 ( 0.7) | 9 ( 0.8) | 0.79 (0.29–2.18) | 0.624 |
| AC/CC | 216 (19.7) | 223 (20.5) | 0.95 (0.77–1.19) | |
| C allele | 224 (10.2) | 232(10.6) | ||
| E2F1 rs3213183 | ||||
| GGa | 552 (50.4) | 533 (48.9) | 1.00 (reference) | |
| AG | 445 (40.6) | 457 (41.9) | 0.95 (0.79–1.14) | |
| AA | 99 (9.0) | 100 (9.2) | 0.93 (0.68–1.27) | 0.522 |
| AG/AA | 544 (49.6) | 557 (51.1) | 0.95 (0.79–1.13) | |
| A allele | 643 (29.3) | 657 (30.1) | ||
| E2F1 rs3213180 | ||||
| GGa | 905 (82.6) | 873 (80.1) | 1.00 (reference) | |
| GC | 180 (16.4) | 211 (19.4) | 0.84 (0.67–1.06) | |
| CC | 11 ( 1.0) | 6 ( 0.5) | 1.63 (0.57–4.62) | 0.311 |
| GC/CC | 191 (17.4) | 217 (19.9) | 0.86 (0.69–1.08) | |
| C allele | 202 (0.09) | 223 (0.10) | ||
| E2F2 rs3218121 | ||||
| GGa | 933(85.1) | 926(84.9) | 1.00 ( reference ) | |
| GA | 158(14.4) | 160(14.7) | 0.95 (0.74–1.22) | 0.776 |
| AA | 5 (0.5) | 4 (0.4) | 1.18 (0.28–4.90) | |
| GA/AA | 163 (14.9) | 164 (15.1) | 0.96 (0.75–1.23) | |
| A allele | 168 (7.6) | 168 (7.7) | ||
| E2F2 rs2742976 | ||||
| GG | 413 (37.7) | 431 (39.5) | 1.00 (reference) | |
| GT | 538 (49.1) | 510 (46.8) | 1.06 (0.87–1.28) | |
| TT | 145 (13.2) | 149 (13.7) | 1.03 (0.78–1.36) | 0.721 |
| GT/TTa | 683 (62.3) | 659 (60.5) | 1.05 (0.88–1.26) | |
| T allele | 828 (37.8) | 808 (37.1) | ||
| E2F2 rs6667575 | ||||
| GG | 573 (52.3) | 579 (53.1) | 1.00 (reference) | |
| GA | 431 (39.3) | 424 (38.9) | 1.03 (0.85–1.24) | |
| AA | 92 (8.4) | 87 (8.0) | 1.13 (0.81–1.57) | 0.514 |
| GA/AAa | 523 (47.7) | 511 (46.9) | 1.04 (0.88–1.25) | |
| A allele | 615 (28.1) | 598 (27.4) | ||
| E2F2 rs3218203 | ||||
| CCa | 731 (66.7) | 719 (66.0) | 1.00 (reference) | |
| CG | 323 (29.5) | 333 (30.5) | 0.94 (0.77–1.14) | 0.855 |
| GG | 42 ( 3.8) | 38 ( 3.5) | 1.14 (0.71–1.81) | |
| CG/GG | 365 (33.3) | 371 (34.0) | 0.96 (0.80–1.15) | |
| G allele | 407 (18.6) | 409 (18.8) | ||
| E2F2 rs3218148 | ||||
| AAa | 347 (31.6) | 326 (29.9) | 1.00 (reference) | |
| AG | 517 (47.2) | 550 (50.5) | 0.87 (0.71–1.06) | 0.850 |
| GG | 232 (21.2) | 214 (19.6) | 1.01 (0.78–1.29) | |
| AG/GG | 749 (68.3) | 764 (70.1) | 0.91 (0.75–1.10) | |
| G allele | 981 (44.8) | 978 (44.9) | ||
| E2F2 rs3218211 | ||||
| CCa | 314 (28.7) | 286 (26.2) | 1.00 (reference) | |
| CT | 530 (48.3) | 551 (50.6) | 0.86(0.70–1.06) | 0.382 |
| TT | 252 (23.0) | 253 (23.2) | 0.90 (0.70–1.16) | |
| CT/TT | 782 (71.4) | 805 (73.8) | 0.87 (0.72–1.06) | |
| T allele | 1034 (47.2) | 1056 (48.5) | ||
| E2F2 rs3218123 | ||||
| GG | 828 (75.5) | 831 (76.2) | 1.00 (reference) | |
| GT | 243 (22.2) | 228 (20.9) | 1.00 (0.81–1.24) | |
| TT | 25 ( 2.3) | 31 (2.9) | 0.83 (0.47–1.42) | 0.724 |
| GT/TTa | 268 (24.5) | 259 (23.8) | 0.98 (0.80–1.21) | |
| T allele | 293 (13.4) | 290 (13.3) | ||
Assumed risk genotypes.
The observed genotype frequency among the control subjects were in agreement with Hardy-Weinberg equilibrium (p2 +2pq + q2 =1) (P = 0.541 for E2F1 rs3213180, P = 0.257 for E2F1 rs3213182, P = 0.494 for E2F1 rs3213183, P = 0.539 for E2F2 rs3218121, P = 0.144 for E2F2 rs2742976, P = 0.127 for E2F2 rs3218148, P = 0.400 for E2F2 rs3218203, P = 0.392 for E2F2 rs6667575, P = 0.328 for E2F2 rsrs3218211, P = 0.120 for E2F2 rs3218123)
Adjusted OR (95% CI) for age, sex, smoking status, alcohol use, and family history of cancer first-degree relatives in logistic regression models
Association and Stratification Analysis of Combined Effects of E2F1 and E2F2 Genotypes and SCCHN Risk
Considering possible combined effects from different variants or genotypes and potential locus-locus interactions of E2F1 and E2F2 SNPs on risk of SCCHN, we combined three E2F1 SNPs and seven E2F2 SNPs as the number of the putative risk genotypes (i.e., more frequent in the cases than in the controls: rs3213182 AA, rs3213183 GG, rs3213180 GG, rs321318121 GG, rs2742976 GT+TT, rs6667575 GA+AA, rs3218203 CC, rs3218148 AA, rs3218211 CC, and rs3218123 GT+TT). As shown in Table 4, when we divided the combined genotypes into three groups by the number of risk genotypes (i.e., 0–4 risk genotypes, 5–8 risk genotypes, and 9–10 risk genotypes), we found that a difference in the distribution of the combined variant genotypes between the cases and controls was statistically significant (P = 0.023) and that this difference was significantly associated with SCCHN risk in a dose-response manner (compared with those with 0–4 risk genotypes, adjusted OR = 1.04, 95% CI = 0.86–1.26 for those with 5–8 risk genotypes and adjusted OR = 1.62, 95% CI = 1.14–2.30 for those with 9–10 risk genotypes; P for trend = 0.045). We also observed the combined genotypes associated with risk of SCCHN in those carrying 9–10 risk genotypes, compared with those carrying 0–4 risk genotypes in subgroups of the stratified analysis; specifically, the combined effects were more pronounced among younger adults (adjusted OR = 1.74; 95% CI = 1.07–2.85), men (adjusted OR = 1.48; 95% CI = 1.00–2.20), never smoker (adjusted OR = 1.85; 95% CI = 1.07–3.17), never drinker (adjusted OR = 2.19; 95% CI = 1.22–3.95), individuals with family history of cancer first-degree relatives (adjusted OR = 1.64; 95% CI = 1.05–2.57), and oropharyngeal cancer (adjusted OR = 1.76; 95% CI = 1.17–2.64). To correct for potential overfitting caused by the combined ten SNPs in a logistic regression model, we randomly permuted case-control status of all subjects 1000 times, and we observed similar associations of the combined ten SNPs with SCCHN risk in the permuted (null) data similar to the original data (Supplement Table 2).
Table 4.
Association and Stratification Analysis of the Combined Genotypes of E2F1 and E2F2 Polymorphisms and the Risk of Squamous Cell Carcinoma of the Head and Neck
| Variables | n, case/control | Number of combined risk genotypes of of E2F1 and E2F2 Polymorphismc
|
P trendd | ||||
|---|---|---|---|---|---|---|---|
| %(case/control)
|
Adjusted OR (95% CI)d
|
Adjusted OR (95% CI)d
|
|||||
| 0–4 | 5–8 | 9–10 | 0–4 vs. 5–8 | 0–4 vs. 9–10 | |||
| Totala | 1096/1090 | 31.0/33.5 | 59.5/60.1 | 9.5/6.4 | 1.04 (0.86–1.26) | 1.62 (1.14–2.30) | 0.045 |
| Tumor site | |||||||
| Oropharynx | 557/1090 | 28.6/33.5 | 61.2/60.1 | 10.2/6.4 | 1.14 (0.90–1.44) | 1.76 (1.17–2.64) | 0.016 |
| Non-oropharynxb | 539/1090 | 33.6/33.5 | 57.7/60.1 | 8.7/6.4 | 0.92 (0.72–1.18) | 1.43 (0.90–2.26) | 0.556 |
| Age | |||||||
| ≤ median (57 years) | 585/572 | 30.1/32.7 | 60.7/61.4 | 9.2/5.9 | 1.07 (0.82–1.39) | 1.74 (1.07–2.85) | 0.083 |
| > median (57 years) | 511/518 | 32.1/34.4 | 58.1/58.7 | 9.8/7.0 | 1.03 (0.78–1.35) | 1.47 (0.89–2.42) | 0.261 |
| Sex | |||||||
| Male | 826/832 | 31.2/32.6 | 59.1/60.7 | 9.7/6.7 | 1.00 (0.80–1.24) | 1.48 (1.00–2.20) | 0.191 |
| Female | 270/258 | 30.4/36.4 | 60.7/58.1 | 8.9/5.4 | 1.21 (0.82–1.79) | 2.09 (0.98–4.48) | 0.074 |
| Smoking status | |||||||
| Never | 304/535 | 30.9/33.3 | 58.2/60.4 | 10.9/6.4 | 1.03 (0.76–1.41) | 1.85 (1.07–3.17) | 0.109 |
| Ever | 792/555 | 31.1/33.7 | 60.0/59.8 | 9.0/6.5 | 1.04 (0.82–1.33) | 1.41 (0.90–2.21) | 0.238 |
| Drinking status | |||||||
| Never | 297/473 | 31.7/36.2 | 58.3/58.4 | 10.1/5.5 | 1.16 (0.84–1.59) | 2.19 (1.22–3.95) | 0.027 |
| Ever | 799/617 | 30.8/31.4 | 60.0/61.4 | 9.3/7.1 | 0.99 (0.78–1.25) | 1.32 (0.86–2.04) | 0.414 |
| Family history of cancer in first-degree relatives | |||||||
| Yes | 662/678 | 30.2/32.2 | 59.8/61.7 | 10.0/6.2 | 1.02 (0.80–1.30) | 1.64 (1.05–2.57) | 0.125 |
| No | 434/412 | 32.3/35.7 | 59.0/57.5 | 8.8/6.8 | 1.11 (0.82–1.50) | 1.53 (0.87–2.66) | 0.173 |
The distribution of three groups of the combined genotypes between the cases and controls for total samples was statistically significant (P = 0.023).
included oral cavity, larynx and hypopharynx.
The number represents the numbers of risk genotypes: the risk genotypes used for the calculation were rs3213182 AA, rs3213183 GG, rs3213180 GG, rs3218121 GG, rs2742976 GT+TT, rs6667575 GA+AA, rs3218203 CC, rs3218148 AA, rs3218211 CC, rs3218123 GT+TT genotypes.
Adjusted OR (95% CI) for age, sex, smoking status, alcohol use, family history of cancer first-degree relatives in logistic regression models.
Age at SCCHN Onset
In the case-only analysis of 1096 patients, the median age at SCCHN onset was 57 years for both those with 0–4 risk genotypes (a range of 22–89 years) and those with 5–10 risk genotypes (a range of 18–85 years). No significant difference in age at onset was observed for subgroups of sex, smokers, and drinkers, and tumor sites (data not shown). However, in never smoking patients, those with the combined 5–10 risk genotypes had a younger age at onset (a median age of 52 years) than those with the 0–4 risk genotypes (a median age of 54 years) (Log rank, P = 0.038) (Figure 2A). In never drinking patients, those with the combined 5–10 risk genotypes also had a younger age at onset (a median age 55 years) than those with the 0–4 risk genotypes (a median age of 56 years) (Log rank, P = 0.045) (Figure 2B). In both never smoking and never drinking patients, those with the combined 5–10 risk genotypes had a younger age at onset (a median age of 53 years) than those with the 0–4 risk genotypes (a median age 56 years) (Log rank, P = 0.034) (Figure 2C).
Figure 2.
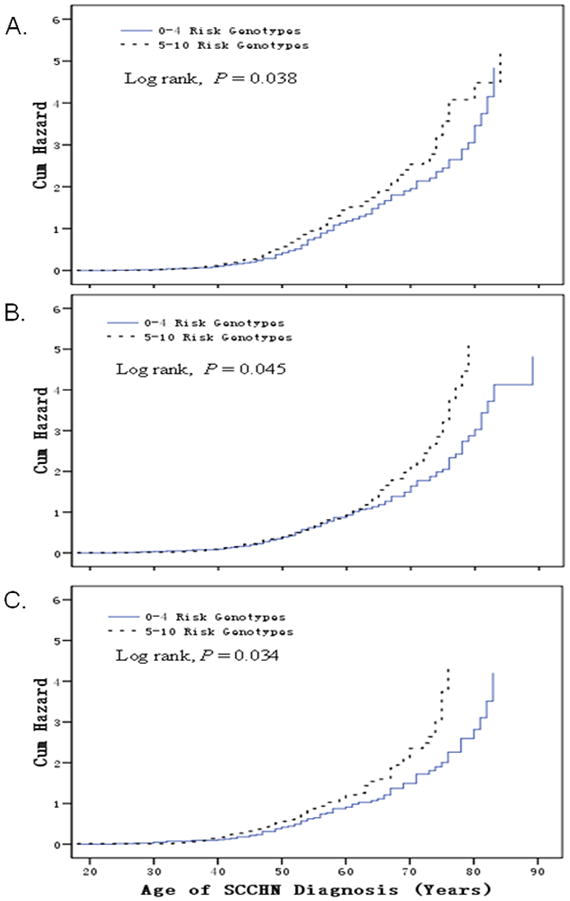
Kaplan-Meier curves: cumulative risk of combined genotypes of rs3213182 AA, rs3213183 GG, rs3213180 GG, rs3218121 GG, rs2742976 GT+TT, rs6667575 GA+AA, rs3218203 CC, rs3218148 AA, rs3218211 CC, rs3218123 GT+TT for age at onset of SCCHN. Patients with 5–10 risk genotypes were associated with earlier onset of SCCHN, compared with those with 1–4 risk genotypes in never smokers (median age 52 yr vs. 54 yr, Long rank, P = 0.038, Figure 2A), in never drinkers (median age 55 yr vs. 56 yr, Long rank, P = 0.045, Figure 2B), and in never smokers and never drinkers (median age 53 yr vs. 56 yr, Long rank, P = 0.034, Figure 2C).
DISCUSSION
We investigated the associations of ten selected SNPs of E2F1 and E2F2 with risk of SCCHN in non-Hispanic whites in a hospital-based case-control study. We found that none of the ten SNPs in E2F1 and E2F2 alone had a significant main effect on risk of SCCHN in our study population. However, our results suggested a combined effect of the ten SNPs on risk of SCCHN in a genotype-dose response manner. Specifically, the combined effect was more pronounced among subgroups of men, non-smokers, non-drinkers, individuals with an early age at onset or with a family history of cancer in first-degree relatives, and patients with oropharyngeal cancer. Furthermore, we observed that those with 5–10 risk genotypes had an earlier age at SCCHN onset than those with the 0–4 risk genotypes in never smoking and/or drinking patients. Given the role of E2F1 and E2F2 genes in activating gene expression and influencing the p53 activity, it is biologically plausible that the E2F1 and E2F2 SNPs may modulate risk of SCCHN.
E2F1 and E2F2 as key transcription factors can interact with other genes in both the Rb-E2Fs pathway and other biological pathways that are involved in proliferation, cell cycle progression, and apoptosis [12,15,23]. For example, it has been shown that the retinoblastoma protein can restrict cells from entering the S phase by binding to and sequestering of the E2F activators in the model of mammalian cell cycle control [24]. Our finding of no main effect of each of E2F1 or E2F2 variants or variant genotypes on risk of SCCHN is consistent with previously published studies on other cancers. One case-control study showed that a SNP rs760607 (E2F2 −5368 A>G) in the promoter region of E2F2 had no main effect on risk of breast cancer [20]. However, conflicting results do exist for other cancers. For example, one study found that E2F2 rs760607 SNP had no association with ovarian cancer risk in the replication set, though an association with ovarian cancer risk was reported for the E2F2 rs760607 in a dominant model and for the E2F2 rs3218203 SNP in a recessive model in the discovery set [19]. However, no reported studies have investigated the association of E2F1 and E2F2 SNPs with SCCHN risk, nor any published studies have evaluated the combined effects of E2F1 and E2F2 variant genotypes on cancer risk. By analyzing ten SNPs simultaneously in our study, we found that the number of putative risk genotypes was associated with risk of SCCHN in genotype dose-response manner.
It is well known that genetic susceptibility contributes to the risk of developing cancer, which is also supported by some evidence in the present study, such as an early age onset, elevated risk in subgroups without apparent exposure to tobacco or alcohol, and among individuals with a family history of cancer in first-degree relatives, all indicating an important role of genetic background in the study population. Several selected SNPs (i.e. rs2742976 (E2F2 −289 G >T), rs6667575 (E2F2 −1808 G >A), and rs3218123 (E2F2 −143 G >T) are located in the E2F2 promoter region. Computer prediction analysis has shown that the −1808, −289 and −143 regions in the 5′ upstream gene sequence in E2F2 contain putative transcription factor binding sites for GATA-1,2, STATx, SRY, and Elk-1 (Table 1). It has been proposed that polymorphisms in the promoter region could destroy transcriptional regulation factors, leading to altered transcription [25]. Micro-RNAs (miRNAs) can also regulate the E2F activity, and miRNAs dysregulation has been implicated in malignancy. One study suggested that the E2F family proteins can target miRNAs by a negative feedback loop [23]. For instance, E2F1, E2F2 and E2F3 directly bind to the promoter of the miR-17~92 cluster activating its transcription, in turn, which negatively modulate translation of E2F1–3 mRNAs by binding sites in their 3′-untranslated regions [26,27]. Likewise, miR-330-3p exhibits its tumor suppressive activity through E2F1-mediated repression of Akt activation [28]. Both E2F1 rs3213180 and E2F2 rs3218211 are located in the 3′UTR and have miRNA binding sites as predicted, and E2F1 rs3213182 and rs3213183 are located in the 3′ near gene. Although we did not observe any their main effects on SCCHN risk, these variants could contribute in part to the observed joint effects on SCCHN risk. It is also likely that other ‘non-functional’ variants, such as tagSNPs in introns, may be markers for untyped functional SNPs and may be in LD with functional or disease causing variants in the genome [29].
There has been a trend for an increased risk in the number of young patients who developed SCCHN without recognized risk factors (i.e., tobacco and alcohol use) [30,31]. It has been reported that never-smoker patients with SCCHN tended to be younger than smokers with SCCHN [32], In the present study, we found that the combined risk genotypes of the ten SNPs were associated with an increased risk of SCCHN among younger adults, men, never smokers or never drinkers in our study population. Furthermore, we found that in 1096 SCCHN patients, those with the combined 5–10 risk genotypes had 1 years earlier at SCCHN onset than those with 0–4 risk genotypes in never drinkers, 2 year earlier in never smokers and 3 years earlier in never smokers and never drinkers. These indicated that SCCHN risk in never smoking and never drinking individuals was likely attributed to genetic factors compared with that in either never smokers or never drinkers, although at least 75% of SCCHN are attributable to a combination of cigarette smoking and alcohol drinking [33]. Therefore, our findings suggested that E2F1 and E2F2 genetic variants may play important roles in head and neck carcinogenesis.
Human papillomavirus (HPV) infection is a recognized risk factor for SCCHN [5]. A number of studies indicated that HPV-related SCCHN occurs predominantly at the oropharynx [34]. HPV16 seropositivity was associated with a 30-fold increased risk of oropharyngeal cancer among light drinkers or never smokers [35]. It has been found that HPV type 16 E7 can bind to E2F1 and activate E2F1-driven transcription through a retinoblastoma protein-independent manner [36]. Longworth et al. found that activating E2F2 transcription through its interaction with histone deacetylases (HDACs) can facilitate HPV31 E7 replication [37]. Indeed, we observed a positive association with risk of oropharyngeal cancer, which supports the likelihood of HPV infection-related SCCHN [34,38], and thus genetic variants in E2F1 and E2F2 may have modulated HPV-induced carcinogenesis in young individuals.
The strengths of our study included relatively large study sample size, our ethnically homogeneous study population, and unbiased genotyping data distributions in the control subjects as suggested by their agreement with Hardy-Weinberg equilibrium. However, there are still some limitations in our study. Firstly, small sample size in subgroup analyses may have attenuated statistical power, and therefore positive findings in stratification analysis need further confirmation in larger studies. Secondly, our study was hospital-based, which likely had recall bias and selection bias. However, the genotype frequencies of eight variants in our controls are similar to those for European populations published in the Hapmap website (http://hapmap.ncbi.nlm.nih.gov/cgi-perl/gbrowse/hapmap24_B36/) (data not shown), though the frequencies of rs3213180, rs3218123 and rs3218211 genotypes have not been reported for European populations in the Hapmap website. For example, the frequencies of the GG, GT, and TT genotypes of the rs2742976 among our 1090 Caucasians control subjects were 39.5%, 46.8%, and 13.7%, respectively, compared with those of Hapmap-CEU: 37.8%, 47.7%, and 14.4%, respectively. Hence, selection bias in our study, if any, was unlikely to be substantial.
In summary, our results suggested that any one of the E2F1 or E2F2 variants alone may not have a substantial effect on SCCHN risk, but a joint effect of the combined risk genotypes (i.e., rs3213182 AA, rs3213183 GG, rs3213180 GG, rs3218121 GG, rs2742976 GT+TT, rs6667575 GA+AA, rs3218203 CC, rs3218148 AA, rs3218211 CC, rs3218123 GT+TT) may contribute to risk of SCCHN, and this increased risk was more pronounced among the younger adults (≤ 57 years old), men, never smokers, never drinkers, individuals with a family history of cancer in the first-degree relatives, and patients with oropharyngeal cancer. In addition, those with the combined 5–10 risk genotypes had an earlier age at SCCHN onset than those with the 0–4 risk genotypes, particularly in never smoking and/or drinking patients. These results indicated the important role of the E2F1 or E2F2 variants in head and neck carcinogenesis, but our findings need to be validated by larger studies.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Health grants R01 CA131274 and R01 ES011740 (Q. Wei), ES015587 (D.G. Johnson), P50 CA097007 (Scott Lippman), and P30 CA016672 (The University of Texas M. D. Anderson Cancer Center). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. We thank Jiachun Lu for statistical assistance, Margaret Lung and Jessica Fiske for their assistance in recruiting the subjects and collecting the questionnaire information and Jianzhong He, Kejing Xu, and Min Zhao and for their assistance in blood processing and DNA extraction.
Abbreviations
- CI
confidence interval
- E2F1
E2F transcription factor 1
- E2F2
E2F transcription factor 2
- LD
linkage disequilibrium
- MAF
minor allele frequency
- OR
odds ratio
- PCR
polymerase chain reaction
- SCCHN
Squamous cell carcinoma of the head and neck
- TFBS
transcription factor binding site
- miRNA
Micro ribonucleic acid
- 5′ UTR
5′ untranslated regions
- 3′UTR
3′ untranslated regions
Footnotes
COMPETING INTERESTS
The authors declare that they have no competing interests.
References
- 1.Siegel R, Ward E, Brawley O, Jemal A Cancer statistics. The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA: a cancer journal for clinicians. 2011;61(4):212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Purdue MP, Hashibe M, Berthiller J, et al. Type of alcoholic beverage and risk of head and neck cancer--a pooled analysis within the INHANCE Consortium. Am J Epidemiol. 2009;169(2):132–142. doi: 10.1093/aje/kwn306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee YC, Boffetta P, Sturgis EM, et al. Involuntary smoking and head and neck cancer risk: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol Biomarkers Prev. 2008;17(8):1974–1981. doi: 10.1158/1055-9965.EPI-08-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hashibe M, Brennan P, Chuang SC, et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol Biomarkers Prev. 2009;18(2):541–550. doi: 10.1158/1055-9965.EPI-08-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Petrini M, Ritta M, Schena M, et al. Head and neck squamous cell carcinoma: role of the human papillomavirus in tumour progression. The new microbiologica. 2006;29(1):25–33. [PubMed] [Google Scholar]
- 6.Sturgis EM, Zheng R, Li L, et al. XPD/ERCC2 polymorphisms and risk of head and neck cancer: a case-control analysis. Carcinogenesis. 2000;21(12):2219–2223. doi: 10.1093/carcin/21.12.2219. [DOI] [PubMed] [Google Scholar]
- 7.Li G, Sturgis EM, Wang LE, et al. Association of a p73 exon 2 G4C14-to-A4T14 polymorphism with risk of squamous cell carcinoma of the head and neck. Carcinogenesis. 2004;25(10):1911–1916. doi: 10.1093/carcin/bgh197. [DOI] [PubMed] [Google Scholar]
- 8.Chen K, Zhao H, Hu Z, et al. CASP3 polymorphisms and risk of squamous cell carcinoma of the head and neck. Clin Cancer Res. 2008;14(19):6343–6349. doi: 10.1158/1078-0432.CCR-08-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niu J, Huang YJ, Wang LE, Sturgis EM, Wei Q. Genetic polymorphisms in the PTPN13 gene and risk of squamous cell carcinoma of head and neck. Carcinogenesis. 2009;30(12):2053–2058. doi: 10.1093/carcin/bgp265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang YJ, Niu J, Liu Z, Wang LE, Sturgis EM, Wei Q. The functional IGFBP7 promoter −418G>A polymorphism and risk of head and neck cancer. Mutat Res. 702(1):32–39. doi: 10.1016/j.mrgentox.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li G, Liu Z, Sturgis EM, Chamberlain RM, Spitz MR, Wei Q. CYP2E1 G1532C, NQO1 Pro187Ser, and CYP1B1 Val432Leu polymorphisms are not associated with risk of squamous cell carcinoma of the head and neck. Cancer Epidemiol Biomarkers Prev. 2005;14(4):1034–1036. doi: 10.1158/1055-9965.EPI-04-0814. [DOI] [PubMed] [Google Scholar]
- 12.Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer. 2009;9(10):738–748. doi: 10.1038/nrc2718. [DOI] [PubMed] [Google Scholar]
- 13.Polager S, Ginsberg D. E2F - at the crossroads of life and death. Trends Cell Biol. 2008;18(11):528–535. doi: 10.1016/j.tcb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Whibley C, Pharoah PD, Hollstein M. p53 polymorphisms: cancer implications. Nat Rev Cancer. 2009;9(2):95–107. doi: 10.1038/nrc2584. [DOI] [PubMed] [Google Scholar]
- 15.DeGregori J, Johnson DG. Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis. Curr Mol Med. 2006;6(7):739–748. doi: 10.2174/1566524010606070739. [DOI] [PubMed] [Google Scholar]
- 16.Attwooll C, Lazzerini Denchi E, Helin K. The E2F family: specific functions and overlapping interests. EMBO J. 2004;23(24):4709–4716. doi: 10.1038/sj.emboj.7600481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen HZ, Tsai SY, Leone G. Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat Rev Cancer. 2009;9(11):785–797. doi: 10.1038/nrc2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pusapati RV, Weaks RL, Rounbehler RJ, McArthur MJ, Johnson DG. E2F2 suppresses Myc-induced proliferation and tumorigenesis. Mol Carcinog. 49(2):152–156. doi: 10.1002/mc.20584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cunningham JM, Vierkant RA, Sellers TA, et al. Cell cycle genes and ovarian cancer susceptibility: a tagSNP analysis. Br J Cancer. 2009;101(8):1461–1468. doi: 10.1038/sj.bjc.6605284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Justenhoven C, Pierl CB, Haas S, et al. Polymorphic loci of E2F2, CCND1 and CCND3 are associated with HER2 status of breast tumors. International journal of cancer. 2009;124(9):2077–2081. doi: 10.1002/ijc.24198. [DOI] [PubMed] [Google Scholar]
- 21.Wang LE, Hu Z, Sturgis EM, et al. Reduced DNA repair capacity for removing tobacco carcinogen-induced DNA adducts contributes to risk of head and neck cancer but not tumor characteristics. Clin Cancer Res. 16(2):764–774. doi: 10.1158/1078-0432.CCR-09-2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Rowland C, Xiromerisiou G, et al. Neither replication nor simulation supports a role for the axon guidance pathway in the genetics of Parkinson’s disease. PLoS One. 2008;3(7):e2707. doi: 10.1371/journal.pone.0002707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Emmrich S, Putzer BM. Checks and balances: E2F-microRNA crosstalk in cancer control. Cell cycle (Georgetown, Tex) 9(13):2555–2567. doi: 10.4161/cc.9.13.12061. [DOI] [PubMed] [Google Scholar]
- 24.Chong JL, Wenzel PL, Saenz-Robles MT, et al. E2f1–3 switch from activators in progenitor cells to repressors in differentiating cells. Nature. 2009;462(7275):930–934. doi: 10.1038/nature08677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun T, Gao Y, Tan W, et al. A six-nucleotide insertion-deletion polymorphism in the CASP8 promoter is associated with susceptibility to multiple cancers. Nature genetics. 2007;39(5):605–613. doi: 10.1038/ng2030. [DOI] [PubMed] [Google Scholar]
- 26.Woods K, Thomson JM, Hammond SM. Direct regulation of an oncogenic micro-RNA cluster by E2F transcription factors. The Journal of biological chemistry. 2007;282(4):2130–2134. doi: 10.1074/jbc.C600252200. [DOI] [PubMed] [Google Scholar]
- 27.Sylvestre Y, De Guire V, Querido E, et al. An E2F/miR-20a autoregulatory feedback loop. The Journal of biological chemistry. 2007;282(4):2135–2143. doi: 10.1074/jbc.M608939200. [DOI] [PubMed] [Google Scholar]
- 28.Chaussepied M, Ginsberg D. Transcriptional regulation of AKT activation by E2F. Molecular cell. 2004;16(5):831–837. doi: 10.1016/j.molcel.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Wang M, Wang M, Yuan L, et al. A novel XPF −357A>C polymorphism predicts risk and recurrence of bladder cancer. Oncogene. 29(13):1920–1928. doi: 10.1038/onc.2009.484. [DOI] [PubMed] [Google Scholar]
- 30.Toner M, O’Regan EM. Head and neck squamous cell carcinoma in the young: a spectrum or a distinct group? Part 1 Head and neck pathology. 2009;3(3):246–248. doi: 10.1007/s12105-009-0135-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toner M, O’Regan EM. Head and neck squamous cell carcinoma in the young: a spectrum or a distinct group? Part 2 Head and neck pathology. 2009;3(3):249–251. doi: 10.1007/s12105-009-0137-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muscat JE, Richie JP, Jr, Thompson S, Wynder EL. Gender differences in smoking and risk for oral cancer. Cancer research. 1996;56(22):5192–5197. [PubMed] [Google Scholar]
- 33.Blot WJ, McLaughlin JK, Winn DM, et al. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer research. 1988;48(11):3282–3287. [PubMed] [Google Scholar]
- 34.Gillespie MB, Rubinchik S, Hoel B, Sutkowski N. Human papillomavirus and oropharyngeal cancer: what you need to know in 2009. Current treatment options in oncology. 2009;10(5–6):296–307. doi: 10.1007/s11864-009-0113-5. [DOI] [PubMed] [Google Scholar]
- 35.Applebaum KM, Furniss CS, Zeka A, et al. Lack of association of alcohol and tobacco with HPV16-associated head and neck cancer. Journal of the National Cancer Institute. 2007;99(23):1801–1810. doi: 10.1093/jnci/djm233. [DOI] [PubMed] [Google Scholar]
- 36.Hwang SG, Lee D, Kim J, Seo T, Choe J. Human papillomavirus type 16 E7 binds to E2F1 and activates E2F1-driven transcription in a retinoblastoma protein-independent manner. The Journal of biological chemistry. 2002;277(4):2923–2930. doi: 10.1074/jbc.M109113200. [DOI] [PubMed] [Google Scholar]
- 37.Longworth MS, Wilson R, Laimins LA. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. The EMBO journal. 2005;24(10):1821–1830. doi: 10.1038/sj.emboj.7600651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Begum S, Gillison ML, Nicol TL, Westra WH. Detection of human papillomavirus-16 in fine-needle aspirates to determine tumor origin in patients with metastatic squamous cell carcinoma of the head and neck. Clin Cancer Res. 2007;13(4):1186–1191. doi: 10.1158/1078-0432.CCR-06-1690. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.