

Background: Different oncogenes or a reduction in the activity of the PP2A tumor suppressor substitute for the expression of SV40 small t-antigen in cell transformation.
Results: These oncogenes and PP2A converge at p27.
Conclusion: p27 levels reflect the signaling milieu controlled by oncogenic activation and PP2A.
Significance: Understanding what p27 levels reflect in transformed cells is crucial for understanding its significance as a biomarker.
Keywords: Cancer Biology, Cell Growth, Cell Signaling, Gene Knockout, PP2A, Ingenuity Pathway Analysis, Cell Transformation, p27
Abstract
SV40 small t-antigen (ST) collaborates with SV40 large T-antigen (LT) and activated rasv12 to promote transformation in a variety of immortalized human cells. A number of oncogenes or the disruption of the general serine-threonine phosphatase protein phosphatase 2A (PP2A) can replace ST in this paradigm. However, the relationship between these oncogenes and PP2A activity is not clear. To address this, we queried the connectivity of these molecules in silico. We found that p27 was connected to each of those oncogenes that could substitute for ST. We further determined that p27 loss can substitute for the expression of ST during transformation of both rodent and human cells. Conversely, knock-in cells expressing the degradation-resistant S10A and T187A mutants of p27 were resistant to the transforming activities of ST. This suggests that p27 is an important target of the tumor-suppressive effects of PP2A and likely an important target of the multitude of cellular oncoproteins that emulate the transforming function of ST.
Introduction
The original Hahn and Weinberg (1) experiments identified a minimal suite of genetic events required for transformation of human fibroblasts and epithelial cells (2). Cell transformation describes a set of phenotypes present (to varying degrees) among cancer-derived or cancer-like cells, but absent from normal cells. These include morphologic changes, growth in low serum, ability to grow at reduced seeding densities, ability to ignore confluence signals (foci formation), the ability to ignore attachment signals (growth in soft agar), and the ability to grow as xenografts in immunocompromised mice. The key point is that cells lacking all of these phenotypes are considered untransformed, albeit they may be immortal, whereas the presence of at least one phenotype is taken as an indication of transformation. Experimentally, these phenotypes typically are observed on a continuum in which growth in soft agar and growth as a xenograft are considered completely transformed, albeit these do not always correlate (3). That said, a particular genetic configuration may indicate that a particular oncogene/tumor suppressor contributes to specific transformation-associated phenotypes, but not all of them.
Activated Ras combined with the inactivation of the Rb and p53 pathways and the expression of telomerase is sufficient to transform cells. However, complete transformation to anchorage-independent growth and growth as a xenograft in immunocompromised mice also requires the inhibition of the serine-threonine phosphatase PP2A.4 Inhibition of PP2A could be accomplished by the loss of one of the scaffolding A subunits, the loss of one of the regulatory B subunits, or the expression of the SV40 viral oncogene, small t-antigen (ST). Alternatively, expression of a diverse assortment of oncogenes including MYCT58A, activated PI3K, and activated AKT and RAC1 could also replace ST, presumably bypassing the need to down-regulate the phosphatase (4). This raised the possibility that PP2A may generally oppose signaling through these pathways (4–7), or alternatively, that PP2A and these signaling pathways may exert opposite effects on a single target.
Consistent with the latter possibility, Ingenuity Pathway analysis identified CDKN1B, the CDK inhibitor p27, as a common target of the gene products that could replace ST. p27 turnover is dependent on the phosphorylation of this protein, which can be reversed by PP2A. Furthermore, reducing p27 in human mesothelioma cells or in mouse fibroblasts conspired with the inactivation of the Rb and p53 pathways and the maintenance of long telomeres to transform these cells to anchorage-independent growth and growth in xenografts. Consequently, we propose that p27 levels represent a nexus of signaling input that cooperates with Rb inactivation to unshackle the restraints on cell proliferation leading to complete cell transformation. This might explain its value as a prognostic marker in multiple human tumors.
EXPERIMENTAL PROCEDURES
Cell Culture and Viral Infection
Primary MEFs were isolated and cultured as described (8). SV40- and DL888-infected human mesothelial cells have been described previously (9). MEFs and mesothelial cells were grown in DME medium supplemented with 10% FBS, 2 mm l-glutamine, penicillin, and streptomycin.
For suspension culture, asynchronously growing cells were trypsinized and transferred to dishes coated with 0.9% agarose as described previously (1.2 × 105 cells/ml). Cells were incubated at 37 °C.
Passage 1 primary MEFs were infected with SV40 or DL888 at a multiplicity of infection of 50 in a minimal volume of medium for 1 h at 37 °C, with rocking every 10 min. Complete medium was then added to each plate. The medium was changed 24 h after infection, and cells were passaged when they reached ∼80% confluence. Cell lines of all genotypes were established for eight passages and used in transformation assays as described below.
To knock down p27, human mesothelial cells were infected with a lentivirus expressing shRNA p27-2 (CCTGTGTACATAACTCTGTAA) or p27-3 (CCGACGATTCTTCTACTCAAA) (Open Biosystems). Infected cells were selected by treatment with G418 (Sigma) at a concentration of 450 μg/ml. Assays were performed on cells after 8–12 passages in selection medium.
Immunoblotting
To determine the amount of SV40 LT and ST (PAb419), phospho-Ser-10-p27, and phospho-Thr-187-p27, proteins were extracted in a buffer containing 100 mm HEPES-KOH, pH 7.5, 500 mm NaCl, 10 mm EDTA, 0.2% Triton X-100, 1 mm DTT, and 1 mm PMSF as described (10). Proteins were extracted in Nonidet P-40 radioimmune precipitation buffer for analysis of cyclin A2 (H-432) and p27 (C-19) as described previously (10). Following SDS-polyacrylamide gel electrophoresis of extracts (10–80 μg), proteins were transferred to PVDF membranes and blotted as described previously (10). All antibodies were purchased from Santa Cruz Biotechnology except for PAb419 (Ab-1;EMD Biosciences-Calbiochem) and phospho-Thr-187-p27 (Zymed Laboratories Inc.).
Foci Formation
The ability of SV40- or DL888-infected cells to form foci in a confluent monolayer was tested as described previously (8). Briefly, 1 × 102 cells of each cell line were mixed with 3 × 105 WT primary MEFs and plated in triplicate 60-mm dishes. Cells were fed every 2–3 days for 14 days, when the plates were stained with crystal violet, and foci were counted.
Soft Agar Growth
The ability of cells to grow in an anchorage-independent manner was measured by plating them in soft agar. The wells of a 6-well dish were coated with 2 ml of a 1:1 mixture of 2× complete MEF medium and 1.2% SeaPlaque agarose (Cambrex) (final 0.6% agarose). Asynchronously growing cultures of 8 × 104 to 1.6 × 105 uninfected primary MEFs or infected cell lines were suspended in a 1:1 mixture of 2× medium and 0.6% SeaPlaque agarose (final 0.3% agarose). This cell suspension was layered onto each of four agarose-coated wells (1.6 × 105 cells/well) and incubated at room temperature until solid (>2 h). The cultures were then incubated at 37 °C and fed weekly with 1–2 ml of 1× medium. For human mesothelial cells transduced with shRNA lentiviruses, G418 was included in each layer of agarose at a final concentration of 450 μg/ml. After 5–6 weeks of growth, images of each well were taken using a stereomicroscope and IPLab or Volocity software, and only those colonies visible to the naked eye were counted.
Tumor Allografts in Immunocompromised Mice
SV40- and DL888-infected cell lines were injected into nude mice to determine their tumorigenicity. Cells were mixed in a 1:1 ratio (v/v) with Matrigel (BD Pharmingen) and injected into the right flank of male nude mice (2 × 106 or 5 × 106 cells/animal). Tumor growth was monitored for 20 weeks or until the animal had to be euthanized when tumor size exceeded 100 cm3.
Analysis of Effects of ST and PP2A on p27 Phosphorylation
200 ng of His-tagged human p27 was phosphorylated by incubation with 200 ng of purified cyclin B1-CDK1 complex as described (11) and [γ-32P]-ATP at 30 °C for 30 min. After boiling for 5 min to inactivate the kinase, precipitated material was removed by centrifugation for 10 min at 13,000 rpm. The supernatant containing phospho-p27 was transferred in aliquots to fresh tubes, and increasing amounts of purified PP2A complexes (Upstate Biotech Millipore) were added. After incubation at 30 °C for 45 min, reactions were stopped by the addition of SDS sample buffer, and the samples were separated by SDS-PAGE. Dried gels were analyzed by autoradiography. Alternatively, the experiment was performed in the presence of cold ATP, and p27 phosphorylation status was determined by immunoblotting with phospho-specific antibodies.
RESULTS
CDKN1B Is a Common Target for Oncogenes That Replace ST Function in Cell Transformation Assays
ST has two genetically defined activities required for transformation. First, ST can displace the B subunit from the PP2A complex and inhibit its activity (12–14). Accordingly, mutants of ST that cannot bind PP2A are unable to contribute to anchorage-independent growth (15–18), and chemical or genetic inhibition of PP2A can partially overcome the absence of ST in transformation assays (19). Moreover, PP2A can dephosphorylate and inactivate several oncogenes, and co-expression of constitutively active AKT1 and RAC1 together, or RALA, can also replace ST in human cell transformation (7, 20–22). ST can also affect the transcription of a number of genes, including cyclin A2 (23, 24), and expression of a stabilized Myc protein, MycT58A, can also replace ST.
To first investigate whether these oncogenes might act through a single target that is regulated in a phosphorylation-dependent manner, we performed in silico experiments using the bioinformatics software Ingenuity Pathway Analysis. This enabled us to define a network of proteins anchored by the four oncoproteins that can substitute for ST (Myc, RAC1, AKT1, and RalA) and by cyclin A2 (CCNA2), a representative of the transcriptional program controlled by ST (23, 24). We then expanded this network in an unbiased manner by unsupervised additions of 20 directly interacting molecules at a time through three iterations, until an anchor was connected to greater than 30 other components in the network. This limit allowed us to keep the network to a usable size and reduces the possibility that signaling cross-talk would obfuscate the primary connectivity relationships.
Rather than applying statistical considerations to this type of aggregate network analysis, which depends on literature citation rather than quantitative measurement and does not account for cell type specificity, we simply asked how connected each of these products were to the anchors. We identified 18 gene products that were directly linked to at least two of the anchor nodes (Fig. 1A). All the gene products and their connections to the anchors are indicated in Fig. 1B. RhoA, TSC2, LOC643751, CDKN1B, and TAF1B appeared in the top quartile of this list (Fig. 1C). LOC643751 was directly linked to RAC1 and Myc and linked to AKT1 through a single intermediate, PRKCZ, but it could not be linked through any single intermediate to either RalA or CCNA2. CDKN1B (p27) was connected directly to Myc, CCNA2, and AKT1, connected to RAC1 indirectly through RhoA, and connected to RalA indirectly through TSC2. TAF1B was directly linked to CCNA2 and Myc but could not be linked to any other node through any single intermediate. Only the nexus of TSC2, RhoA, and p27 was linked to all anchors directly or through a single intermediate.
FIGURE 1.

Network analysis connecting those gene products genetically implicated in human cell transformation. A, using Ingenuity Pathway Analysis, a network was seeded with five anchors (shown in orange), including the four oncoproteins (Myc, RAC1, AKT1, and RalA) whose functions collaborate with LT, hTERT, and RASV12 in transformation assays and a representative ST-regulated PP2A-independent target (CCNA2). Proteins in the lower three quartiles of connectivity between two or more nodes are shaded yellow, and those in the upper quartile are shaded green. Thick black lines indicate the nexus of RhoA, TSC2, and CDKN1B, which link either directly or through a single intermediate with all five anchors. B, Venn diagram of network analysis. Primary groups in black (Myc, RalA, or the combination of RAC1 and AKT1) have been superimposed on a fourth group in red (cyclin A2). The gene products are placed in their respective overlaps based on the connections. C, this graph represents the number of connections for each of the genes that linked to at least two of the nodes that anchored our pathway analysis. The number of connections each gene product shares with other components in the network are indicated on the ordinate (connectivity).
Observations reported in the literature place p27 downstream of TSC2 and RhoA. The growth-suppressive activity of TSC2 depends on p27 (25, 26), and the ability of RHOA to inhibit the translation of p27 mRNA contribute to its transforming activity (8). RalA can also control nuclear localization of p27 (27). On the other hand, Myc and AKT1 can directly suppress the accumulation of, or the nuclear localization of, the CDK inhibitor p27 (28–35), and CCNA2 presents p27 to the SCFskp2 ubiquitin-ligase (11). Thus, p27 is a single target downstream of RalA (through TSC2), RAC1 (through RhoA), Myc, AKT1, and CCNA2.
p27 Insufficiency Replaces ST Function in Human and Mouse Cell Transformation
Accordingly, we sought to experimentally test whether p27 loss could substitute for ST in cell transformation similarly to what had been reported for the other oncogenes. We first looked in human mesothelial cells that support a persistent noncytopathic infection with SV40. In keeping with studies of other human cell types, immortalized mesothelial cells expressing both hTERT and LT were transformed to anchorage-independent growth by subsequent infection with SV40 or transfection with a plasmid expressing ST alone, but were not transformed by infection with a mutant SV40 virus, DL888, that lacks ST expression (9). Similarly, these mesothelial cells could be transformed either by expressing a combination of activated AKT1 and RAC1 (7) or by expressing c-MYCT58A alone instead of ST.5 Importantly, p27 expression increased in DL888-infected cells relative to the amount in wild type SV40-infected cells when plated in suspension (Fig. 2A), indicating that ST was needed to reduce the accumulation of p27 during human mesothelial cell transformation.
FIGURE 2.
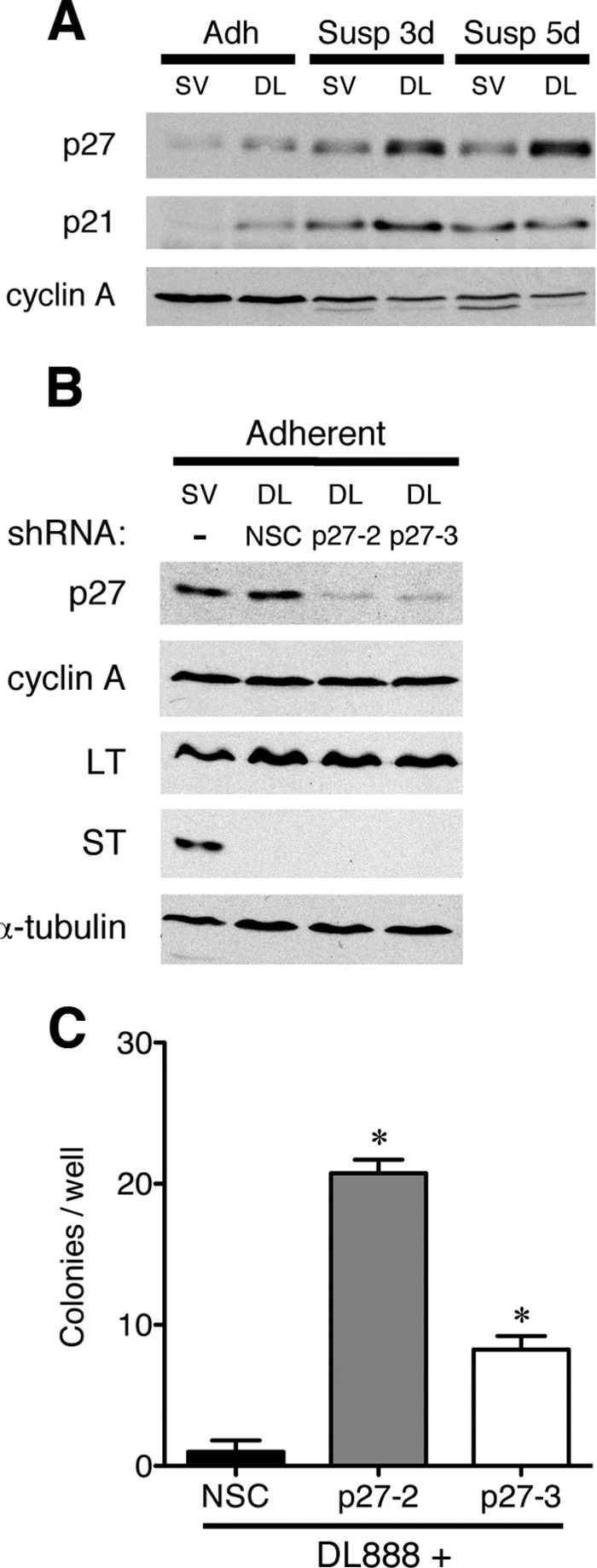
p27 deficiency can cooperate with LT to induce anchorage-independent growth in human mesothelial cells. A, SV40 (SV)- and DL888 (DL)-infected hTERT immortalized human mesothelial cells were cultured normally (adh) or grown in suspension (susp) for the times indicated. The expression of p27, p21, and cyclin A was determined by immunoblot. B, DL888-infected cells were transduced with a lentivirus expressing either an NSC shRNA or a p27-specific (p27-2 and p27-3) shRNA, and pools of cells were selected with G418. Protein extracts were prepared, and the levels of p27, cyclin A, LT, ST, and α-tubulin were determined by immunoblotting. Similar results were obtained from two independent pools of cells. C, soft agar growth. DL888-infected cells transduced with NSC, p27-2, or p27-3 lentivirus were plated in soft agar, and colonies visible to the eye were counted after 5–6 weeks of growth. The graph shows the average number of colonies per well from at least three independent experiments and is representative of the results obtained with two independent pools of cells. Statistical significance was determined by Student's t test. Error bars indicate S.D.
To determine whether the increased amount of p27 observed in the DL888-infected cells contributed to impaired transformation, we superinfected the DL888-infected cells with a lentivirus expressing either of two shRNAs directed against human p27CDKN1B (p27-2 and p27-3) or a nonsilencing shRNA control (NSC) and selected pools of transduced cells with G418. As shown in Fig. 2B, the amount of p27 was similar in the SV40-infected and DL888-NSC-infected cells, but was lower in the DL888-infected cells that were superinfected with the p27-specific shRNA constructs. Knocking down p27 did not affect accumulation of LT or cyclin A in DL888-infected cells (Fig. 2B).
To determine whether these cells were transformed, we then examined their ability to grow in an anchorage-independent manner. The cells expressing the control shRNA did not grow in soft agar, whereas a significant number of cells expressing either p27 shRNA construct did (Fig. 2C). The transformation efficiency of cells with less p27 increased approximately 10–20-fold. Statistically, the difference between the p27-2- and p27-3-expressing cell lines was not significant. Therefore, reduced expression of p27 could at least partially substitute for ST function in human cell transformation.
However, manipulating gene expression by shRNA vectors has limitations. The use of genetically engineered p27 knock-out mouse cells allows us to avoid any complications associated with inefficient shRNA knockdown, and the availability of p27 knock-in mutant cell lines allows us to examine the role of p27 phosphorylation in transformation. Furthermore, unlike human cells that require more genetic manipulations to be transformed, primary mouse cells can be transformed by the expression of LT and ST alone. Mouse cells immortalized by 3T3 culture or genetically by Ink4a/Arf deletion are less dependent on ST for cell transformation (data not shown).
We infected three independently derived clones of passage 1 wild type and p27 knock-out (p27D51/D51) primary MEFs with either wild type SV40, expressing both LT and ST, or a mutant virus, DL888, which expresses only LT at a multiplicity of infection of 50. At this multiplicity of infection, the ability of DL888 and SV40 to induce foci formation in a confluent monolayer was equivalent (Fig. 3A) (36, 37). However, although SV40 was ∼10-fold better at inducing growth in soft agar when compared with DL888 in wild type cells (Fig. 3B) (36, 37), the number of colonies induced by DL888 and SV40 was equivalent in p27-deficient cells (Fig. 3B). The genotype of the virus and the cells did not grossly affect the expression of LT (Fig. 3C). Similar results were seen with two other viruses that contained mutations in ST, C97S and C103S (data not shown) (17).
FIGURE 3.

p27 loss collaborates with LT to promote anchorage-independent growth. A, p27 deficiency does not abrogate the confluence-dependent checkpoint in cells expressing LT. Foci were visualized by crystal violet staining. The graph displays the number of foci (mean and S.D.) per plate in three independent experiments with each line. The image is representative of one such experiment. B, p27 deficiency can complement anchorage-independent growth in ST-deficient cells. Representative images of colony growth after 5 weeks are shown. The graph displays the number of colonies (mean and S.D.) per well for three independent WT and p27D51/D51 cell lines repeated three times. The Student's t test was used to determine the significance. C, first passage primary MEFs isolated from three independently derived WT or p27D51/D51 embryos (denoted A, B, and C) were infected with SV40 (SV) or DL888 (DL), and the expression of LT and ST was determined by immunoblotting. Lamin B is a loading control.
When grown in an adherent manner, neither the type of virus nor the p27 status affected the doubling time (data not shown) nor the percentage of S-phase cells (Fig. 4A, black bars), nor did the type of virus or p27 status grossly affect cell cycle exit when the cells were plated for 26 h in suspension (Fig. 4A, white bars). Conversely, when cultured in suspension, equivalent amounts of cyclin A-associated kinase activity were detected in p27 knock-out cells infected with DL888 and wild type and p27 knock-out cells infected with SV40, and this was higher than the level observed in wild type cells infected with DL888 (Fig. 4B). Nevertheless, the amount of activity was lower than that observed in cycling cells (Fig. 4B). When grown in suspension, there was more p27 in the wild type cells infected with DL888 than in the wild type cells infected with SV40 (Fig. 4B). Cyclin A levels were modestly decreased in suspended cells as well, with a greater decrease in those cells infected with DL888 than with SV40, consistent with the transcriptional effect ST has on this promoter. Thus, p27 deficiency, like ST, could cooperate with LT to support anchorage-independent growth.
FIGURE 4.

Cyclin A kinase activity and p27 levels are changed in an ST-dependent manner in suspended cells. A, SV40- and DL888-infected wild type (WT) and p27 knock-out (p27D51) cells were cultured on plastic (black bars) or in suspension (white bars) for 24 h. Cells were subsequently labeled with either tritiated thymidine for 2.5 h (top) or BrdU (bottom) for 1 h. The values were obtained from the average of two experiments done in triplicate with one of the cell lines; however, all cell lines behaved similarly. Error bars indicate S.D. B, cyclin A- and CDK2-associated histone H1 kinase activity was measured (top). Expression of p27, cyclin A, and CDK2 was determined by immunoblot (IB) (bottom). This image is representative of the data obtained from one of the three cell lines for each genotype; however, all cell lines behaved similarly. IP, immunoprecipitation; SV, SV40; DL, DL888.
p21, another CDK inhibitor in the CIP/KIP family, also increases when human cells were plated in soft agar and when mouse cells were plated in suspension (Fig. 2A) (38). Unlike p27 knock-out cells, primary p21 knock-out MEFs were not efficiently transformed to growth in soft agar by DL888 virus (negative data not shown). Thus, this suggested that the complementation activity was specific to p27 loss and not a consequence of general reduction in the amount of CDK inhibitor.
To determine whether these cell lines were tumorigenic in vivo, we injected cells into immunocompromised mice and followed their growth (Table 1). SV40-infected wild type and both SV40-infected and DL888-infected p27D51/D51 cells were able to form tumors in nude mice, but DL888-infected wild type cells were not. Collectively, these results show that p27 loss can substitute for the function of ST in SV40-induced anchorage-independent growth and tumorigenicity.
TABLE 1.
Frequency of tumor formation following injection of cells into immunocompromised mice
| Cell line | Tumor frequencya |
|
|---|---|---|
| 2 × 106 | 5 × 106 | |
| WT SV40 | 4/5 | 3/5 |
| WT DL888 | 1/5 | 0/5 |
| p27D51/D51 SV40 | 4/5 | 4/5 |
| p27D51/D51 DL888 | 5/5 | 5/5 |
| p27S10A/S10A SV40 | 0/4 | 1/5 |
| p27T187A/T187A SV40 | 0/5 | 1/5 |
a 2 × 106 or 5 × 106 cells mixed with Matrigel (1:1 volume) were injected into the right flank of male nude mice, and tumor growth was monitored for 20 weeks.
PP2A Can Dephosphorylate p27
Phosphorylation of either serine 10 or threonine 187 can reduce the amount of nuclear p27 and promote its turnover. Thus, cells expressing ST are expected to have more phosphorylated p27 due to its ability to inhibit phosphatases. A number of groups reported that the B56γ regulatory subunit of PP2A plays a role in the G1-to-S transition and in cell transformation. Furthermore, ST binding to the A/C dimer of PP2A can block the association of the B56 regulatory and targeting subunits (12).
Thus, we first asked whether PP2A interacted with p27; however, we were unable to coprecipitate endogenous p27 and PP2A (negative data not shown). Nevertheless, when we overexpress the different B subunits in 293T cells, p27 coprecipitated with the B56γ1 subunit, but not the B56α, B56β, B56γ3, B56δ, and B56ϵ subunits. On the other hand, cyclin G coprecipitated with the B56δ and B56ϵ subunits (Fig. 5A).
FIGURE 5.

PP2A and p27. A, 500 μg of extract from 293T cells transfected with HA-tagged versions of the different B56 isoforms, indicated above each lane, was immunoprecipitated (IP) with anti-HA, and the presence of p27 and cyclin G was determined by immunoblot (IB). This image is representative of two independent transfections. The expression of the B56 isoforms was determined in the top panel by immunoblotting. B, PP2A can dephosphorylate serine 10 and threonine 187. Top panel, [32P]-labeled recombinant His-tagged human p27 was incubated with increasing amounts of purified PP2A. Reactions were stopped by the addition of sample buffer, and proteins were separated by SDS-PAGE followed by autoradiography. This experiment was repeated twice with similar results. Bottom panel, the experiment was performed as above except that cold ATP was used instead of radiolabeled ATP and phosphorylation was detected by immunoblotting with phospho-specific antibodies to Ser-10-phosphorylated p27 (pS10-p27) or Thr-187-phosphorylated p27 (pT187-p27). This experiment was repeated twice with similar results. C, NIH3T3 cells were serum-starved for 24 h and infected in the absence of serum with either an empty adenoviral vector (CMV) or one expressing ST. The cells were harvested 24 h later, and the expression of ST, p27, cyclin A (cycA), and the Ser-10-phosphorylated(pS10-p27) and Thr-187-phosphorylated p27 (pT187-p27) isoforms was measured by immunoblotting. This image is representative of three independent experiments. MOI, multiplicity of infection.
We also asked whether purified PP2A could remove p27 phosphorylation in vitro. To assess the ability of the catalytically active core of PP2A to dephosphorylate p27, we first phosphorylated recombinant human p27 in vitro with purified cyclin B1-CDK1 in the presence of [γ-32P]ATP, and after heat inactivation of the kinase, we recovered the substrate and incubated it with recombinant PP2A. PP2A was able to dephosphorylate both the Ser-10 and the Thr-187 positions (Fig. 5B).
Finally, we asked whether ST expression would stabilize phosphorylated p27 in serum-starved cells. To accomplish this, we transfected cells with a vector expressing ST from the CMV promoter and looked at the accumulation of p27 phospho-Thr-187 and p27 phospho-Ser-10 by immunoblot 24 h later. Clearly, ST increased the amount of cyclin A and p27 phosphorylated on Thr-187, but not the amount of p27 phosphorylated on Ser-10 or the total amount of p27 (Fig. 5C). There was no significant change in the amount of cells engaged in S-phase of the cell cycle (data not shown). Collectively, these data suggest that PP2A activity can affect the accumulation of Thr-187 phosphorylated p27 in vivo. Such phosphorylation promotes p27 turnover, and thus, it is not surprising that the level of p27 was greater in cells infected with DL888 when compared with the level in the cells infected with SV40 when plated in suspension (Figs. 2A and 4B) or even when grown on plastic (Figs. 2A and 6A).
FIGURE 6.

Nonphosphorylatable mutants of p27 prevent anchorage-independent growth following infection with SV40. A, p27T187A/T187A and p27S10A/S10A MEFs were established by infection with SV40 (SV) or DL888 (DL), and the expression of LT, ST, and p27 was determined by immunoblot. Lamin B was used as a loading control. The shift in LT mobility observed in the T187A knock-in cell line is an occasional artifact we observe with this extraction buffer. The molecular nature of this shift is not known. B, foci formation was determined as described in the legend for Fig. 3. C, SV40-infected p27T187A/T187A and p27S10A/S10A cells were analyzed for their ability to grow in soft agar as described in the legend for Fig. 3. Error bars indicate S.D.
Stabilized p27 Inhibits Transformation Measured by Anchorage-independent Growth, but Not When Measured by Foci Formation
To determine whether stabilizing p27 would inhibit transformation, we obtained p27T187A/T187A and p27S10A/S10A knock-in MEFs (a generous gift from Jim Roberts) and asked whether these cells were more resistant to transformation by SV40. Mice expressing the T187A allele still develop lung and colon tumors, albeit the progression of intestinal adenomas to carcinoma is impaired. On the other hand, mice expressing the S10A allele are refractory to Ras transformation and tumor-resistant. Nuclear p27 levels were higher in both of these cell lines, yet they proliferated at rates similar to wild type cells as described previously (39, 40).
These cells were infected with SV40 or DL888, and pools of infected cells were collected after eight passages. Immunoblotting revealed that these p27 mutations did not affect the expression of LT (Fig. 6A). p27 levels were higher in the knock-in MEFs relative to wild type cells, with more pronounced stabilization in the p27T187A/T187A cell lines (Fig. 6A).
As before, we assessed the transformation properties of these cells by foci formation and soft agar growth. We found that the number of foci was comparable between SV40-infected wild type and SV40-infected p27T187A/T187A cells (Fig. 6B), indicating that the stabilization of p27 by this mutation did not grossly affect foci formation. In contrast, the number of foci formed by SV40-infected p27S10A/S10A cells was reduced ∼3-fold, and these were generally smaller than those formed by SV40-infected wild type cells (Fig. 6B). More importantly, mutation of either p27 phosphorylation site prevented growth in soft agar following SV40 infection (Fig. 6C). Furthermore, neither SV40-infected p27T187A/T187A nor SV40-infected p27S10A/S10A cells were tumorigenic in nude mice (Table 1). Together these results suggest that the ability to phosphorylate p27 serine 10 and threonine 187 was crucial for cell transformation by SV40.
DISCUSSION
In summary, we demonstrated that the ST expression can increase phosphorylation of p27 and that the loss of p27 can replace the transformation activity associated with expression of ST. Our network analysis indicates that the oncogenes or manipulations of PP2A that replace ST in cell transformation assays converge at a nexus containing TSC2, RhoA, and p27. To determine whether p27 deficiency led to the inappropriate activation of these oncogenes, we compared gene expression arrays obtained from three p27 wild type SV40-infected cell lines and three p27 knock-out DL888-infected cell lines grown in suspension by gene set enrichment analysis using six different Myc signatures and one RhoA, RAC1, and AKT1 signature (data not shown). The AKT1-responsive gene signature was up-regulated in the KO DL888 cells, and two of the six Myc signatures were up-regulated in the WT SV40 cells. No significant enrichment of the other four Myc signatures or RAC or RhoA signatures were found. Thus, we concluded that anchorage-independent growth in p27 KO cells not expressing ST was not due to compensatory changes in Myc, RAC1, or RhoA activity, but we could not formally exclude a role for AKT signaling. Furthermore, although we cannot exclude the possibility that PP2A activity can contribute to transformation by modulating cell type-dependent signaling pathways, our data suggest that it can affect accumulation of p27 and that this may alter the signaling milieu.
Although the signaling pathways that can replace ST are cell type-specific (41), reduced p27 levels are common in tumors of diverse origin. Thus, it seems likely that reduced p27 levels in tumors are a good readout of the tumor signaling milieu, reflecting the extent of imbalance between oncogenic signaling and serine-threonine phosphatase activity. Thus, immunohistochemical staining of p27 can be a widely used simple, cheap, and robust measurement to assess the nature of the overall signaling environment, and perhaps this explains its value as a prognostic marker independent of cell proliferation.
When we began this study, the relationship between PP2A and the oncogenes that could replace ST in the transformation assay was unclear. It is generally accepted that the ability of ST to reduce PP2A activity allows for increased oncogenic signaling, through Myc or AKT1, depending on the cell type and oncogenic environment thus driving oncogenesis (5–7). However, in silico analysis suggested that these oncogenic pathways could converge at the regulation of p27, a protein whose accumulation can also be controlled directly by PP2A through phosphorylation-dependent nuclear exclusion and/or ubiquitination. Consequently, the signaling inputs important for cell transformation, as measured by anchorage-independent growth and ability to grow as a xenograft, converge at a nexus of p27.
The transforming equivalence of ST and the loss of p27 reported here in cultured cells are also seen in a mouse model. Prostate cancer progression in mice that express LT from the rat probasin promoter (W10) is accelerated when this transgene is crossed into a p27-deficient background (42). The survival and tumor incidence of the W10p27D51/D51 mice are remarkably similar to those of TRAMP mice, which express both LT and ST from the same promoter, with virtually all of the mice dying with poorly differentiated cancer by 6 months of age (43). In contrast, two independent strains of mice expressing LT from different promoters, W10 and LEDY (42, 44), have remarkably similar survival profiles with 50% of the animals still alive at 1 year, and most of those with moderately or well differentiated cancer. Thus, this model supports the relevance of p27 as a critical target for ST in tumorigenesis.
Forty years of empirical data establish that many primary rodent cells are more transformable than their human counterparts. The conditions for oncogenic transformation are cell type-specific (1, 41), and within a single cell type, not all lines are equally transformable (45); however, the number of genetic events needed to transform human cells is usually greater than that of mouse cells. This difference may be associated with how mouse and human cells respond to oncogenic stress, specifically whether these events lead to apoptosis or senescence, the sensitivity of the spindle assembly checkpoint, molecular mechanisms controlling genomic integrity, and telomere biology (1, 41, 46–49). However, all cells, mouse and human, must unshackle themselves from checkpoints governing entry and exit from the cell cycle.
Deregulation of p27-dependent G1 control mechanisms in mice demonstrably impacts the sensitivity of transformed cells to apoptosis (50), DNA repair (51), and mitotic checkpoints (52) without grossly altering the proliferation indices. Given the association of low p27 with poor prognosis and genome instability in a variety of human tumors, again not associated with gross changes in proliferative index, the roles that p27 can play in mouse are probably comparable in human tumors. Nevertheless, deregulation of specific cell cycle regulators in mice does not always display the cell type specificity seen in humans. For example, Rb loss in mice leads to pituitary tumors, whereas retinoblastoma and sarcoma are common in humans. Even when Rb loss is combined with other genetic events in mice that drive retinoblastoma, the cell of origin is different from the one observed in humans (reviewed in Ref. 53). This may be an impediment if an investigator wants to study how the regulator participates in a specific disease, but not if they are trying to understand and define molecular mechanisms by which these proteins participate in any disease. In fact, it may be a grand opportunity to delve into the cell type-specific contextual clues that impact on the role of these proteins.
Nevertheless, two findings from our work need to be reconciled with other evidence regarding the role of PP2A in p27 and cancer. First, although Porrás et al. (23) showed that ST was required to increase p27 proteolysis and Chen et al. (19) reported that B56γ was important for cell transformation associated with ST expression, Lee et al. (54) reported that it was specifically the B56γ3 subunit of PP2A that interacted with p27 and promoted the G1-to-S transition. We were unable to recapitulate the interaction of B56γ3 with p27, but we were able to show that B56γ1 was able to interact. The reason for this difference is not clear; however, regardless of which interaction is correct, the evidence supports the conclusion that PP2A acts, at least in part, by directly affecting p27 levels.
More disconcerting to us was the conclusion that Rangarajan et al. (41) reached concerning the relationship of PP2A to transformation of primary mouse fibroblasts. They suggested that PP2A inactivation by ST was not necessary for transformation of normal murine fibroblasts when LT and RASV12 were expressed. This provocative conclusion contradicts the original highly regarded identification of ST antigen and its role in murine cell transformation (36, 37) and seemingly calls into question our use of the mouse model to probe ST/PP2A/p27 genetic and biochemical interactions. Although immortalization of mouse cells or disruption of the ARF/p53 pathways can obfuscate the requirement for ST, this is not seen with primary cells immediately infected with SV40 viruses and the mutants. Our network analysis suggests that if PP2A and oncogenes were converging independently on p27, any relationship between ST or PP2A activity in transformation of primary murine cells would be dependent on the dose or the strength of the primary oncogenic signal. Thus, at higher levels of oncogenic signaling, a requirement for inhibiting PP2A would not be necessary as the cells would be able to control p27 expression through transcriptional, translational, or post-translational mechanisms.
Thus, in conclusion, we showed that eliminating p27 can replace the role of ST in transformation of primary mouse embryo fibroblasts and in an immortalized human mesothelioma cell line. The Ingenuity Pathway Analysis network suggested that oncogenes that could replace ST antigen function could converge to lower p27 expression or activity. Thus, although we cannot eliminate the idea that PP2A acts as a tumor suppressor by reducing flux through signaling pathways, our data suggest the equally compelling idea that PP2A activity and oncogenic signaling are integrated by the accumulation of p27, a strong marker of the evolution of tumor aggressiveness.
Acknowledgments
We thank Prasad Jallepalli, David Cobrinik, Mary Alpaugh, John Petrini, Travis Stracker, Neal Rosen, David Solit, David Shaffer, Robert Bowman, and David Liu (Memorial Sloan-Kettering Cancer Center) and Rebecca Katzman and Kathleen Rundell (Northwestern University) for help with experiments and discussions throughout the course of these studies. We also thank James Roberts (Fred Hutchinson Cancer Research Center) for generous gift of S10A and T187A cells for these studies. The Memorial Sloan-Kettering Cancer Center was supported by a core grant from the National Institutes of Health.
This work was supported, in whole or in part, by National Institutes of Health Grant CA89563 from the NCI (to A. K.). This work was also supported by funding from The Golfers Against Cancer Foundation (to A. K.).
R. Katzman and K. Rundell, personal communication.
- PP2A
- protein phosphatase 2A
- ST
- small t-antigen
- LT
- large T-antigen
- MEF
- mouse embryonic fibroblast
- Rb
- retinoblastoma
- NSC
- nonsilencing shRNA control.
REFERENCES
- 1. Hahn W. C., Weinberg R. A. (2002) Modeling the molecular circuitry of cancer. Nat. Rev. Cancer 2, 331–341 [DOI] [PubMed] [Google Scholar]
- 2. Ahuja D., Sáenz-Robles M. T., Pipas J. M. (2005) SV40 large T-antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 24, 7729–7745 [DOI] [PubMed] [Google Scholar]
- 3. Seger Y. R., García-Cao M., Piccinin S., Cunsolo C. L., Doglioni C., Blasco M. A., Hannon G. J., Maestro R. (2002) Transformation of normal human cells in the absence of telomerase activation. Cancer Cell 2, 401–413 [DOI] [PubMed] [Google Scholar]
- 4. Westermarck J., Hahn W. C. (2008) Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol. Med. 14, 152–160 [DOI] [PubMed] [Google Scholar]
- 5. Junttila M. R., Puustinen P., Niemelä M., Ahola R., Arnold H., Böttzauw T., Ala-aho R., Nielsen C., Ivaska J., Taya Y., Lu S. L., Lin S., Chan E. K., Wang X. J., Grènman R., Kast J., Kallunki T., Sears R., Kähäri V. M., Westermarck J. (2007) CIP2A inhibits PP2A in human malignancies. Cell 130, 51–62 [DOI] [PubMed] [Google Scholar]
- 6. Millward T. A., Zolnierowicz S., Hemmings B. A. (1999) Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem. Sci. 24, 186–191 [DOI] [PubMed] [Google Scholar]
- 7. Skoczylas C., Fahrbach K. M., Rundell K. (2004) Cellular targets of the SV40 small t-antigen in human cell transformation. Cell Cycle 3, 606–610 [PubMed] [Google Scholar]
- 8. Vidal A., Millard S. S., Miller J. P., Koff A. (2002) Rho activity can alter the translation of p27 mRNA and is important for RasV12-induced transformation in a manner dependent on p27 status. J. Biol. Chem. 277, 16433–16440 [DOI] [PubMed] [Google Scholar]
- 9. Yu J., Boyapati A., Rundell K. (2001) Critical role for SV40 small t-antigen in human cell transformation. Virology 290, 192–198 [DOI] [PubMed] [Google Scholar]
- 10. Yeh N., Miller J. P., Gaur T., Capellini T. D., Nikolich-Zugich J., de la Hoz C., Selleri L., Bromage T. G., van Wijnen A. J., Stein G. S., Lian J. B., Vidal A., Koff A. (2007) Cooperation between p27 and p107 during endochondral ossification suggests a genetic pathway controlled by p27 and p130. Mol. Cell. Biol. 27, 5161–5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu X. H., Nguyen H., Halicka H. D., Traganos F., Koff A. (2004) Noncatalytic requirement for cyclin A-cdk2 in p27 turnover. Mol. Cell. Biol. 24, 6058–6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cho U. S., Morrone S., Sablina A. A., Arroyo J. D., Hahn W. C., Xu W. (2007) Structural basis of PP2A inhibition by small t-antigen. PLoS Biol. 5, e202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Scheidtmann K. H., Mumby M. C., Rundell K., Walter G. (1991) Dephosphorylation of simian virus 40 large T-antigen and p53 protein by protein phosphatase 2A: inhibition by small t-antigen. Mol. Cell. Biol. 11, 1996–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang S. I., Lickteig R. L., Estes R., Rundell K., Walter G., Mumby M. C. (1991) Control of protein phosphatase 2A by simian virus 40 small t-antigen. Mol. Cell. Biol. 11, 1988–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hahn W. C., Dessain S. K., Brooks M. W., King J. E., Elenbaas B., Sabatini D. M., DeCaprio J. A., Weinberg R. A. (2002) Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol. Cell. Biol. 22, 2111–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mateer S. C., Fedorov S. A., Mumby M. C. (1998) Identification of structural elements involved in the interaction of simian virus 40 small tumor antigen with protein phosphatase 2A. J. Biol. Chem. 273, 35339–35346 [DOI] [PubMed] [Google Scholar]
- 17. Mungre S., Enderle K., Turk B., Porrás A., Wu Y. Q., Mumby M. C., Rundell K. (1994) Mutations which affect the inhibition of protein phosphatase 2A by simian virus 40 small t-antigen in vitro decrease viral transformation. J. Virol. 68, 1675–1681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sontag E., Fedorov S., Kamibayashi C., Robbins D., Cobb M., Mumby M. (1993) The interaction of SV40 small tumor antigen with protein phosphatase 2A stimulates the map kinase pathway and induces cell proliferation. Cell 75, 887–897 [DOI] [PubMed] [Google Scholar]
- 19. Chen W., Possemato R., Campbell K. T., Plattner C. A., Pallas D. C., Hahn W. C. (2004) Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell 5, 127–136 [DOI] [PubMed] [Google Scholar]
- 20. Sablina A. A., Chen W., Arroyo J. D., Corral L., Hector M., Bulmer S. E., DeCaprio J. A., Hahn W. C. (2007) The tumor suppressor PP2A Aβ regulates the RalA GTPase. Cell 129, 969–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yeh E., Cunningham M., Arnold H., Chasse D., Monteith T., Ivaldi G., Hahn W. C., Stukenberg P. T., Shenolikar S., Uchida T., Counter C. M., Nevins J. R., Means A. R., Sears R. (2004) A signaling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 6, 308–318 [DOI] [PubMed] [Google Scholar]
- 22. Zhao J. J., Gjoerup O. V., Subramanian R. R., Cheng Y., Chen W., Roberts T. M., Hahn W. C. (2003) Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell 3, 483–495 [DOI] [PubMed] [Google Scholar]
- 23. Porrás A., Bennett J., Howe A., Tokos K., Bouck N., Henglein B., Sathyamangalam S., Thimmapaya B., Rundell K. (1996) A novel simian virus 40 early-region domain mediates transactivation of the cyclin A promoter by small t-antigen and is required for transformation in small t-antigen-dependent assays. J. Virol. 70, 6902–6908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moreno C. S., Ramachandran S., Ashby D. G., Laycock N., Plattner C. A., Chen W., Hahn W. C., Pallas D. C. (2004) Signaling and transcriptional changes critical for transformation of human cells by simian virus 40 small tumor antigen or protein phosphatase 2A B56γ knockdown. Cancer Res. 64, 6978–6988 [DOI] [PubMed] [Google Scholar]
- 25. Soucek T., Rosner M., Miloloza A., Kubista M., Cheadle J. P., Sampson J. R., Hengstschläger M. (2001) Tuberous sclerosis causing mutants of the TSC2 gene product affect proliferation and p27 expression. Oncogene 20, 4904–4909 [DOI] [PubMed] [Google Scholar]
- 26. Soucek T., Yeung R. S., Hengstschläger M. (1998) Inactivation of the cyclin-dependent kinase inhibitor p27 upon loss of the tuberous sclerosis complex gene-2. Proc. Natl. Acad. Sci. U.S.A. 95, 15653–15658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kfir S., Ehrlich M., Goldshmid A., Liu X., Kloog Y., Henis Y. I. (2005) Pathway- and expression level-dependent effects of oncogenic N-Ras: p27Kip1 mislocalization by the Ral-GEF pathway and Erk-mediated interference with Smad signaling. Mol. Cell. Biol. 25, 8239–8250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bouchard C., Thieke K., Maier A., Saffrich R., Hanley-Hyde J., Ansorge W., Reed S., Sicinski P., Bartek J., Eilers M. (1999) Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 18, 5321–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fujita N., Sato S., Katayama K., Tsuruo T. (2002) Akt-dependent phosphorylation of p27Kip1 promotes binding to 14-3-3 and cytoplasmic localization. J. Biol. Chem. 277, 28706–28713 [DOI] [PubMed] [Google Scholar]
- 30. Keller U. B., Old J. B., Dorsey F. C., Nilsson J. A., Nilsson L., MacLean K. H., Chung L., Yang C., Spruck C., Boyd K., Reed S. I., Cleveland J. L. (2007) Myc targets Cks1 to provoke the suppression of p27Kip1, proliferation, and lymphomagenesis. EMBO J. 26, 2562–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liang J., Zubovitz J., Petrocelli T., Kotchetkov R., Connor M. K., Han K., Lee J. H., Ciarallo S., Catzavelos C., Beniston R., Franssen E., Slingerland J. M. (2002) PKB/Akt phosphorylates p27, impairs nuclear import of p27, and opposes p27-mediated G1 arrest. Nat. Med. 8, 1153–1160 [DOI] [PubMed] [Google Scholar]
- 32. O'Hagan R. C., Ohh M., David G., de Alboran I. M., Alt F. W., Kaelin W. G., Jr., DePinho R. A. (2000) Myc-enhanced expression of Cul1 promotes ubiquitin-dependent proteolysis and cell cycle progression. Genes Dev. 14, 2185–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perez-Roger I., Kim S. H., Griffiths B., Sewing A., Land H. (1999) Cyclins D1 and D2 mediate Myc-induced proliferation via sequestration of p27Kip1 and p21Cip1. EMBO J. 18, 5310–5320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shin I., Yakes F. M., Rojo F., Shin N. Y., Bakin A. V., Baselga J., Arteaga C. L. (2002) PKB/Akt mediates cell cycle progression by phosphorylation of p27Kip1 at threonine 157 and modulation of its cellular localization. Nat. Med. 8, 1145–1152 [DOI] [PubMed] [Google Scholar]
- 35. Viglietto G., Motti M. L., Bruni P., Melillo R. M., D'Alessio A., Califano D., Vinci F., Chiappetta G., Tsichlis P., Bellacosa A., Fusco A., Santoro M. (2002) Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27Kip1 by PKB/Akt-mediated phosphorylation in breast cancer. Nat. Med. 8, 1136–1144 [DOI] [PubMed] [Google Scholar]
- 36. Bouck N., Beales N., Shenk T., Berg P., di Mayorca G. (1978) New region of the simian virus 40 genome required for efficient viral transformation. Proc. Natl. Acad. Sci. U.S.A. 75, 2473–2477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sleigh M. J., Topp W. C., Hanich R., Sambrook J. F. (1978) Mutants of SV40 with an altered small t-protein are reduced in their ability to transform cells. Cell 14, 79–88 [DOI] [PubMed] [Google Scholar]
- 38. Vidal A., Zacharoulis S., Guo W., Shaffer D., Giancotti F., Bramley A. H., de la Hoz C., Jensen K. K., Kato D., MacDonald D. D., Knowles J., Yeh N., Frohman L. A., Rafii S., Lyden D., Koff A. (2005) p130Rb2 and p27kip1 cooperate to control mobilization of angiogenic progenitors from the bone marrow. Proc. Natl. Acad. Sci. U.S.A. 102, 6890–6895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Besson A., Gurian-West M., Chen X., Kelly-Spratt K. S., Kemp C. J., Roberts J. M. (2006) A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev. 20, 47–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Timmerbeul I., Garrett-Engele C. M., Kossatz U., Chen X., Firpo E., Grünwald V., Kamino K., Wilkens L., Lehmann U., Buer J., Geffers R., Kubicka S., Manns M. P., Porter P. L., Roberts J. M., Malek N. P. (2006) Testing the importance of p27 degradation by the SCFskp2 pathway in murine models of lung and colon cancer. Proc. Natl. Acad. Sci. U.S.A. 103, 14009–14014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rangarajan A., Hong S. J., Gifford A., Weinberg R. A. (2004) Species- and cell type-specific requirements for cellular transformation. Cancer Cell 6, 171–183 [DOI] [PubMed] [Google Scholar]
- 42. Shaffer D. R., Viale A., Ishiwata R., Leversha M., Olgac S., Manova K., Satagopan J., Scher H., Koff A. (2005) Evidence for a p27 tumor-suppressive function independent of its role regulating cell proliferation in the prostate. Proc. Natl. Acad. Sci. U.S.A. 102, 210–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Greenberg N. M., DeMayo F., Finegold M. J., Medina D., Tilley W. D., Aspinall J. O., Cunha G. R., Donjacour A. A., Matusik R. J., Rosen J. M. (1995) Prostate cancer in a transgenic mouse. Proc. Natl. Acad. Sci. U.S.A. 92, 3439–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kasper S., Sheppard P. C., Yan Y., Pettigrew N., Borowsky A. D., Prins G. S., Dodd J. G., Duckworth M. L., Matusik R. J. (1998) Development, progression, and androgen-dependence of prostate tumors in probasin-large T-antigen transgenic mice: a model for prostate cancer. Lab. Invest. 78, 319–333 [PubMed] [Google Scholar]
- 45. Akagi T., Sasai K., Hanafusa H. (2003) Refractory nature of normal human diploid fibroblasts with respect to oncogene-mediated transformation. Proc. Natl. Acad. Sci. U.S.A. 100, 13567–13572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Akagi T. (2004) Oncogenic transformation of human cells: shortcomings of rodent model systems. Trends Mol. Med. 10, 542–548 [DOI] [PubMed] [Google Scholar]
- 47. Akagi T., Hanafusa H. (2004) Human diploid fibroblasts are refractory to oncogene-mediated transformation. Cell Cycle 3, 257–258 [DOI] [PubMed] [Google Scholar]
- 48. Fink L. S., Roell M., Caiazza E., Lerner C., Stamato T., Hrelia S., Lorenzini A., Sell C. (2011) 53BP1 contributes to a robust genomic stability in human fibroblasts. Aging 3, 836–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haller K., Kibler K. V., Kasai T., Chi Y. H., Peloponese J. M., Yedavalli V. S., Jeang K. T. (2006) The N terminus of rodent and human MAD1 confers species-specific stringency to spindle assembly checkpoint. Oncogene 25, 2137–2147 [DOI] [PubMed] [Google Scholar]
- 50. Carneiro C., Jiao M. S., Hu M., Shaffer D., Park M., Pandolfi P. P., Cordon-Cardo C., Koff A. (2003) p27 deficiency desensitizes Rb−/− cells to signals that trigger apoptosis during pituitary tumor development. Oncogene 22, 361–369 [DOI] [PubMed] [Google Scholar]
- 51. See W. L., Miller J. P., Squatrito M., Holland E., Resh M. D., Koff A. (2010) Defective DNA double-strand break repair underlies enhanced tumorigenesis and chromosomal instability in p27-deficient mice with growth factor-induced oligodendrogliomas. Oncogene 29, 1720–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Payne S. R., Zhang S., Tsuchiya K., Moser R., Gurley K. E., Longton G., deBoer J., Kemp C. J. (2008) p27kip1 deficiency impairs G2/M arrest in response to DNA damage, leading to an increase in genetic instability. Mol. Cell. Biol. 28, 258–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xu X. L., Fang Y., Lee T. C., Forrest D., Gregory-Evans C., Almeida D., Liu A., Jhanwar S. C., Abramson D. H., Cobrinik D. (2009) Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell 137, 1018–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee T. Y., Lai T. Y., Lin S. C., Wu C. W., Ni I. F., Yang Y. S., Hung L. Y., Law B. K., Chiang C. W. (2010) The B56γ3 regulatory subunit of protein phosphatase 2A (PP2A) regulates S-phase-specific nuclear accumulation of PP2A and the G1-to-S transition. J. Biol. Chem. 285, 21567–21580 [DOI] [PMC free article] [PubMed] [Google Scholar]