

Background: Base J regulates Pol II transcription.
Results: JBP1 and -2 stimulate the first step of base J synthesis: hydroxylation of thymidine.
Conclusion: JBP are Fe2+/2-OG-dependent dioxygenases sensitive to physiologically relevant O2 tensions.
Significance: These results predict that JBPs can act as oxygen sensors regulating trypanosome gene expression and adaption to different host niches.
Keywords: DNA Enzymes, Glycobiology, Hydroxylase, Leishmania, Nucleic Acid Synthesis, Transcription Regulation, Trypanosome, Dioxygenase, Thymidine Hydroxylase
Abstract
We have recently demonstrated that O-linked glucosylation of thymine in trypanosome DNA (base J) regulates polymerase II transcription initiation. In vivo analysis has indicated that base J synthesis is initiated by the hydroxylation of thymidine by proteins (JBP1 and JBP2) homologous to the Fe2+/2-oxoglutarate (2-OG)-dependent dioxygenase superfamily where hydroxylation is driven by the oxidative decarboxylation of 2-OG, forming succinate and CO2. However, no direct evidence for hydroxylase activity has been reported for the JBP proteins. We now demonstrate recombinant JBP1 hydroxylates thymine specifically in the context of dsDNA in a Fe2+-, 2-OG-, and O2-dependent manner. Under anaerobic conditions, the addition of Fe2+ to JBP1/2-OG results in the formation of a broad absorption spectrum centered at 530 nm attributed to metal chelation of 2-OG bound to JBP, a spectroscopic signature of Fe2+/2-OG-dependent dioxygenases. The N-terminal thymidine hydroxylase domain of JBP1 is sufficient for full activity and mutation of residues involved in coordinating Fe2+ inhibit iron binding and thymidine hydroxylation. Hydroxylation in vitro and J synthesis in vivo is inhibited by known inhibitors of Fe2+/2-OG-dependent dioxygenases. The data clearly demonstrate the JBP enzymes are dioxygenases acting directly on dsDNA, confirming the two-step J synthesis model. Growth of trypanosomes in hypoxic conditions decreases JBP1 and -2 activity, resulting in reduced levels of J and changes in parasite virulence previously characterized in the JBP KO. The influence of environment upon J biosynthesis via oxygen-sensitive regulation of JBP1/2 has exciting implications for the regulation of gene expression and parasite adaptation to different host niches.
Introduction
β-d-Glucopyranosyloxymethyluracil (base J)2 is a hyper-modified DNA base found in eukaryotes. This DNA modification is evolutionarily conserved within members of the kinetoplastid family, namely Trypanosoma brucei, Trypanosoma cruzi, and Leishmania, where J replaces about 1% of the total T in the genome and is predominantly present in repetitive DNA sequences, such as telomeric repeats (for review, see Ref. 1). However, more recently, we localized a minor fraction of J to chromosome-internal regions coinciding with RNA polymerase II (Pol II) transcription initiation and termination sites (2). Loss of base J synthesis at these chromosome-internal regions in T. cruzi led to increased Pol II transcription initiation and corresponding changes in gene expression and parasite virulence (3, 4). Thus, base J represents a novel epigenetic modification of kinetoplastid DNA involved in regulating gene expression.
Indirect evidence (for review, see Ref. 5) indicates J is synthesized in a two-step pathway (Fig. 1). Step one involves the hydroxylation of thymine in DNA by a thymidine hydroxylase (TH) enzyme, forming 5-hydroxymethyluracil (hmU). This intermediate is then glucosylated by a glucosyltransferase forming base J. Although the glucosyltransferase has not been identified, two proteins involved in the first step (JBP1 and JBP2) (6, 7) have been characterized. Both JBP1 and JBP2 (8, 9) contain a putative TH domain at the N terminus that has led to the designation of these enzymes belonging to the new TET/JBP subfamily of dioxygenases that require Fe2+ and 2-oxoglutarate (2-OG) for activity (10, 11). Family members are typically identified on a structural level by the presence of a jelly roll β-helix sheet that contains four key conserved residues involved in the binding of Fe2+ and 2-OG and are essential for catalytic activity (see for review, see Ref. 12). Mutation of these conserved residues within the TH domain of JBP1 and JBP2 ablates enzyme function in J synthesis in vivo, supporting their classification as Fe2+/2-OG dioxygenases (8, 9). Deletion of either JBP1 or JBP2 from bloodstream form T. brucei resulted in a 20- and 8-fold reduction in J levels, respectively (13, 14). The simultaneous deletion of both generated a cell line unable to synthesize the modified base unless the cells are fed hmU, confirming their importance in the first step of J synthesis (9). Deletion of either JBP1 or JBP2 in T. cruzi and JBP2 in Leishmania tarentolae results in similar reductions in J levels as seen in T. brucei mutants (4, 13). Attempts to delete both JBPs from L. tarentolae and T. cruzi have failed to date, leading to the proposal that base J is essential in these organisms. However, detailed characterization of the T. cruzi JBP1 KO cell line has elucidated an important function of base J in trypanosomes. Anti-J ChIP analysis indicated that whereas the remaining J in the T. cruzi JBP1 KO is located in telomeric DNA, base J is lost from the divergent and convergent strand-switch regions involved in Pol II initiation and termination, respectively. The loss of base J at transcription start sites coincides with a decrease in nucleosome abundance, increased histone acetylation, and increased Pol II occupancy at promoter regions (3, 4). This increase in Pol II recruitment correlates with an increased rate of transcription initiation and changes in gene expression (3). These studies indicate the importance of epigenetic regulation of Pol II transcription via DNA modification and chromatin structure in kinetoplastids as well as provide a mechanism for J regulation of trypanosome gene expression. Thus, characterization of the enzyme(s) regulating the hydroxylation of specific T-residues along the chromosome is critical for understanding the control of trypanosome gene expression.
FIGURE 1.
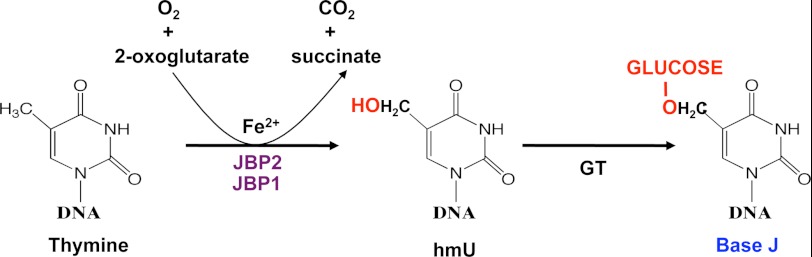
Proposed mechanism of JBP catalyzed thymidine hydroxylation; the first step of J synthesis. Base J is thought to be synthesized in DNA by a two-step mechanism involving thymidine hydroxylation and glycosylation. According to this model, JBP1 and JBP2 are members of the Fe2+/2-OG dioxygenase family that utilize 2-OG and O2 as co-substrate to hydroxylate T-residues in dsDNA, releasing succinate and CO2 as byproducts. The intermediate hmU is then glycosylated by an unknown glucosyltransferase (GT) forming base J.
The Fe2+/2-OG-dependent dioxygenase enzyme family encompasses a large group of enzymes that catalyze the hydroxylation of a diverse variety of substrates, including but not limited to DNA, protein, RNA, and lipid (for review, see Ref. 12). Most dioxygenases utilize Fe2+ as a cofactor and 2-OG and oxygen (O2) as co-substrates with the reaction coupling oxidative decarboxylation of 2-OG to the hydroxylation of substrate. Succinate and carbon dioxide are released as byproducts. The requirement of enzyme activity for molecular O2 has suggested many of these dioxygenases function as direct O2 sensors (14, 15). They may also be activated by the co-substrate 2-OG, a Krebs cycle intermediate, ascorbate, and Fe2+ and inhibited by the product succinate. Members of this dioxygenase enzyme family catalyze the demethylation of Nϵ-methyl lysine residues in histones and of N-methylated nucleic acids as well as hydroxylation of 5-methyl cytosine in DNA and 5-methoxycarbonylmethyluridine at the wobble position of tRNA. As such, dioxygenase enzymes have a broad range of biological roles, including DNA repair and O2 sensing and regulating gene expression (16, 17).
A distant homolog of the JBP1/2 TH domain was recently identified in the mammalian protein TET1, which is found fused to the histone methyltransferase MLL gene during acute myeloid leukemia (11, 18). TET1 (and the related TET2 and TET3 proteins) has been shown to convert 5-methylcytosine in DNA to 5-hydroxymethylcytosine. This conversion may play an important role in the epigenetic control of gene expression in mammals (11, 18). Based on the sequence similarity, JBP and TET proteins have been grouped together in the TET/JBP subfamily of dioxygenases (10).
Although the characterization of the JBP enzymes in vivo has proven to be useful in confirming their importance in J biosynthesis and elucidating the function of base J, TH activity has not yet been demonstrated for JBP in vitro. Here we develop an in vitro assay to demonstrate that JBP hydroxylation of thymine residues in dsDNA is dependent on Fe2+, 2-OG, and O2. We show rJBP1 binds Fe2+, and mutation of the two conserved metal binding ligands of dioxygenases results in the loss of Fe2+ binding and inability to hydroxylate thymidine. Competitive inhibitors of 2-OG significantly reduce JBP activity both in vivo and in vitro. Taken together, this data confirm the identity of the JBPs as Fe2+/2-OG-dependent dioxygenases and provide direct evidence for the two-step J-biosynthesis pathway of modifying T-residues in kinetoplastid DNA. Furthermore, the O2 requirement for JBP1-stimulated hydroxylation in vitro and J synthesis in vivo and the corresponding changes in T. cruzi virulence suggest the oxygen regulation of JBP enzyme activity to be important in allowing the parasite to adapt to changing host conditions during its lifecycle.
EXPERIMENTAL PROCEDURES
Cell Culture
Blood stream form T. brucei cell line 221a of strain 427 were cultured as described previously (7). Promastigote L. major cells were grown at 26 °C in M199 media supplemented with 10% FBS as described (19). Y strain T. cruzi wild-type (WT) and JBP1 double knock-out epimastigotes were grown in liver infusion tryptose medium containing 10% fetal bovine serum as previously described (4). Epimastigotes were differentiated to infective metacyclics as described (20). For in vivo inhibition of TH activity, cells were grown in the presence of dimethyloxoglycine (DMOG) (Frontier Scientific), 2,4-pyridinedicarboxylic acid hydrate (Acros Organics), or DMSO as indicated in Fig. 4 (inhibitor stocks were made up in 25% DMSO). As a control, parasites were grown in medium with comparable DMSO concentrations as cultures with the highest level of diluted inhibitor.
FIGURE 4.

Thymidine hydroxylase activity of JBP1 and JBP2 is reduced by dioxygenase inhibitors. The in vitro TH assay was performed using JBP1 in the presence/absence of 0.5 mm DMOG (A) and 1 mm succinate (B). Error bars represent the S.D. of triplicate experiments. Data are expressed as % activity relative to the control reaction. C, T. brucei cells were grown in the presence or absence of 0.5 mm DMOG or 0.5 mm 2,4-pyridinedicarboxylic acid hydrate for 6 days, and DNA was isolated for anti-J dot blot analysis. Samples were 2-fold serially diluted. The same blots were hybridized with a tubulin probe to control for DNA loading. DMSO is provided as a control where parasites were grown in medium with comparable DMSO concentrations as cultures with highest level of diluted inhibitor. Similar DMOG treatment was also performed in L. major (D) and T. cruzi (E).
Preparation of Recombinant JBP1
Recombinant His-tagged L. tarentolae JBP1 (Lt-JBP1) was produced essentially as previously described (8) with the following modifications. The Lt-JBP1 vector was transformed to BL21-DE3 T1R (21). Protein production was induced when cells reached an optical density of 0.6–0.9 with 0.3 mm isopropyl 1-thio-β-d-galactopyranoside at 16 °C for 16 h. rJBP1 was purified over a Talon resin column eluted with 600 mm NaCl, 50 mm HEPES, and 200 mm imidazole followed by dialysis in 50 mm HEPES (pH 8.0), 100 mm NaCl, and 5 mm EDTA for 3 h. Protein was then subject to further dialysis (ON) in 50 mm HEPES and 100 mm NaCl. Purified protein, >95% pure as judged by SDS-PAGE, was concentrated by Centricon.
Analysis of Fe2+ Binding to JBP1 Using UV-Visible Spectroscopy
Fe2+ binding analysis was carried out using 250 μm rJBP1, 100 mm dithionite, 240 μm Fe2SO4, 1 mm 2-OG, and 1 mm ascorbic acid essentially as previously described (22). All solutions were made anaerobic by several rounds of vacuum degassing and flushing with argon using a vacuum manifold and sealed serum vials. The reaction mixture was then read from 200 to 800 nm in a Shimadzu UV-visible spectrophotometer using a quartz cuvette fitted with a stopper and purged with argon. After blanking against JBP1, we recorded spectra for samples to which anaerobic aliquots of Fe2+ and 2-OG were added. No significant absorption between 400 and 750 nm was detected for the reaction buffer containing Fe2+ and 2-OG without JBP1.
TH Assay
Reaction conditions were 50 mm HEPES (pH 8.0), 50 mm NaCl, 8 mm ascorbic acid, 4 mm 2-OG, 1 mm Fe2SO4, 1 mm ATP, 20 μg/ml BSA, 0.5 mm DTT, 3.5 μg of telomeric duplex DNA substrate, and 2.2 μm JBP1 in a total reaction volume of 50 μl. DNA duplexes were made by boiling complementary telomere oligos (top strand 5′-TGGGATTTGGATTTGGATTTGGAT-3′; bottom strand 5′-ATCCCAATCCCAATCCCAATCCCA3′) for 5 min and allowed to cool overnight. Samples were incubated at 37 °C for 30 min, and DNA was isolated using the Qiagen nucleotide removal kit. Detection of 5-hydroxymethyl-2′-deoxyuridine (hmdU) was performed by immunoprecipitation of 32P-labeled mononucleotides using a hmdU-specific goat-polyclonal antibody (Abcam) as previously described (9) with the following modifications. DNA was digested with 0.2 units of bovine spleen phosphodiesterase (SPD) and 50 units of micrococcal nuclease (MNase) (Worthington) overnight at 37 °C, and nucleotides were phosphorylated with T4 polynucleotide kinase (New England Biolabs). Nucleotide substrate (3.5 μg) was incubated with antibody for 2 h at room temperature with agitation. One hundred microliters of magnetic beads (Genscript) were then added for an additional 2 h. After stringent washing, beads were added to scintillation fluid, and cpm was measured. Assays were performed in triplicate.
Hypoxia
To grow cells in a hypoxic environment, 50-ml culture flasks were placed in anaerobic chambers (BD Biosciences) with an Aneropak pouch (Mitsubishi Gas Chemical Co.). Generation of hypoxic environment was confirmed using indicator strip. For growing cells at 1 or 3% O2, culture flasks were placed in anaerobic chambers and gas-exchanged by an evacuation-replacement procedure using premixed 1 or 3% O2 with 5% CO2 and balanced nitrogen. A continuous flow of gas was then applied for at least 5 min followed by daily gassing. To maintain hypoxic environment, hypoxia-treated cells were harvested and lysed inside the anaerobic chamber (Coy) to avoid reoxygenation. Samples were then processed for RNA/DNA under atmospheric O2 conditions.
T. cruzi Invasion Assay
T. cruzi epimastigotes grown under hypoxic conditions as described above for 10 days were transferred to hypoxic Grace's insect media (Invitrogen; made anaerobic by vacuum degassing and flushing with argon) for 14 days to differentiate to infective metacyclics as previously described (4). Epimastigotes that were grown at 21% O2 were differentiated in a similar procedure but under atmospheric O2 conditions. Differentiated metacyclic parasites (from 21% O2 and hypoxic conditions) were then purified, equal numbers of infective metacyclic parasites were incubated with vero cells for 3 h, and parasite invasion was quantitated as previously described at 21% O2 conditions (4). Briefly, vero cells were plated on 13-mm round coverslips at a density of 3 × 104 cells in Eagle's Minimum Essential Medium with 10% fetal calf serum and cultivated in 24-well plates for 24 h at 37 °C in a 5% CO2 atmosphere and 21% oxygen. Coverslips with attached cells were then washed three times with phosphate-buffered saline (PBS) to remove the unattached cells and debris. Purified metacyclic forms were used for invasion assays. Purified metacyclics from each growth condition were centrifuged to remove the cell debris and seeded onto the Vero cells in equal numbers (5 × 106 parasites/well). After 3 h at 37 °C, the interaction was stopped by removing the parasites, and washing the cells three times with PBS. Monolayers were fixed and stained with Giemsa stain. Invasion was quantified using two methods. The percentage of infected cells was calculated by counting the number of intracellular parasites per coverslip and expressed as the number of parasites per 100 Vero cells. We also determined the average number of parasites per infected cell by counting the number of parasites in ∼90 infected Vero cells for each experiment.
Detection of hmdU by Mass Spectrometry
After the incubation of dsDNA substrate with or without JBP1 enzyme in the TH assay as described above, the purified DNA was digested to nucleosides and subjected to liquid chromatography-mass spectrometry (LC-MS) analysis. The enzymatic digestion of the duplex DNA substrate was conducted following previously described procedures with some modifications (23). Nuclease P1 (4 units), phosphodiesterase 2 (0.005 units), erythro-9-(2-hydroxy-3-nonyl) adenine (20 nmol), and a 20-μl solution containing 300 mm sodium acetate (pH 5.6) and 10 mm zinc chloride were added to the DNA and incubated at 37 °C for 48 h. To the digestion mixture were then added alkaline phosphatase (2 units), phosphodiesterase 1 (0.005 units), and 40 μl of 0.5 m Tris-HCl (pH 8.9). The digestion was continued at 37 °C for 2 h and subsequently neutralized with formic acid. To the mixture were then added 1.5 pmol of isotopically labeled hmdU (24). The enzymes in the digestion mixture were subsequently removed by chloroform extraction twice, and the resulting aqueous layer was subjected to off-line HPLC enrichment of hmdU. A 4.6 × 250-mm Alltima HP C18 column (5 μm in particle size, Grace Davison, Deerfield, IL) was used for the enrichment of hmdU from the enzymatic digestion products of DNA. The mobile phases were 10 mm ammonium formate in water (solution A) and methanol (solution B), and the flow rate was 1 ml/min. A gradient of 42 min at 0% B, 1 min at 0–5% B, 22 min at 5% B, 5 min at 5–20% B, and 10 min at 20% B was employed. A typical HPLC trace is shown in supplemental Fig. S3A. The HPLC fractions eluting at 18.0–24.0 min were pooled for hmdU. The collected fractions were dried in the SpeedVac, redissolved in H2O, and subjected for LC-MS/MS analysis.
For LC-MS/MS analysis, a 3.0 × 100-mm Hypersil GOLD column (particle size, 5 μm, Thermo Scientific) was used for the separation of the fractions containing hmdU, and the flow rate was 50 μl/min. A solution of 0.1% (v/v) formic acid in water (solution A) and a solution of 0.1% (v/v) formic acid in methanol (solution B) were employed as mobile phases, and a gradient of 35 min 0–70% at B, 1 min 70–0% at B, and 14 min at 0% B was used for the separation. The effluent from the LC column was directed to a TSQ Vantage triple quadrupole mass spectrometer (Thermo Fisher Scientific), which was set up for monitoring the fragmentation of the [M-H]− ions of the unlabeled and labeled hmdU in the multiple-reaction monitoring mode. The S-lens radio frequency amplitude was maintained at 97 V, the capillary temperature was maintained at 270 °C, and the vaporizer temperature was 153 °C. The sheath gas flow rate was 20 arbitrary units, the auxiliary gas flow rate was 0.3 arbitrary units, the spray voltage was 2.7 kV, and the scan time was 100 ms. The multiple-reaction monitoring transitions were m/z 257 → 124 and m/z 260 → 126 for unlabeled and labeled hmdU, respectively (supplemental Fig. S3, C and D). The collision energy was set at 17 V.
Determination of Genomic Level of J
To quantify the genomic J levels, DNA was isolated and utilized in the anti-J DNA immunoblot assay as described (25). Briefly, 2-fold serially diluted genomic DNA was blotted to nitrocellulose followed by incubation with anti-J antisera. Bound antibodies were detected by a secondary goat anti-rabbit antibody conjugated to HRP (horseradish peroxidase) and visualized by ECL (enhanced chemiluminescence). The membrane was stripped and hybridized with a probe for the β-tubulin gene to control for DNA loading.
Quantitative Reverse Transcription-PCR
Total RNA was obtained using Qiagen RNeasy kits according to the manufacturer's instructions. First-strand cDNA was synthesized from 1 μg of total RNA using an iScript cDNA synthesis kit (Bio-Rad) per the manufacturer's instructions. Heat-inactivated cDNA reaction mixtures were finally treated with RNase H at 37 °C for 45 min. Quantification of selected genes were performed on an iCycler with an iQ5 multicolor real-time PCR detection system (Bio-Rad). Primer sequences used in the analysis are available upon request. The reaction mixture contained 5 pmol of forward and reverse primer, 2× iQ SYBR Green super mix (Bio-Rad), and 2 μl of template cDNA. Standard curves were prepared for each gene using 10-fold dilutions of known quantity (15 ng/μl) of WT DNA. The quantities were calculated using iQ5 optical detection system software. Each sample was normalized to 24S rRNA (T. cruzi) or tubulin (L. major). Statistical analysis was performed using Student's t test. A value of p < 0.05 was considered significant.
RESULTS
JBP Is Fe2+/2-OG-dependent Dioxygenase Hydroxylating Thymine in dsDNA to hmU
We have previously demonstrated the ability to detect hmU-modified DNA using an anti-hmU pulldown assay that is highly specific for hmU over base J or unmodified thymidine (Ref. 9 and supplemental Fig. S1A). We now utilize this hmU detection system to set up an in vitro TH assay, incubating recombinant JBP1 with a 24-mer duplex oligodeoxynucleotide in the presence of Fe2+, 2-OG, and ascorbic acid. As predicted, incubation of JBP1 with duplex DNA resulted in the formation of hmU (Fig. 2C). The addition of JBP1 caused a significant increase in cpm, reflective of the production of hmU by >9-fold over background (no JBP1). A titration of increasing amounts of JBP1 protein corresponded with an increase in hmU formation, the peak of which was used for subsequent assays (supplemental Fig. S1B). To confirm the species of product generated in our in vitro TH reaction, we measured hmdU in the DNA substrate upon incubation with JBP1 using LC-MS/MS with the stable isotope-dilution method. In this LC-MS/MS method, we monitored the m/z transitions for unlabeled and labeled hmdU (26), respectively, in the multiple-reaction monitoring mode. We found that the level of hmdU in the DNA substrate after incubation with JBP1 was ∼7 lesions/104 dT, which was ∼7-fold higher than that in the DNA from the control reaction (Fig. 2D). Interestingly, the degree of hydroxylation in the presence of JBP1 over the negative control measured by LC-MS/MS is very similar to the increase detected by the hmU pulldown assay. These results demonstrate that JBP1 can induce the oxidation of thymine in duplex DNA to give hmU in vitro. The data also confirm the use of our anti-hmU pulldown assay for following in vitro hmU production.
FIGURE 2.

rJBP1 binds Fe2+ and stimulates hmU formation in vitro. A, shown is a schematic diagram of JBP1 proteins analyzed in the in vitro thymidine hydroxylase assay. The proposed thymidine hydroxylase domain (TH), homologous to members of the Fe2+/2-OG dependent dioxygenase family, is indicated by the white box. The J binding domain (J-DBD) is indicated by the dark box. The arrowhead within the J-DBD indicates the location of the essential residue for J-DNA binding (35). The black box at the N terminus indicates the His tag. The amino acid positions corresponding to the Lt JBP1 gene sequence are indicated along the top. The functionally conserved amino acid signature among the dioxygenase family is indicated below the WT JBP1, with residues involved in coordinating Fe2+ and 2-OG highlighted. In the JBP1 H189A/H239A mutant, the two metal binding ligands were mutated to alanine. JBP 1–450 represents a truncated version of the WT JBP1. B, JBP1 binds Fe2+ and 2-OG. JBP1 samples (250 μm) in 25 mm imidazole buffer (pH 7.0) were made anaerobic in a sealed cuvette and scanned, a mixture of 2-OG and ascorbate was added (final concentrations of 500 and 100 μm, respectively), and scans were recorded before and after adjusting to 2 mm ferrous ammonium sulfate. Difference absorption (Abs) spectra (with the spectrum of JBP1 subtracted) are depicted for the JBP1/2-OG and JBP1/2-OG/Fe2+ of WT JBP1 and JBP1/2-OG/Fe2+ of the JBP1 H189A/H239A mutant (Mutant). C, the TH assay was performed as described under “Experimental Procedures” using WT JBP1 (JBP1) and JBP1 H189A/H239A mutant. cpm counts, indicative of hmU formation, were read for each sample. All experiments were performed in triplicate, and error bars are representative of S.D. *, p < 0.001. D, LC MS/MS was performed to detect the levels of hmdU in duplex DNA after incubation in the absence or presence of JBP1. Data are expressed as the number of hmdU molecules formed per 104 thymidines. E, the TH assay was performed as in C but using JBP1 1–450. *, p < 0.01. F, the TH assay was performed using equal amounts of WT JBP1 and the 1–450 truncation to compare activity. Data are expressed as % activity relative to the WT-JBP1 control. G, equal amounts of the indicated protein (based on Bradford assay) were run on an 10% SDS-page gel and Coomassie-stained to confirm even loading.
Utilizing the hmU pulldown assay, we demonstrate that the TH domain is within the N terminus of JBP1. The N-terminal half of JBP1 (amino acids 1–450) is capable of hydroxylating thymidine (Fig. 2, A and E) and is as active as the full-length enzyme (Fig. 2, F and G). Thymine in the context of single-stranded DNA substrate, rather than duplex DNA, is not hydroxylated (Fig. 3A), demonstrating that the TH activity of JBP1 is specific for double-stranded DNA substrate. Consistent with other members of the Fe2+/2-OG-dependent dioxygenase family, optimal JBP1 activity requires O2, Fe2+, and 2-OG, with a significant reduction in thymidine hydroxylation seen when the assay is performed in the absence of each (Fig. 3, B–D). As observed for other dioxygenases (27), the TH activity of JBP1 was stimulated by ascorbate (data not shown), potentially reflecting the regeneration of Fe2+ from Fe3+ (28).
FIGURE 3.

JBP1 is an Fe2+/2-OG-dependent dioxygenase enzyme. The TH assay was performed using WT JBP1 in the presence of either single stranded (ss) or double stranded (ds) substrate (A) or the presence or absence of O2 (B), 2-OG (C), or Fe2+ (D). Error bars represent S.D. of triplicate experiments. *, p < 0.01.
The requirement for both Fe2+ and 2-OG for optimal thymidine hydroxylation along with the stimulation by ascorbate strongly supports the proposal that JBP1 is a member of the dioxygenase family. Direct evidence that JBP1 binds Fe2+ and 2-OG was provided by examining the absorption spectrum of the anaerobic protein in the presence and absence of Fe2+ and 2-OG. The protein with bound Fe2+ and cofactor had an absorption peak at 530 nm (Fig. 2B), similar to the transition observed in other family members. This chromophore is due to a weak charge transfer from Fe2+ to 2-OG and is a spectroscopic signature of Fe2+/2-OG-dependent dioxygenases (12). The two highly conserved histidine metal binding ligands in the core jelly roll domain of Fe2+/2-OG enzymes have been shown to be essential for Fe2+ binding and hydroxylase function (12, 17). Consistent with this finding, mutation of residues H189A and H239A in JBP1 ablated J synthesis in vivo (8). We see here that in vitro Fe2+ binding and thymidine hydroxylation is ablated in the H189A/H239A mutant, demonstrating that Fe2+ binding is critical for TH activity of JBP1 (Fig. 2, B and C). Taken together, we have unequivocally shown that JBP1 is a Fe2+/2-OG-dependent dioxygenase, requiring Fe2+, O2, and 2-OG for activity. The data also support the previous sequence and in vivo analyses, indicating the TH domain at the N terminus of JBP1 and JBP2.
Dioxygenase Inhibitors Reduce JBP Activity Both in Vitro and in Vivo
Many inhibitors of Fe2+/2-OG dioxygenases have been well characterized (17). Two of these characterized inhibitors, DMOG and 2,4-pyridinedicarboxylic acid hydrate, are competitive inhibitors of 2-OG. These compounds have been shown to be effective inhibitors of several members of the Fe2+/2-OG dioxygenases (17) including (but by no means limited to) the DNA modifying enzymes such as fat mass and obesity-associated protein (FTO) (29). When the JBP1 TH assay was performed in the presence of either DMOG or 2,4-pyridinedicarboxylic acid hydrate, we saw a highly significant inhibition of JBP1 activity (Fig. 4A and data not shown). Also consistent with other dioxygenases, we saw inhibition with succinate by product inhibition (Fig. 4B).
As base J is the downstream product of thymidine hydroxylation in vivo, we predicted that the treatment of J containing kinetoplastids with these cell-permeable inhibitors would result in a reduction in J biosynthesis. Indeed the treatment of T. brucei, L. major, and T. cruzi with DMOG caused a significant inhibition in J synthesis (Fig. 4, C—E and supplemental Fig. 2). The ability to generate a cell line that lacks base J in T. brucei after the inhibition of JBP TH activity with DMOG is consistent with our ability to delete both enzymes and the apparent nonessential nature of base J in this parasite (9). The generation of J null cells through DMOG feeding of T. brucei also demonstrates that both JBP1 and JBP2 are 2-OG dioxygenase enzymes. Similarly, DMOG reduces J synthesis in the JBP1KO and JBP2 KO T. cruzi cell lines (data not shown).
JBP Activity Is Regulated by Oxygen Levels in Vivo
Given that O2 is critical for JBP1 to hydroxylate thymidine in vitro (Fig. 3B), we hypothesized that O2 levels would impact J synthesis in vivo. To address this, we grew T. cruzi and L. major at atmospheric O2 concentrations (21%) and under low O2 (1%) or hypoxic (<0.1%) conditions. Consistent with inhibition of JBP TH activity and loss of J after DNA replication, we saw significant reductions in J synthesis in T. cruzi when grown under 1% O2 and <1% hypoxic conditions (Fig. 5A). We did not detect any change in mRNA levels of the JBP enzymes in hypoxia, and J synthesis was fully restored within 30 min of re-exposure of the cells to 21% O2 (Fig. 5, A and B). This supports the reduction in J biosynthesis being at the level of inhibition of TH activity rather than a down-regulation of the synthesis machinery and that JBP-stimulated thymidine hydroxylation and thus J synthesis occurs independently of DNA replication. Reduced O2 levels affected the enzymatic activity of both JBP1 and JBP2 in T. cruzi, as significant reductions in J levels were seen in both JBP1KO and JBP2 KO cell lines (Fig. 5C). Low O2 had similar effects on JBP function in L. major (supplemental Fig. S4, A and B). Our inability to grow T. brucei in 3% or less O2 conditions (data not shown) precluded similar analyses of JBP function in this parasite.
FIGURE 5.

JBP as oxygen sensors in vivo regulating J synthesis and virulence. A, T. cruzi epimastigotes were grown in atmospheric (21% O2), 1% O2, and hypoxic (<0.1 O2) conditions for 10 days. A sample of cells grown at <0.1% O2 was re-exposed to 21% O2 for 30 min (R). DNA was isolated, and J levels were assessed by anti-J dot blot. Blots were stripped and probed for tubulin as a loading control. B, RNA was isolated from the same T. cruzi cells, and the relative amounts of mRNA for JBP1 and JBP2 were calculated by quantitative RT-PCR as described under “Experimental Materials.” Black bars, 21%; white bars, <0.1% O2. Values were normalized to 24 S control. C, T. cruzi cells that have either JBP1 (JBP1KO) or JBP2 (JBP2KO) deleted from the genome were grown in the indicated oxygen conditions, and levels of J were detected as described in A. D and E, epimastigotes grown in 21% or <0.1% O2 were then differentiated into metacyclics and purified, and equal numbers of differentiated metacyclic parasites were allowed to invade mammalian cells in the presence of 21% O2 as described under “Experimental Procedures.” Slides were then Giemsa-stained and assessed for the presence of intracellular parasites. Data are expressed as the number of parasites per 100 cells (D) and the number of parasites per infected cell (E). Error bars represent the S.D. of triplicate experiments. *, p < 0.0001.
We have previously demonstrated that reduced J levels in T. cruzi results in increased Pol II recruitment to promoter regions and an increase in the rate of transcription initiation, which in turn causes global changes in gene expression (3, 4). Notably, surface proteins involved in parasite pathogenesis (including members of the trans-sialidase and mucin gene family) are significantly affected during reduced J levels. Accordingly, we measured a 5-fold increase in the ability of T. cruzi to invade the mammalian cell after the 20-fold reduction in base J (4). Therefore, we hypothesized that the 5-fold reduction in J levels associated with cells grown in hypoxic conditions would also result in an enhanced invasion phenotype. When T. cruzi epimastigotes were grown under hypoxia for 10 days and differentiated to infective metacyclics, and the mammalian cell invasion assay was performed in 21% O2, we saw a significant increase in both the percent of host cells infected as well as the number of parasites within each cell compared with parasites grown and differentiated under 21% O2 (Fig. 5, D and E). Taken together, the data suggest the JBP TH enzymes may act as O2 sensors, allowing the parasite to respond to changes in O2 levels, and regulate J synthesis, gene expression, and (among other phenotypes) virulence.
DISCUSSION
Sequence analysis indicated that JBP1/2 contain a Fe2+/2-OG-dependent dioxygenase-like domain at the N termini, including the presence of four conserved catalytic residues, critical in binding nonheme iron, 2-OG, and enzyme activity (12). The recent identification and characterization of the TET enzymes catalyzing the hydroxylation of 5-MeC in DNA strengthened the identification of JBPs as dioxygenases (11), characterizing them as members of the new TET/JBP subfamily of Fe2+/2-OG dioxygenase (10, 11). However, no direct analysis of JBP thymidine hydroxylation has been demonstrated despite a clear in vivo gene knock out and mutagenesis studies demonstrating the importance of JBP1 and JBP2 in J biosynthesis (8, 9, 30). The oxidation of thymidine residues by a thymidine hydroxylase is unusual. Thymine hydroxylase enzymes that oxidize the free base are known (31, 32) but thymidine hydroxylases that oxidize the base in DNA have not previously been characterized. Here we utilize an in vitro thymidine hydroxylase assay and in vivo inhibition studies to provide direct evidence for hydroxylation of T-residues in DNA during J synthesis by JBP1 and JBP2 and identify them as members of the Fe2+/2-OG family of dioxygenases. We also now provide conclusive evidence for the two-step J biosynthesis mechanism (Fig. 1) by demonstrating the ability of JBP to modify T-residues specifically in dsDNA substrate in vitro and regulating J synthesis in a DNA replication-independent manner in vivo.
The in vitro TH assay clearly demonstrates JBP1 is a Fe2+/2-OG dioxygenase that requires Fe2+, 2-OG, and O2 for hydroxylating thymine in the context of dsDNA. Extension of these analyses in vivo indicates both JBP1 and JBP2 utilize similar co-factors for T-hydroxylation and J synthesis. The sensitivity of J synthesis in various WT and JBP KO kinetoplastid cell lines to 2-OG inhibitors and hypoxia confirms that both JBPs are Fe2+/2-OG dioxygenase enzymes. This is further supported by the ability of both JBP1 and JBP2 to stimulate hmU modification of genomic DNA when expressed in Escherichia coli.3 The rapid generation of a cell line that completely lacks base J upon DMOG treatment of T. brucei supports the nonessential nature of the modified base in this species as demonstrated by our previous generation of a J-null trypanosome by deleting both JBP1 and JBP2 (9). The inability to delete both JBP1 and JBP2 in T. cruzi (3, 4) and inability to delete JBP1 from L. tarentolae (33) have suggested the essential nature of J in these species. Future studies will utilize DMOG to elucidate the essential nature and function of J in these and other J-containing organisms.
The hydroxylation activity of JBP1 is specific for dsDNA, correlating with the specificity of J-DNA binding domain at the C terminus of JBP1 (34, 35). Inactivation of the N-terminal TH domain has no effect on J-DNA binding (8). Thus, JBP1 binding to J-DNA is independent from TH activity. We demonstrate here that the TH domain at the N terminus is functional in vitro without the C-terminal J-DNA binding domain. However, it is possible that in vivo TH activity of the JBPs is stimulated by their C-terminal domain. In fact, the low level of in vitro conversion of thymidine residues suggests that the ability of JBP1 to bind J residues in dsDNA may enhance TH activity. This “propagation” activity may represent an essential function for JBP1 in vivo (7, 35). We have previously demonstrated that JBP1 and JBP2 operate optimally in different chromatin environments; JBP1 at internal regions involved in Pol II transcription and JBP2 within telomeric-localized repetitive DNA (2). Presumably this functional difference is due to distinct C-terminal domains on each enzyme, including the SWI/SNF2 chromatin remodeling domain in JBP2 (2, 5, 7). The apparent low activity of JBP1-stimulated T-hydroxylation on synthetic DNA may reflect the optimal substrate for these enzymes in vivo is chromatin or, as mentioned above specifically for JBP1, the stimulatory effects of initial levels of J. Furthermore, we cannot rule out the possibility that additional factors stimulate JBP recruitment and/or TH activity in vivo.
As documented for other Fe2+/2-OG oxygenases, our analyses indicated a role of intracellular levels of 2-OG and succinate in regulating thymidine hydroxylation by JBP. The requirement of 2-OG and inhibition by succinate in JBP1-stimulated TH activity indicate the 2-OG/succinate ratio within the parasite as a critical factor for J synthesis and may help explain the lack of base J in the procyclic T. brucei life stage (36). It is well characterized that procyclic T. brucei cells, in contrast to the long-slender bloodstream life stage that contains base J, produce high concentrations of succinate representing incompletely oxidized product of aerobic fermentation (37, 38). The pathway(s) leading to lifecycle-specific production of succinate are not well understood, but the unique absence of a tricarboxylic acid cycle in bloodstream form T. brucei is presumably a key factor (39, 40). Our data suggest that the developmental increase in succinate within the parasite residing in the tsetse fly (and associated shift in the 2-OG/succinate ratio) would prevent optimal hydroxylation of thymidine by the Fe2+/2-OG dioxygenases, reducing J synthesis and presumably leading to the eventual down-regulation of JBP expression. Interestingly, upon re-expression of identical levels of JBP1 enzyme in the procyclic and bloodstream form trypanosome, an 800-fold decrease in the succinate/2-OG ratio in bloodstream versus procyclic parasites corresponds to an ∼15-fold increase in J synthesis in bloodstream forms.4 The response of JBP TH activity and J synthesis to levels of O2, 2-OG, and succinate implicate a role for metabolic flux as well as the environment to which the parasite is exposed in epigenetic regulation of gene expression in these human pathogens.
The functional dependence on O2 has implicated a number of Fe2+/2-OG dependent dioxygenase enzymes to operate as O2 sensors, playing crucial roles in cellular function (14). Perhaps the best studied is prolyl hydroxylase, which is an Fe2+/2-OG dioxygenase responsible for hydroxylation of the transcription factor, hypoxia-inducible factor on its proline residues at positions 402 and 564 (41). Hydroxylated hypoxia-inducible factor-α is then recognized by the VHL subunit of the E3(UBC)-ubiquitin ligase followed by degradation within the 26 S proteasome. However, hypoxia renders the prolyl hydroxylase inactive, and therefore, hypoxia-inducible factor-α is stabilized, allowing translocation to the nucleus, where it dimerizes with hypoxia-inducible factor-β, regulating gene transcription (for review, see Ref. 41 and 42). These changes in gene transcription include genes that allow cell adaptation and survival in a hypoxic environment, such as genes involved in glycolysis or angiogenesis (43).
Oxygen occurs in varying concentrations within the human host, ranging from 0 to 21% (anoxic and aerobic environments) (44). Not only do O2 levels vary between human tissues but also an O2 gradient within the tissue depending upon the proximity to blood vessels and to the O2 consumptive activity of the cell. As a consequence, in contrast to human cells, pathogens have evolved to adapt and prevail in a diverse array of oxygen concentrations. This adaptation is especially important for pathogens, such as T. cruzi, that experience varying oxygen concentrations from high levels on the skin to low levels within various tissues (i.e. gut and muscle) of the human host and insect vector.
We have shown that in T. cruzi, decreased J levels after the loss of JBP function result in increased Pol II recruitment and transcription initiation and corresponding genome wide changes in gene expression, including genes involved in pathogenesis (4). This in turn has direct effects on the host-parasite relationship, with increased mammalian cell invasion and delayed egress (4). These data combined with the analysis of JBP TH activity presented here suggest JBP1/2 can act as O2 sensors in vivo to modulate J biosynthesis and gene expression, allowing the parasite to adapt to changing conditions within the mammalian host. To examine this hypothesis, we exposed T. cruzi parasites to hypoxia. To rule out the potential effects of the mammalian cell hypoxia-inducible factor response to hypoxia, the invasion assay itself was performed at 21% O2. Therefore, rapid recovery of TH activity and J synthesis upon re-exposure of the cells to 21% O2 (Fig. 5) during the 3-h T. cruzi invasion assay may have reduced the effects of hypoxia. Regardless, the data clearly demonstrate a parasite response to low O2 in both levels of J synthesis and virulence phenotypes we have previously directly linked to epigenetic regulation by J (4). Therefore, we propose that the thymidine hydroxylase enzymes, JBP1 and JBP2, can act as O2 sensors allowing the parasite to respond to changes in O2 levels and regulate J synthesis, thus providing at least one mechanism of hypoxic signaling regulating gene expression and, among other potential phenotypes, virulence. Although we have not fully characterized the sensing capability of JBP by measuring the Km for O2, we do demonstrate significantly reduced enzyme activity (i.e. J synthesis) upon shifting the parasite to physiologically relevant low oxygen tensions. Although previous analysis of JBP loss of function mutants supports the proposed model of hypoxia regulation of T. cruzi virulence via JBP, we acknowledge that the current data do not rule out other potential O2 sensing pathways influencing gene expression and corresponding cellular response. This is especially relevant when we consider trypanosomes encode putative JmjC domain-containing dioxygenase enzymes that may serve as histone lysine demethylases. To better describe the mechanistic link between hypoxic signaling and JBP function, future experiments will explore the relationship between length of time parasites are grown under different levels of O2 and corresponding changes in J synthesis, chromatin structure, Pol II occupancy, transcription initiation, and gene expression profiles.
The ability of O2 to modulate epigenetic regulation of gene expression is an important factor to consider when evaluating transcriptome profiles of various parasite life stages. All studies thus far grow kinetoplastids under atmospheric 21% O2, conditions that are far removed from physiological normoxia. Because changes in levels of base J significantly affect the transcription profile of T. cruzi and J levels are responsive to O2 levels, we believe care and consideration should be given when evaluating transcriptional profiling of kinetoplast parasites grown at 21% O2, as this may well not reflect the “true” transcriptome profile of the parasite life stage. The epigenetic control of gene expression and its regulation through O2 levels and other metabolites present exciting new possibilities for Pol II regulatory mechanisms in early-divergent organisms where transcriptional regulation is poorly understood.
Acknowledgments
We especially thank Bill Lanzollotta, John Demmick and other members of the Lanzilotta laboratory for assistance and use of their anaerobic chamber and spectrophotometer. Robert Arnold provided assistance in the initial hypoxia experimental design. We also thank Marion Marshall and Timothy Southern for preliminary work on development of hmU IP assay and rJBP1 protein purification. We also thank Steve Hajduk, Rudo Kieft, Piet Borst, and John Harrington for useful comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants 2R56AI063523-07A1 (to R. S.) and R01 CA101864 (to Y. W.).

This article contains supplemental Figs. 1–4.
T. Southern, M. Marshall, and R. Sabatini, unpublished results.
L. J. Cliffe and R. Sabatini, unpublished data.
- base J
- β-d-glucopyranosyloxymethyluracil
- 2-OG
- 2-oxoglutarate
- DMOG
- dimethyloxoglycine
- TH
- thymidine hydroxylase
- JBP
- base J-binding protein
- rJBP1
- recombinant JBP1
- hmU
- 5-hydroxymethyluridine
- hmdU
- 5-hydroxymethyl-2′deoxyuridine
- Pol
- polymerase
- TET
- ten-eleven translocation-1 protein.
REFERENCES
- 1. Borst P., Sabatini R. (2008) Base J. Discovery, biosynthesis, and possible functions. Annu. Rev. Microbiol. 62, 235–251 [DOI] [PubMed] [Google Scholar]
- 2. Cliffe L. J., Siegel T. N., Marshall M., Cross G. A., Sabatini R. (2010) Two thymidine hydroxylases differentially regulate the formation of glucosylated DNA at regions flanking polymerase II polycistronic transcription units throughout the genome of Trypanosoma brucei. Nucleic Acids Res. 38, 3923–3935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ekanayake D., Sabatini R. (2011) Epigenetic regulation of polymerase II transcription initiation in Trypanosoma cruzi. Modulation of nucleosome abundance, histone modification, and polymerase occupancy by O-linked thymine DNA glucosylation. Eukaryot. Cell 10, 1465–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ekanayake D. K., Minning T., Weatherly B., Gunasekera K., Nilsson D., Tarleton R., Ochsenreiter T., Sabatini R. (2011) Epigenetic regulation of transcription and virulence in Trypanosoma cruzi by O-linked thymine glucosylation of DNA. Mol. Cell. Biol. 31, 1690–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sabatini R. C., L. J., Vainio S., Borst P. (2008) in DNA and RNA Modification Enzymes; Comparative Structure, Mechanism, Function, Cellular Interactions. and Evolution (Grosjean H., ed) pp. 120–131, Landes Biosciences, Austin, TX [Google Scholar]
- 6. Cross M., Kieft R., Sabatini R., Wilm M., de Kort M., van der Marel G. A., van Boom J. H., van Leeuwen F., Borst P. (1999) The modified base J is the target for a novel DNA-binding protein in kinetoplastid protozoans. EMBO J. 18, 6573–6581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DiPaolo C., Kieft R., Cross M., Sabatini R. (2005) Regulation of trypanosome DNA glycosylation by a SWI2/SNF2-like protein. Mol. Cell 17, 441–451 [DOI] [PubMed] [Google Scholar]
- 8. Yu Z., Genest P. A., ter Riet B., Sweeney K., DiPaolo C., Kieft R., Christodoulou E., Perrakis A., Simmons J. M., Hausinger R. P., van Luenen H. G., Rigden D. J., Sabatini R., Borst P. (2007) The protein that binds to DNA base J in trypanosomatids has features of a thymidine hydroxylase. Nucleic Acids Res. 35, 2107–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cliffe L. J., Kieft R., Southern T., Birkeland S. R., Marshall M., Sweeney K., Sabatini R. (2009) JBP1 and JBP2 are two distinct thymidine hydroxylases involved in J biosynthesis in genomic DNA of African trypanosomes. Nucleic Acids Res. 37, 1452–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Iyer L. M., Tahiliani M., Rao A., Aravind L. (2009) Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 8, 1698–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tahiliani M., Koh K. P., Shen Y., Pastor W. A., Bandukwala H., Brudno Y., Agarwal S., Iyer L. M., Liu D. R., Aravind L., Rao A. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hausinger R. P. (2004) Fe(II)/α-ketoglutarate-dependent hydroxylases and related enzymes. Crit. Rev. Biochem. Mol. Biol. 39, 21–68 [DOI] [PubMed] [Google Scholar]
- 13. Vainio S., Genest P. A., ter Riet B., van Luenen H., Borst P. (2009) Evidence that J-binding protein 2 is a thymidine hydroxylase catalyzing the first step in the biosynthesis of DNA base J. Mol. Biochem. Parasitol. 164, 157–161 [DOI] [PubMed] [Google Scholar]
- 14. Ozer A., Bruick R. K. (2007) Non-heme dioxygenases. Cellular sensors and regulators jelly rolled into one? Nat. Chem. Biol. 3, 144–153 [DOI] [PubMed] [Google Scholar]
- 15. Kaelin W. G., Jr., Ratcliffe P. J. (2008) Oxygen sensing by metazoans. The central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402 [DOI] [PubMed] [Google Scholar]
- 16. Loenarz C., Schofield C. J. (2008) Expanding chemical biology of 2-oxoglutarate oxygenases. Nat. Chem. Biol. 4, 152–156 [DOI] [PubMed] [Google Scholar]
- 17. Rose N. R., McDonough M. A., King O. N., Kawamura A., Schofield C. J. (2011) Inhibition of 2-oxoglutarate-dependent oxygenases. Chem. Soc. Rev. 40, 4364–4397 [DOI] [PubMed] [Google Scholar]
- 18. Ito S., D'Alessio A. C., Taranova O. V., Hong K., Sowers L. C., Zhang Y. (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal, and inner cell mass specification. Nature 466, 1129–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kapler G. M., Coburn C. M., Beverley S. M. (1990) Stable transfection of the human parasite Leishmania major delineates a 30-kb region sufficient for extrachromosomal replication and expression. Mol. Cell. Biol. 10, 1084–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilson D. M., 3rd, Takeshita M., Demple B. (1997) Abasic site binding by the human apurinic endonuclease, Ape, and determination of the DNA contact sites. Nucleic Acids Res. 25, 933–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sigman D. S., Kuwabara M. D., Chen C. H., Bruice T. W. (1991) Nuclease activity of 1,10-phenanthroline-copper in study of protein-DNA interactions. Methods Enzymol. 208, 414–433 [DOI] [PubMed] [Google Scholar]
- 22. Ryle M. J., Padmakumar R., Hausinger R. P. (1999) Stopped-flow kinetic analysis of Escherichia coli taurine/α-ketoglutarate dioxygenase. Interactions with α-ketoglutarate, taurine, and oxygen. Biochemistry 38, 15278–15286 [DOI] [PubMed] [Google Scholar]
- 23. Wang J., Yuan B., Guerrero C., Bahde R., Gupta S., Wang Y. (2011) Quantification of oxidative DNA lesions in tissues of Long-Evans cinnamon rats by capillary high performance liquid chromatography-tandem mass spectrometry coupled with stable isotope-dilution method. Anal. Chem. 83, 2201–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hong H., Cao H., Wang Y., Wang Y. (2006) Identification and quantification of a guanine-thymine intrastrand cross-link lesion induced by Cu(II)/H2O2/ascorbate. Chem. Res. Toxicol. 19, 614–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Leeuwen F., Wijsman E. R., Kieft R., van der Marel G. A., van Boom J. H., Borst P. (1997) Localization of the modified base J in telomeric VSG gene expression sites of Trypanosoma brucei. Genes Dev. 11, 3232–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Frelon S., Douki T., Ravanat J. L., Pouget J. P., Tornabene C., Cadet J. (2000) High performance liquid chromatography-tandem mass spectrometry measurement of radiation-induced base damage to isolated and cellular DNA. Chem. Res. Toxicol. 13, 1002–1010 [DOI] [PubMed] [Google Scholar]
- 27. Clifton I. J., McDonough M. A., Ehrismann D., Kershaw N. J., Granatino N., Schofield C. J. (2006) Structural studies on 2-oxoglutarate oxygenases and related double-stranded β-helix fold proteins. J. Inorg. Biochem. 100, 644–669 [DOI] [PubMed] [Google Scholar]
- 28. Trewick S. C., Henshaw T. F., Hausinger R. P., Lindahl T., Sedgwick B. (2002) Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature 419, 174–178 [DOI] [PubMed] [Google Scholar]
- 29. Gerken T., Girard C. A., Tung Y. C., Webby C. J., Saudek V., Hewitson K. S., Yeo G. S., McDonough M. A., Cunliffe S., McNeill L. A., Galvanovskis J., Rorsman P., Robins P., Prieur X., Coll A. P., Ma M., Jovanovic Z., Farooqi I. S., Sedgwick B., Barroso I., Lindahl T., Ponting C. P., Ashcroft F. M., O'Rahilly S., Schofield C. J. (2007) The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science 318, 1469–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kieft R., Brand V., Ekanayake D. K., Sweeney K., DiPaolo C., Reznikoff W. S., Sabatini R. (2007) JBP2, a SWI2/SNF2-like protein, regulates de novo telomeric DNA glycosylation in bloodstream form Trypanosoma brucei. Mol. Biochem. Parasitol. 156, 24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holme E., Lindstedt G., Lindstedt S., Tofft M. (1970) 7-Hydroxylation of thymine in a Neurospora strain coupled to oxidative decarboxylation of 2-ketoglutarate. Biochim. Biophys. Acta 212, 50–57 [DOI] [PubMed] [Google Scholar]
- 32. Simmons J. M., Müller T. A., Hausinger R. P. (2008) Fe(II)/α-ketoglutarate hydroxylases involved in nucleobase, nucleoside, nucleotide, and chromatin metabolism. Dalton Trans. 38, 5132–5142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Genest P. A., ter Riet B., Dumas C., Papadopoulou B., van Luenen H. G., Borst P. (2005) Formation of linear inverted repeat amplicons after targeting of an essential gene in Leishmania. Nucleic Acids Res. 33, 1699–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sabatini R., Meeuwenoord N., van Boom J. H., Borst P. (2002) Recognition of base J in duplex DNA by J-binding protein. J. Biol. Chem. 277, 958–966 [DOI] [PubMed] [Google Scholar]
- 35. Heidebrecht T., Christodoulou E., Chalmers M. J., Jan S., Ter Riet B., Grover R. K., Joosten R. P., Littler D., van Luenen H., Griffin P. R., Wentworth P., Jr., Borst P., Perrakis A. (2011) The structural basis for recognition of base J containing DNA by a novel DNA binding domain in JBP1. Nucleic Acids Res. 39, 5715–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van Leeuwen F., Dirks-Mulder A., Dirks R. W., Borst P., Gibson W. (1998) The modified DNA base β-d-glucosyl-hydroxymethyluracil is not found in the tsetse fly stages of Trypanosoma brucei. Mol. Biochem. Parasitol. 94, 127–130 [DOI] [PubMed] [Google Scholar]
- 37. Tielens A. G., van Hellemond J. J. (2009) Surprising variety in energy metabolism within Trypanosomatidae. Trends Parasitol. 25, 482–490 [DOI] [PubMed] [Google Scholar]
- 38. Cazzulo J. J. (1992) Energy metabolism in Trypanosoma cruzi. Subcell. Biochem. 18, 235–257 [DOI] [PubMed] [Google Scholar]
- 39. Fairlamb A. H., Henderson G. B., Cerami A. (1986) The biosynthesis of trypanothione and N1-glutathionylspermidine in Crithidia fasciculata. Mol. Biochem. Parasitol. 21, 247–257 [DOI] [PubMed] [Google Scholar]
- 40. Michels P. A., Hannaert V., Bringaud F. (2000) Metabolic aspects of glycosomes in trypanosomatidae. New data and views. Parasitol. Today 16, 482–489 [DOI] [PubMed] [Google Scholar]
- 41. Masson N., Ratcliffe P. J. (2003) HIF prolyl and asparaginyl hydroxylases in the biological response to intracellular O2 levels. J. Cell Sci. 116, 3041–3049 [DOI] [PubMed] [Google Scholar]
- 42. Majmundar A. J., Wong W. J., Simon M. C. (2010) Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 40, 294–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wenger R. H. (2000) Mammalian oxygen sensing, signaling, and gene regulation. J. Exp. Biol. 203, 1253–1263 [DOI] [PubMed] [Google Scholar]
- 44. Ernst J. F., Tielker D. (2009) Responses to hypoxia in fungal pathogens. Cell. Microbiol. 11, 183–190 [DOI] [PubMed] [Google Scholar]