

Background: Improved myofilament Ca2+ sensitivity alleviates defects in thin filament bearing disease-causing mutations.
Results: By engineering the cardiac muscle Ca2+ sensor troponin C, aberrant myofilament Ca2+ sensitivity can be corrected in vitro.
Conclusion: Engineered TnC provides a novel and versatile avenue to reset disease-related myofilament Ca2+ sensitivity.
Significance: Engineered TnC could be a new therapeutic strategy for cardiac muscle diseases.
Keywords: Calcium, Calcium-binding Proteins, Cardiac Muscle, Cardiomyopathy, Troponin, EF-hand
Abstract
Aberrant myofilament Ca2+ sensitivity is commonly observed with multiple cardiac diseases, especially familial cardiomyopathies. Although the etiology of the cardiomyopathies remains unclear, improving cardiac muscle Ca2+ sensitivity through either pharmacological or genetic approaches shows promise of alleviating the disease-related symptoms. Due to its central role as the Ca2+ sensor for cardiac muscle contraction, troponin C (TnC) stands out as an obvious and versatile target to reset disease-associated myofilament Ca2+ sensitivity back to normal. To test the hypothesis that aberrant myofilament Ca2+ sensitivity and its related function can be corrected through rationally engineered TnC constructs, three thin filament protein modifications representing different proteins (troponin I or troponin T), modifications (missense mutation, deletion, or truncation), and disease subtypes (familial or acquired) were studied. A fluorescent TnC was utilized to measure Ca2+ binding to TnC in the physiologically relevant biochemical model system of reconstituted thin filaments. Consistent with the pathophysiology, the restrictive cardiomyopathy mutation, troponin I R192H, and ischemia-induced truncation of troponin I (residues 1–192) increased the Ca2+ sensitivity of TnC on the thin filament, whereas the dilated cardiomyopathy mutation, troponin T ΔK210, decreased the Ca2+ sensitivity of TnC on the thin filament. Rationally engineered TnC constructs corrected the abnormal Ca2+ sensitivities of the thin filament, reconstituted actomyosin ATPase activity, and force generation in skinned trabeculae. Thus, the present study provides a novel and versatile therapeutic strategy to restore diseased cardiac muscle Ca2+ sensitivity.
Introduction
Prokaryotes, eukaryotes, and even viruses utilize Ca2+ and Ca2+-binding proteins to perform specific duties (1–3). The most common motif used by proteins to bind Ca2+ is the EF-hand (4). EF-hand proteins help perform cellular functions by maintaining the structural integrity of multimeric protein complexes, altering protein interactions like switches, or simply buffering Ca2+ (5, 6). Ultimately, these Ca2+-dependent processes are controlled by the Ca2+ signal and the Ca2+ binding properties of the protein (7, 8). A prime example of such behavior is the increased amplitude and decreased duration of cardiac muscle contraction after a surge of adrenaline. In this case, both the Ca2+ transient profile and the Ca2+-dependent response of the protein are complimentarily modulated to affect contraction and relaxation (9, 10).
In the heart, troponin C (TnC)2 is the Ca2+-dependent, switch-like protein that helps regulate force development as an integral part of the contractile machinery (5). Different isoforms of the troponin complex (Tn) within an organism and between different species help to tune the response of TnC to Ca2+ to meet developmental and environmental demands of the heart (11–15). In this regard, TnC does not behave like a simple switch because the Ca2+ binding properties of the regulatory EF-hand of TnC are modulated by interactions with its protein binding partner, troponin I (TnI) (16, 17). The response of TnC to Ca2+ can be further adjusted by additional myofilament proteins (troponin T, actin, tropomyosin, and myosin) and by an assortment of posttranslational modifications to many of these proteins (5, 17–19). Thus, it would appear that TnC acts as a central hub converging information from the myofilament proteins to tune its response to the Ca2+ signal (5). Unfortunately, in many inherited and acquired cardiac diseases, the proper tuning of TnC to Ca2+ is disturbed (18, 20, 21).
Wide assortments of mutations, deletions, truncations, and aberrant posttranslational modifications of numerous myofilament proteins have been associated with various cardiac diseases (21–23). Alterations in the Ca2+ sensitivity of TnC and force development have been commonly observed to be one of many problems that arise in these complex disorders (20, 21, 23–26). Strikingly, for any particular class of inherited cardiomyopathies, the apparent Ca2+ sensitivity of TnC and force development are typically altered in a qualitatively similar manner (20, 22, 25). Furthermore, pharmacological and genetic interventions that rectify the apparent Ca2+ sensitivity of cardiac muscle in transgenic animal models harboring cardiomyopathic genes show promise of alleviating the disease symptoms (27–29). For instance, modulating TnI, TnT, or tropomyosin (each of which can indirectly tune the Ca2+ sensitivity of TnC) counteracted the abnormal cardiac muscle Ca2+ sensitivities and ameliorated the disease symptoms (28–30). Ultimately, correcting the aberrant Ca2+ sensitivity may be part of an integrative approach to improving cardiac function in these complex cardiomyopathies (26).
Due to its central role as the Ca2+ sensor for cardiac muscle contraction, TnC stands out as an obvious and more versatile target to genetically modulate cardiac muscle Ca2+ sensitivity. Unfortunately, there are no pharmacological compounds that just target TnC (31). However, by directly engineering TnC, our laboratory has developed several TnC constructs, which behave as Ca2+ sensitizers or desensitizers in biochemical model systems and in muscle (16, 32, 33). By utilizing different design principles (32, 33), the intrinsic Ca2+ binding properties of TnC can be finely or grossly tuned. We have rationally engineered TnC to test whether the Ca2+ dependence of biochemical and physiological systems harboring disease-associated protein modifications could be reset. We chose to test this idea using three disease-related protein modifications (inherited and acquired) in TnI and TnT that exhibit physiologically abnormal increased or decreased Ca2+ sensitivities. We demonstrate that by specifically adjusting the Ca2+ binding properties of TnC, both the aberrant biochemical and the aberrant physiological Ca2+ sensitivity of the cardiac myofilaments can be corrected.
EXPERIMENTAL PROCEDURES
Materials
Phenyl-Sepharose CL-4B, sodium molybdate dihydrate, and EGTA were purchased from Sigma. IAANS and phalloidin were purchased from Invitrogen. Affi-Gel 15 affinity medium was purchased from Bio-Rad. Malachite green oxalate was purchased from Fisher Scientific.
Mutagenesis
TnC, TnI, and TnT mutants were constructed from their respective pET3a expression plasmids by primer-based site-directed mutagenesis and confirmed by DNA sequence analysis.
Protein Expression and Purification
The plasmid encoding human cardiac TnC was transformed into Escherichia coli BL21(DE3)pLysS cells (Novagen, San Diego, CA), whereas those for TnI and TnT were transformed into RosettaTM(DE3)pLysS cells (Novagen). TnC, TnI, and TnT were purified as described previously (15, 16). Rabbit skeletal actin and bovine ventricular cardiac tropomyosin (Tm) were purified from acetone powders as described previously (34, 35). Rabbit ventricular S1 was isolated from purified myosin after α-chymotrypsin digestion (36).
Fluorescent Labeling
TnCT53C 3 and its constructs were labeled with the environmentally sensitive thiol reactive fluorescent probe IAANS as described previously (17).
Reconstitution of Tn Complex
The Tn complexes were prepared and reconstituted as described previously (17).
Reconstitution of Regulated Thin Filaments
Thin filaments were prepared in a reconstitution buffer containing 10 mm MOPS, 150 mm KCl, 3 mm MgCl2, 1 mm DTT, pH 7.0, as described previously (17).
Steady-state Fluorescence
All steady-state fluorescence measurements were performed using a PerkinElmer Life Sciences LS 55 fluorescence spectrometer at 15 °C. IAANS fluorescence was excited at 330 nm and monitored at 450 nm as microliter amounts of CaCl2 were added to 2 ml of each Tn complex or thin filament in 200 mm MOPS, 150 mm KCl, 3 mm MgCl2, 1 mm DTT, pH 7.0, as described previously (17). The Ca2+ sensitivity was reported as a dissociation constant Kd, representing a mean of at least three titrations. The data were fit with the Hill equation.
Stopped-flow Fluorescent Measurements
Ca2+ dissociation rates were characterized using an Applied Photophysics model SX.20 stopped-flow instrument with a dead time of 1.4 ms at 15 °C. IAANS fluorescence was excited at 330 nm. The IAANS emission was monitored through a 510-nm broad band-pass interference filter for the thin filament. The filters were purchased from Oriel (Stratford, CT). Data traces (an average of 3–5 individual traces) were fit with a single exponential equation to calculate the kinetic rates. The working buffer used for the kinetic measurements was 10 mm MOPS, 150 mm KCl, 1 mm DTT, 3 mm MgCl2 at pH 7.0. 10 mm EGTA was utilized to remove 200 μm Ca2+ from the thin filaments.
Actomyosin S1 ATPase Assay
Reconstituted thin filaments were formed in a buffer containing 5 mm MgCl2, 30 mm MOPS, pH 7.0. The thin filaments were formed using 5 μm actin, 2 μm Tm, and 1.5 μm Tn. 0.2 μm myosin S1 was used in the assay. A final EGTA concentration of 0.5 mm and various amounts of Ca2+ were added to the reaction mixture to form the different pCa values. The reactions were initiated by adding 3 mm ATP, and 15-μl aliquot reaction mixtures were terminated by the addition of 0.2 m ice-cold Δ′-pyrroline-5-carboxylic acid at different time intervals (typically every 4 min). ATPase activity was determined by analyzing the amount of phosphate released in a time course of up to 20 min. The malachite green assay was utilized to quantify the phosphate released during the reaction as described previously (37).
Skinned Muscle Chamber and Apparatus
Trabeculae were “T-clipped” and attached to hooks connected to a servo-controlled DC torque motor (Cambridge Technologies) and an isometric force transducer (model 403A, Cambridge Technologies) located in stainless steel troughs (38). A reticule on the eyepiece of the dissecting microscope was used to measure the width and depth of the trabecula. Cross-sectional area was calculated from the depth and width measurements by assuming an elliptical circumference. The motor and force transducer were set on a three-way positioner that can be moved to adjust the resting sarcomere length to ∼2.2 μm as determined by the first-order diffraction pattern from a HeNe laser directed through the trabeculae. The analog output of the force transducer was digitized using a DaqBoard/2000 and the DaqView software. The temperature of the solution in the troughs was maintained at 15 °C by a thermocouple-controlled Peltier device.
Preparation of Rat Cardiac Trabeculae
All protocols were approved by the Institutional Animal Care and Use Committee. Rat cardiac trabeculae were harvested and prepared from male LBN-F1 rats (175–200 g) as described previously (39). Briefly, rats were anesthetized via intraperitoneal injection of pentobarbital sodium (Nembutal, 50 mg/kg), and the thoracic cavity was opened. Heparin (0.1 ml of 10,000 units/ml stock) was injected intracardially, and right ventricular trabeculae were harvested and placed overnight at 4 °C in a relaxing solution containing 1% Triton X-100. The trabeculae were used within 48 h of harvest. The mean maximal F/cross-sectional area of 23 trabeculae used in this study was 48 ± 3 millinewtons/mm2.
Human Troponin Exchange in Rat Cardiac Trabeculae
After maximal force was measured in the pre-exchanged trabeculae, the trabeculae were shortened by 20% of the resting length and soaked in a Rigor Buffer (10 mm MOPS, 150 mm KCl, 20 mm 2,3-butanedione monoxime, 0.01% NaN3, 0.5 mm DTT, and 3 mm MgCl2) for 30 min. The temperature was then elevated to 25 °C, and the trabeculae were soaked in an exchange buffer consisting of the Rigor Buffer with 7–15 μm human Tn and 500 μm Ca2+ for 2.5 h. In the case of the mock Tn exchange, no Tn was added to the exchange buffer. The exchange buffer was briefly mixed in the chamber every 15 min. After exchange, the trabeculae were stretched back to their original length and transferred to a pCa 9.0 solution with 20 mm 2,3-butanedione monoxime, and the passive tension was measured at 15 °C. The trabeculae were subsequently washed three times in pCa 9.0 solution for 5 min each to remove residual 2,3-butanedione monoxime. Afterward, maximal tension at pCa 4.0 was measured twice to determine the percentage of maximal force recovery. The trabeculae were then randomly contracted in solutions of varying [Ca2+] with a maximal contraction performed in the middle and end to determine rundown. Muscles that exhibited greater than 20% rundown in the maximal force over the course of the force-pCa experiments were excluded.
Quantification of Tn Exchange
The percentage of exchanged Tn was quantified for trabecula that underwent force measurements. After removing the T-clips, the trabeculae were extracted in sample buffer (50 mm Tris-HCl, pH 6.8, containing 2% SDS, 10% glycerol, and 0.1% bromphenol blue) by heating to 80 °C for 6 min with periodic vortexing. Each sample was subsequently clarified by centrifugation. The extracted proteins from an entire trabeculae were separated by SDS-PAGE on a 16 × 18 cm (Hoefer) 12% (29:1) acrylamide gel cooled to 8 °C. The portion of the gel containing TnI was then transferred to a polyvinylidene difluoride membrane, probed with an anti-TnI antibody (C5, Fitzgerald), and detected by Enhanced Chemiluminescence Plus (GE Healthcare). The films were scanned and quantified using an ImageQuant TL and software (GE Healthcare) (40). The variable size of each trabecula resulted in a varied amount of total TnI loading; therefore the amount of exchanged human TnI was expressed as a percentage of the total TnI (exchanged human and remaining rat endogenous TnI).
Statistical Analysis
Statistical significance was determined by analysis of variance followed by a post hoc t test using the statistical analysis software Minitab (State College, PA). Two means were considered to be significantly different when the p value was < 0.05. All data are shown as a mean value ± S.E.
RESULTS
Thin Filament Ca2+ Binding Studies
Our laboratory has developed a fluorescent troponin C, TnCIAANST53C, which minimally affects cTnC function and reports the structural changes that occur in the regulatory domain of cTnC upon Ca2+ binding and dissociation on the thin filament (17, 32). This fluorescent TnC enabled the Ca2+ binding studies reported here.
Restrictive cardiomyopathy (RCM) is characterized by impaired ventricular filling due to an extremely stiff heart (24). Consistent with the diastolic dysfunction, RCM-associated contractile proteins typically sensitize actomyosin ATPase activity and force generation to Ca2+ (18, 24). As shown in Fig. 1, thin filament-bound control TnIAANST53C exhibited a Ca2+ sensitivity of 4.8 ± 0.2 μm (Fig. 1 and Table 1). Consistent with previous studies, thin filament Ca2+ sensitivity increased ∼3-fold when the RCM associated mutation TnI R192H was incorporated into the Tn complex (Fig. 1 and Table 1) (18).
FIGURE 1.
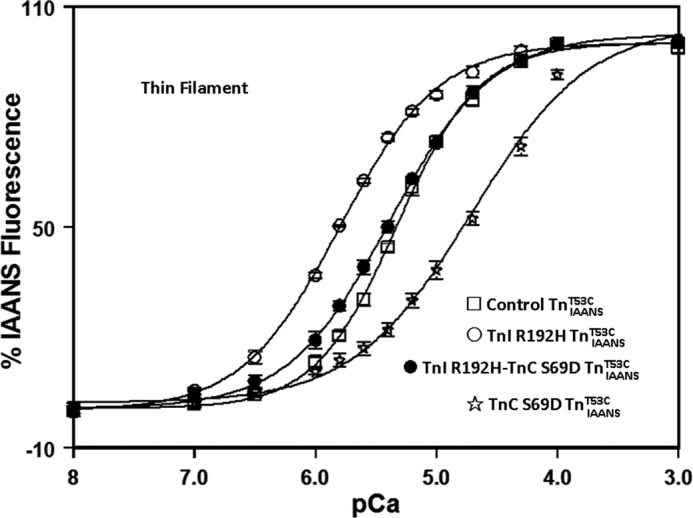
An engineered TnC corrects RCM TnI R192H thin filament Ca2+ sensitivity. The figure shows the Ca2+-dependent changes in IAANS fluorescence for control TnIAANST53C (□), TnI R192H TnIAANST53C (○), TnI R192H-TnC S69D TnIAANST53C (●), and TnC S69D TnIAANST53C (☆) reconstituted thin filaments as a function of pCa.
TABLE 1.
Summary of thin filament Ca2+ sensitivity
Values marked with * are significantly different from the control values (p < 0.05). NA denotes a measurement that is not applicable.
| Tn | pCa50a | Ca2+ sensitivity | Hill coefficient | Relative change in Ca2+ sensitivityb | Ca2+koff (/s) | Relative change in Ca2+koffb |
|---|---|---|---|---|---|---|
| μm | ||||||
| Control | 5.33 ± 0.02 | 4.8 ± 0.2 | 1.28 ± 0.06 | NA | 104.7 ± 0.5 | NA |
| TnI R192H | 5.80 ± 0.003* | 1.59 ± 0.01* | 1.07 ± 0.03* | ↑ 3.0 | 73 ± 2* | ↓1.4 |
| TnI-(1–192) | 6.15 ± 0.03* | 0.71 ± 0.05* | 1.14 ± 0.09 | ↑ 6.7 | 55.6 ± 0.6* | ↓1.9 |
| TnT ΔK210 | 4.81 ± 0.03* | 15 ± 1* | 1.19 ± 0.06 | ↓ 3.2 | 253 ± 7* | ↑2.4 |
| TnI R192H-TnC S69D | 5.39 ± 0.01 | 4.08 ± 0.08 | 1.04 ± 0.03* | ↑ 1.2 | 167 ± 4* | ↑1.6 |
| TnC S69D | 4.71 ± 0.04* | 20 ± 2* | 0.89 ± 0.03* | ↓ 4.1 | 224 ± 3* | ↑2.1 |
| TnI-(1–192)-TnCS69D/D73N | 5.46 ± 0.01 | 3.46 ± 0.07 | 0.93 ± 0.04* | ↑ 1.4 | 240 ± 7* | ↑2.3 |
| TnC S69D/D73N | 4.78 ± 0.07* | 17 ± 2* | 1.12 ± 0.05 | ↓ 3.6 | 252 ± 12* | ↑2.4 |
| TnT ΔK210-TnC M45Q | 6.01 ± 0.02* | 0.98 ± 0.05* | 0.77 ± 0.04* | ↑ 4.8 | 169 ± 7* | ↑1.6 |
| TnTΔK210-TnCM45Q/S69D | 5.38 ± 0.09 | 4.3 ± 0.8 | 0.89 ± 0.05* | ↑ 1.1 | 84 ± 2* | ↓1.2 |
| TnC M45Q/S69D | 5.80 ± 0.05* | 1.6 ± 0.2* | 1.26 ± 0.07 | ↑ 3.0 | 102 ± 2 | ↓1.0 |
| TnC M45Q | 6.23 ± 0.03* | 0.59 ± 0.04* | 1.45 ± 0.05 | ↑ 8.1 | 40 ± 5* | ↓2.6 |
a The Ca2+ concentration at half-maximal fluorescent change.
b ↑ indicates increase in Ca2+ sensitivity or koff, ↓ indicates decrease in Ca2+ sensitivity or koff.
Ca2+ binding to an EF-hand is partially controlled by the number and position of acidic residues within the Ca2+-binding loop (41). Previously, we have shown that the Ca2+ affinity of calmodulin (an EF-hand protein) could be decreased by modulating the position of acidic residues within the Ca2+-binding loop of its N-terminal domain (41). Utilizing a similar strategy, constructs that desensitize TnC to Ca2+ were also generated. One of the engineered TnC constructs, TnC S69D, desensitized thin filament Ca2+ binding ∼4-fold when compared with the control TnIAANST53C (Fig. 1 and Table 1). Excitingly, when combined with TnI R192H, TnI R192H-TnC S69D TnIAANST53C exhibited a thin filament Ca2+ sensitivity and cooperativity that was indistinguishable from the control TnIAANST53C (Fig. 1 and Table 1). Thus, the increased thin filament Ca2+ sensitivity of an RCM mutation can be corrected through an engineered Ca2+-desensitizing TnC.
Dilated cardiomyopathy (DCM) is another subtype of familial cardiomyopathy that is characterized by ventricular dilation and diminished systolic function of the left or both ventricles (25). Contrary to RCM, DCM is typically associated with decreased Ca2+ sensitivity of actomyosin ATPase activity and force generation (22, 25). Fig. 2 shows that the DCM TnT ΔK210 modification decreased thin filament Ca2+ sensitivity ∼3-fold when compared with the control TnIAANST53C (Table 1). A Ca2+-sensitizing TnC will be required to correct the DCM thin filament behavior. By mutating the hydrophobic pocket of the regulatory domain of TnC, Ca2+-sensitizing TnC constructs can be engineered (32, 33). For instance, the TnC M45Q mutation increased thin filament Ca2+ sensitivity ∼8-fold when compared with the control TnIAANST53C (Fig. 2A and Table 1). Upon combining the Ca2+-sensitizing TnC M45Q mutation with the DCM TnT ΔK210 modification, the thin filament Ca2+ sensitivity was ∼5-fold overcorrected (Fig. 2A and Table 1). Interestingly, the resultant change in Ca2+ sensitivity was roughly an additive effect of the two mutations. These data suggest that the Ca2+-sensitizing TnC will need to be precisely tuned to correct the aberrant thin filament Ca2+ sensitivity.
FIGURE 2.

An engineered TnC corrects DCM TnT ΔK210 thin filament Ca2+ sensitivity. A, the Ca2+-dependent changes in IAANS fluorescence for control TnIAANST53C (open squares), TnC M45Q TnIAANST53C (semicircles), TnT ΔK210-TnC M45Q TnIAANST53C (filled squares), and TnT ΔK210 TnIAANST53C (inverted triangles) reconstituted thin filaments as a function of pCa. B, the Ca2+-dependent changes in IAANS fluorescence for control TnIAANST53C (open squares), TnC M45Q/S69D TnIAANST53C (filled diamonds), TnT ΔK210-TnC M45Q/S69D TnIAANST53C (filled inverted triangles), and TnT ΔK210 TnIAANST53C (open inverted triangles) reconstituted thin filaments as a function of pCa.
To temper the strong Ca2+ binding of M45Q, the Ca2+-desensitizing mutation S69D was introduced to fine-tune its Ca2+ sensitivity. TnC M45Q/S69D only sensitized thin filament Ca2+ sensitivity ∼3-fold when compared with the control TnIAANST53C (Fig. 2B and Table 1). When TnC M45Q/S69D was combined with the DCM TnT ΔK210 modification, TnT ΔK210-TnC M45Q/S69D TnIAANST53C exhibited a thin filament Ca2+ sensitivity of 4.3 ± 0.8 μm that was nearly identical to control TnIAANST53C, yet with a slightly reduced cooperativity (Fig. 2B and Table 1). Thus, it is also possible to fine-tune the Ca2+ sensitivity of TnC and correct abnormally desensitized thin filament Ca2+ binding associated with a dilated cardiomyopathy.
Besides inherited cardiac muscle diseases, cardiac muscle Ca2+ sensitivity can also be adversely affected during acquired conditions such as ischemia reperfusion-induced injury (21). Proteolysis of myofilament proteins such as TnI has been proposed to play a key role in human myocardial ischemia/reperfusion injury (21, 42, 43). Consistent with its reported effect of sensitizing both actomyosin ATPase activity and force generation to Ca2+ (21), truncated TnI-(1–192) increased thin filament Ca2+ sensitivity ∼7-fold when compared with TnIAANST53C (Fig. 3 and Table 1). To improve this extremely sensitized Ca2+ binding, we developed another Ca2+-desensitizing TnC, S69D/D73N. TnC S69D/D73N decreased thin filament Ca2+ sensitivity ∼4-fold when compared with the control TnIAANST53C (Fig. 3 and Table 1). When combined with the truncated TnI-(1–192), TnI-(1–192)-TnC S69D/D73N TnIAANST53C exhibited a thin filament Ca2+ sensitivity of 3.46 ± 0.07 μm, which was statistically indistinguishable from that of the control TnIAANST53C, albeit with a slightly reduced cooperativity (Fig. 3 and Table 1). Thus, the hypersensitized thin filament Ca2+ binding associated with an acquired cardiac disease can also be corrected by an engineered TnC.
FIGURE 3.
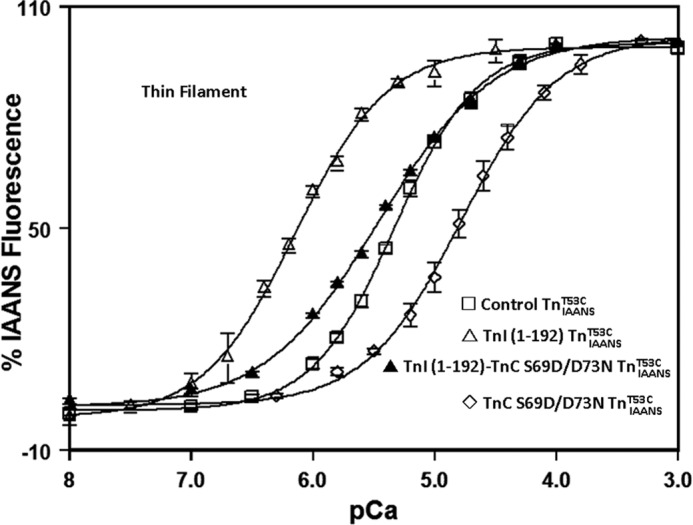
An engineered TnC corrects ischemic TnI-(1–192) thin filament Ca2+ sensitivity. The figure shows the Ca2+-dependent increase in IAANS fluorescence for control TnIAANST53C (□), TnI-(1–192) TnIAANST53C (▵), TnI-(1–192)-TnC S69D/D73N TnIAANST53C (▴), and TnC S69D/D73N TnIAANST53C (♢) reconstituted thin filaments as a function of pCa.
Thin Filament Ca2+ Dissociation Rates
In addition to altering the steady-state Ca2+ binding properties of TnC, disease-related protein modifications have been shown to alter the rate of Ca2+ dissociation from TnC (44–46). Fig. 4 shows that the rate of Ca2+ dissociation from the thin filament reconstituted with control TnIAANST53C occurred at 104.7 ± 0.5/s (Table 1). Both disease-related Ca2+-sensitizing modifications (TnI R192H and TnI-(1–192)) slowed the rate of Ca2+ dissociation 1.4–1.9-fold, whereas the disease-related Ca2+-desensitizing modification (TnT ΔK210) accelerated the rate of Ca2+ dissociation ∼2.4-fold when compared with the control TnIAANST53C (Fig. 4, A–C, and Table 1). Both correcting TnC constructs (TnC S69D and TnC S69D/D73N) engineered against the disease-related Ca2+-sensitizing modifications accelerated the rate of Ca2+ dissociation 2.1–2.4-fold when compared with the control TnIAANST53C (Fig. 4, A and C, and Table 1). Interestingly, the correcting TnC construct M45Q/S69D designed against the disease-related Ca2+-desensitizing modification had a negligible effect on the rate of Ca2+ dissociation (Fig. 4B and Table 1). When combined, all of the correcting TnC constructs were able to reverse the effects of the disease-related protein modification on the rate of Ca2+ dissociation from the thin filament (Fig. 4, A–C, and Table 1).
FIGURE 4.
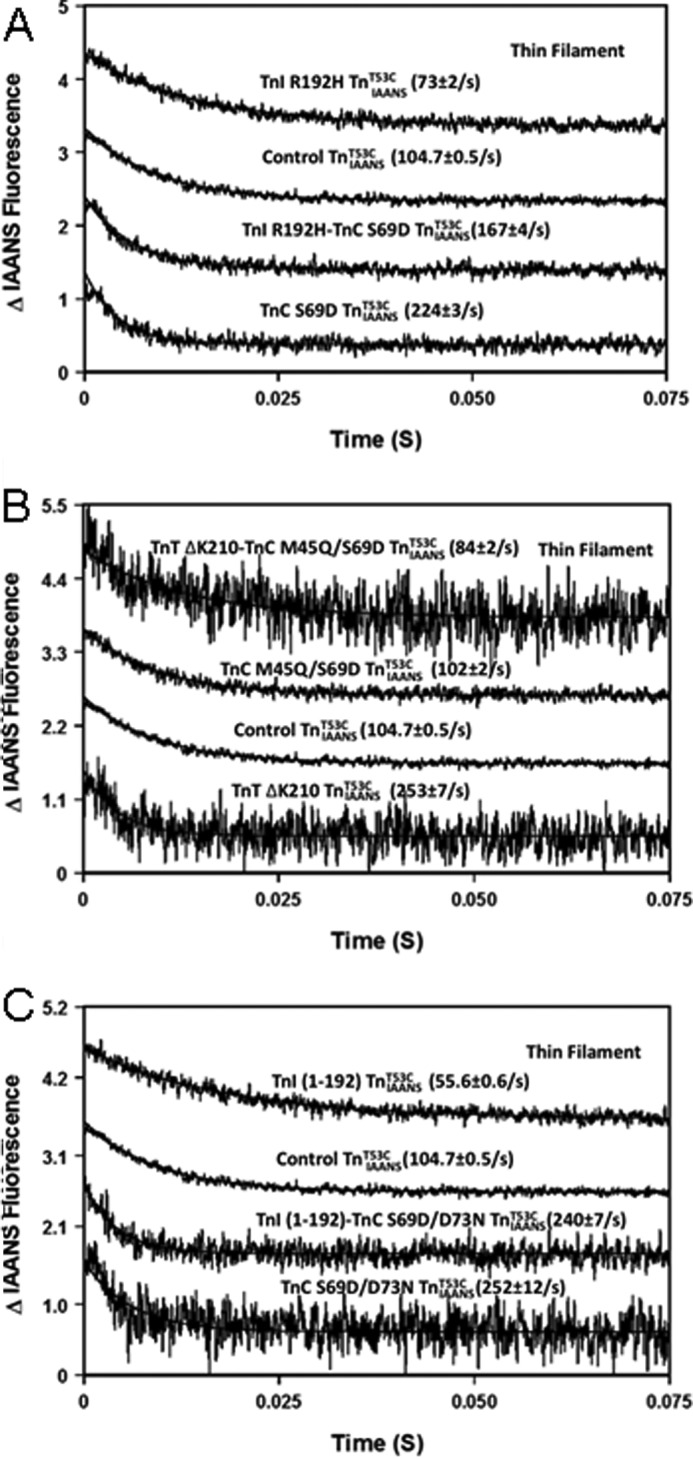
Effect of disease-related protein modifications, correcting TnC and their combinations on rate of Ca2+ dissociation from thin filament. A, the time courses of the decrease in IAANS fluorescence as Ca2+ was removed by EGTA from thin filament-bound control TnIAANST53C, TnI R192H TnIAANST53C, TnC S69D TnIAANST53C, and TnI R192H-TnC S69D TnIAANST53C. B, the time courses of the decrease in IAANS fluorescence as Ca2+ was removed by EGTA from thin filament-bound control TnIAANST53C, TnT ΔK210 TnIAANST53C, TnC M45Q/S69D TnIAANST53C, and TnT ΔK210-TnC M45Q/S69D TnIAANST53C. C, the time courses of the decrease in IAANS fluorescence as Ca2+ was removed by EGTA from thin filament-bound control TnIAANST53C, ischemic TnI(1–192) TnIAANST53C, TnC S69D/D73N TnIAANST53C, and TnI(1–192)-TnC S69D/D73N TnIAANST53C. The data traces have been staggered and normalized for clarity.
Actomyosin S1 ATPase Assay
The thin filament Ca2+ binding studies demonstrated that it was feasible to engineer TnC constructs with appropriately tuned Ca2+ sensitivities to correct both abnormally decreased and abnormally increased Ca2+ binding associated with different cardiac dysfunctions. To further verify the significance of the corrected thin filament Ca2+ binding, the functional assay of thin filament actomyosin S1 ATPase was performed. For control TnIAANST53C, the Ca2+ sensitivity of the actomyosin ATPase activity occurred at 1.6 ± 0.1 μm (Fig. 5 and Table 2). Consistent with the thin filament Ca2+ binding studies, the RCM TnI R192H mutation sensitized the Ca2+-dependent ATPase activity ∼3-fold. TnC S69D desensitized the ATPase activity to Ca2+ ∼4-fold when compared with the control TnIAANST53C (Fig. 5 and Table 2). When combined, TnI R192H-TnC S69D TnIAANST53C exhibited an actomyosin ATPase Ca2+ sensitivity of 1.5 ± 0.3 μm, which was indistinguishable from the control TnIAANST53C but with a slightly reduced cooperativity (Fig. 5 and Table 2). Thus, an engineered Ca2+-desensitizing TnC was able to functionally correct the disease-associated increased Ca2+ sensitivity of the actomyosin ATPase activity. Unfortunately, the DCM mutation TnT ΔK210 exhibited a diminished maximal ATPase activity, whereas the ischemic TnI-(1–192) exhibited an increased basal ATPase activity (data not shown). As a result, the Ca2+-regulated ATPase activity for these mutations could not be measured due to compromised signal amplitudes.
FIGURE 5.
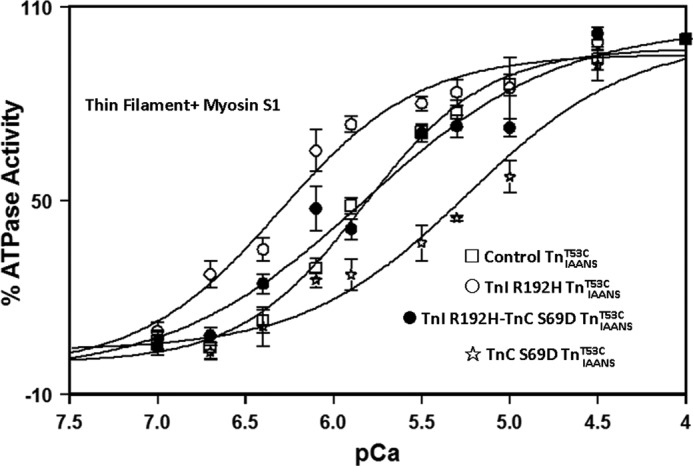
Engineered TnCs functionally correct RCM TnI R192H actomyosin ATPase activity. Normalized actomyosin S1 ATPase activity for thin filaments containing control TnIAANST53C (□), TnI R192H TnIAANST53C (○), TnC S69D TnIAANST53C (☆), and TnI R192H-TnC S69D TnIAANST53C (●) is plotted as a function of pCa in the figure.
TABLE 2.
Summary of actomyosin ATPase activity
Values marked with* are significantly different from the control values (p < 0.05).
| Tn | ATPase pCa50a | ATPase Ca2+ sensitivity | Hill coefficient | Maximal ATPase activityb | Minimal ATPase activityc |
|---|---|---|---|---|---|
| μm | |||||
| Control | 5.81 ± 0.03 | 1.6 ± 0.1 | 1.18 ± 0.09 | 0.037 ± 0.003 | 0.0072 ± 0.0006 |
| TnI R192H | 6.29 ± 0.03* | 0.52 ± 0.04* | 1.1 ± 0.1 | 0.029 ± 0.003 | 0.0110 ± 0.0008* |
| TnC S69D | 5.27 ± 0.07* | 5.6 ± 0.9* | 1.0 ± 0.2 | 0.035 ± 0.003 | 0.010 ± 0.002 |
| TnI R192H-TnC S69D | 5.84 ± 0.09 | 1.5 ± 0.3 | 0.83 ± 0.08* | 0.038 ± 0.002 | 0.012 ± 0.001* |
a The Ca2+ concentration at half-maximal ATPase activity.
b Maximal ATPase activity [mol of phosphate s−1 (mol of S1)−1].
c Minimal ATPase activity [mol of phosphate s−1 (mol of S1)−1].
Skinned Trabecula Force Measurement
Due to the technical limitations of the actomyosin ATPase assay, force-pCa measurements were performed to assess the physiological relevance of correcting TnT ΔK210 and TnI-(1–192) thin filament Ca2+ binding. Recombinant TnIAANST53C complexes were exchanged into rat skinned trabecula to measure the force-pCa2+ relationship. As shown in Fig. 6A and Table 3, the force-pCa50 occurred at 5.67 ± 0.06, 5.76 ± 0.07, and 5.74 ± 0.06 for endogenous, mock-exchanged and control TnIAANST53C-exchanged skinned trabecula, respectively. Thus, the exchange protocol, the IAANS probe, and the mutations associated with the labeling of TnC did not significantly affect the Ca2+ sensitivity of skinned trabecula force generation. However, the Tn exchange protocol appears to reduce both the cooperativity and the maximal force generated (Table 3). Fig. 6B shows that rat TnI and human TnI migrate differently in a SDS acrylamide gel and can be used to determine the efficiency of Tn exchange. Quantification of the Tn exchange by Western blot of TnI demonstrated that ∼76% of the endogenous Tn was replaced by the exogenous Tn (Fig. 6B).
FIGURE 6.
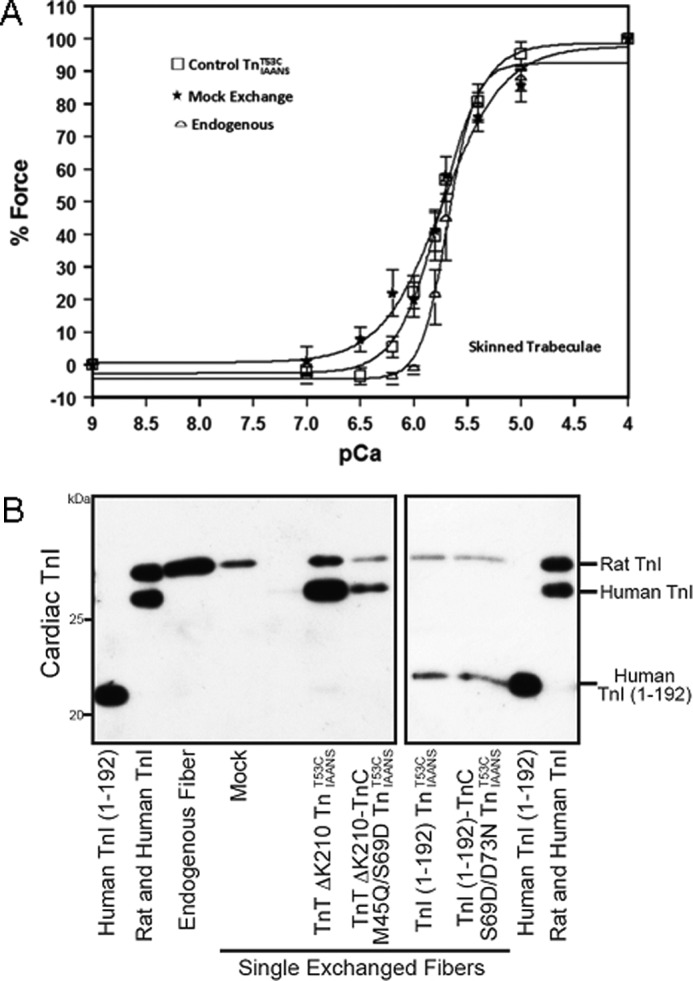
Effect of Tn exchange on skinned cardiac muscle force generation. A, the Ca2+-dependent force generation for skinned trabeculae without Tn exchange (endogenous, □) and without Tn exchange but going through the same exchange protocol (mock exchange, ★) and for skinned trabeculae exchanged with control TnIAANST53C (□). Data traces were individually normalized. B, a Western blot for cardiac TnI demonstrating the presence of experimental human TnI in representative single trabecula from force experiments. Both wild-type and human TnI-(1–192) migrate faster than endogenous rat TnI, allowing for the quantification of human Tn exchanged into trabeculae from functional measurements. Human TnI (1–192), recombinant, purified human TnI-(1–192) fragment; Rat and Human TnI, mixed recombinant wild-type rat (50%) and human purified TnI (50%).
TABLE 3.
Summary of skinned trabecula force generation
Values marked with* are significantly different from the control values (p < 0.05). NA denotes a measurement that is not applicable.
| Tn | pCa50a | Hill Coefficient | % of force recovery | % of active tension at pCa 9.0 |
|---|---|---|---|---|
| Endogenous | 5.67 ± 0.06 | 5 ± 2 | NA | 9 ± 3 |
| Mock exchange | 5.76 ± 0.07 | 1.8 ± 0.3 | 68 ± 5 | 1 ± 1 |
| Control | 5.74 ± 0.06 | 2.2 ± 0.2 | 55 ± 7 | 1 ± 2 |
| TnI-(1–192) | 6.8 ± 0.1* | 1.3 ± 0.1* | 86 ± 8* | 21 ± 4* |
| TnI-(1–192)-TnCS69D/D73N | 5.88 ± 0.05 | 1.5 ± 0.1* | 70 ± 6 | 7 ± 4 |
| TnT ΔK210 | 5.35 ± 0.02* | 2.7 ± 1.0 | 51 ± 4 | 0 ± 1 |
| TnT ΔK210-TnCM45Q/S69D | 5.74 ± 0.04 | 2.4 ± 0.3 | 52 ± 3 | 5 ± 1 |
a The Ca2+ concentration at half-maximal force.
Consistent with the thin filament Ca2+ binding studies, DCM TnT ΔK210 desensitized skinned trabecula force generation to Ca2+ ∼2.5-fold (Fig. 7A and Table 3). When combined with its correcting TnC M45Q/S69D, the Tn-exchanged trabecula exhibited a Ca2+ sensitivity and cooperativity indistinguishable from that of the control, with a pCa50 of 5.74 ± 0.04 (Fig. 7A and Table 3). On the other hand, ischemia-induced truncated TnI-(1–192) considerably sensitized skinned trabecula force generation to Ca2+ ∼8-fold (Fig. 7B and Table 3). Additionally, truncated TnI-(1–192) uniquely raised the Ca2+-independent force at pCa 9.0 by ∼21% (Table 3). When combined with its correcting TnC S69D/D73N, Tn-exchanged trabeculae exhibited a substantially improved Ca2+ sensitivity but not cooperativity, with a pCa50 of 5.88 ± 0.05 (Fig. 7B and Table 3). Furthermore, TnC S69D/D73N was able to ameliorate the elevated Ca2+-independent force at pCa 9.0 caused by TnI-(1–192) (Table 3). Thus, disease-related Ca2+ sensitivity of force generation can be corrected through engineering TnC, too.
FIGURE 7.
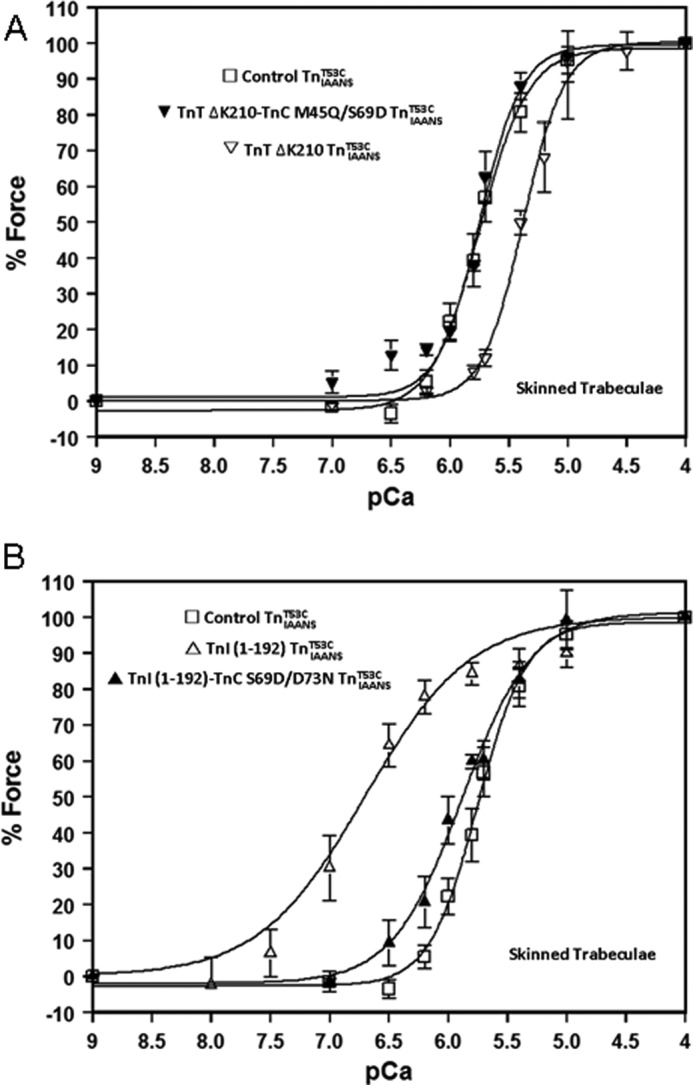
Engineered TnC constructs functionally correct DCM TnT ΔK210 and ischemic TnI-(1–192) skinned cardiac muscle force generation. A, the Ca2+-dependent force generation for skinned trabeculae exchanged with control TnIAANST53C (□), TnT ΔK210 TnIAANST53C (▿), and TnT ΔK210-TnC M45Q/S69D TnIAANST53C (▾). B, the Ca2+-dependent force generation for skinned trabeculae exchanged with control TnIAANST53C (□), TnI-(1–192) TnIAANST53C (▵), and TnI-(1–192)-TnC S69D/D73N TnIAANST53C (▴). Data traces were individually normalized.
DISCUSSION
The goal of the current study was to test the hypothesis that disease-related myofilament Ca2+ sensitivity can be corrected by rationally engineered TnC constructs. RCM TnI R192H, DCM TnT ΔK210, and ischemia-induced truncated TnI-(1–192) were chosen to test the hypothesis because they represent different protein (TnI or TnT) modifications (missense mutation, deletion, or truncation) and disease subtypes (familial or acquired) that can afflict both mouse and humans (27, 47, 48). By engineering TnC with a wide, yet fine-tunable, range of Ca2+ sensitivities, abnormally increased or decreased Ca2+ binding associated with different cardiac dysfunctions can be corrected, and their altered Ca2+ dissociation rates can be reversed. More significantly, disease-associated myofilament function can also be improved by the engineered TnC constructs.
Cardiovascular diseases are diverse and complex. Within the heart, numerous systems can fail, such as cellular coupling (electrical and mechanical), energetics, Ca2+ handling, and Ca2+ sensitivity (26, 31). Furthermore, the cooperativity and kinetics of Ca2+ exchange may be as significant to proper cardiac function as the overall Ca2+ sensitivity itself (5). Thus, targeting a single problem may not be sufficient to cure or even curb heart disease. Rather, an integrative approach may ultimately be necessary (26), of which TnC is one potential target. Although the cooperativity of Ca2+ binding was not always corrected by the engineered TnC constructs, improving the Ca2+ sensitivity may still improve outcome.
Aberrant myofilament Ca2+ sensitivity is commonly observed with multiple cardiac diseases, especially familial cardiomyopathies (22). Although the etiology of the cardiomyopathies remains unclear, experimental evidence shows promise that improving cardiac muscle Ca2+ sensitivity through either pharmacological or genetic approaches can relieve disease-related symptoms (27, 28, 30, 49). Ca2+ sensitizers have attracted growing clinical interest for their potential therapeutic value in treating heart failure and cardiomyopathies that desensitize cardiac muscle to Ca2+ (31). Although new compounds have been discovered, many of the Ca2+ sensitizers typically have deleterious side effects such as inhibiting cAMP phosphodiesterases and ATP-sensitive potassium channels (31). On the other hand, little effort has been put into developing therapeutic compounds that desensitize cardiac muscle to Ca2+. As an alternative to pharmaceuticals, genetic approaches that directly modulate contractile proteins have recently received increasing attention. Excitingly, chimeric tropomyosin, N-terminal truncated TnI, and fetal TnT have all been shown to improve disease-related abnormal cardiac muscle Ca2+ sensitivity and in vivo function (28, 30, 49). However, it is not clear how applicable these proteins will be to correcting the wide assortment of Ca2+-sensitizing and -desensitizing cardiac diseases because it is unknown how to specifically tune their performance.
For the past several decades, researchers have been discovering the rules that govern the Ca2+ binding properties of EF-hand proteins, especially TnC (5, 32, 33, 41, 50–54). By taking advantage of these rules, TnC has been engineered to encompass a wide range of Ca2+ sensitivities, which can accommodate a broad spectrum of disease-related Ca2+ binding (32, 33). Thus, TnC is a more versatile protein to modulate and reset disease-associated myofilament Ca2+ sensitivity. For instance, the ability of TnC to open its buried N-terminal hydrophobic pocket and bind TnI is a major determinant of its apparent Ca2+ sensitivity (Fig. 8) (32, 33). Modifying the network of side chain interactions involved with the opening of the TnI-binding pocket (such as the M45Q mutation; Fig. 8) can substantially affect the apparent Ca2+ sensitivity of TnC. Interestingly, these types of hydrophobic pocket mutations do not directly interact with the ligated Ca2+ ion. On the other hand, the Ca2+ affinity of TnC can be directly altered by manipulating the charge and position of the Ca2+-chelating residues within its Ca2+-binding loop. It would appear that the first and last chelating residues within the loop must be acidic for an EF-hand to bind Ca2+ (50, 51, 55), whereas the internal chelating loop residues can vary substantially and still allow Ca2+ binding (41). Altering the internal chelating loop residues can directly tune the Ca2+ sensitivity of the EF-hand, as was the case for the S69D and D73N mutations of TnC (Fig. 8).
FIGURE 8.

Location of disease-related Tn modifications and engineered TnC mutations in crystal structure of Tn complex. A ribbon representation of the cardiac Tn core domain is shown (PDB 1J1E). TnC is colored in magenta, TnI is colored in blue, and TnT is colored in yellow. The positions of the disease-related Tn modifications such as Lys-210 of TnT, as well as the TnC mutations including Met-45, Ser-69, and Asp-73 are labeled. The C-terminal end of TnI (residues 192–210) was not included in the crystal structure and is absent from the figure.
It is currently unknown how the disease-related proteins alter the Ca2+ sensitivity of TnC. However, the C-terminal region of TnI (residues 188–210) is thought to contribute to the inhibition of the actomyosin interactions during diastole by directly binding to actin-Tm, competing for the binding of TnI with TnC (17, 56). Both alterations of TnI (TnI R192H and TnI-(1–192)) are located within this region of TnI. We hypothesize that these disease-related modifications reduce the affinity of TnI for actin-Tm. In this regard, these protein modifications would facilitate the switching of TnI from actin-Tm to TnC and cause an enhanced apparent sensitivity of TnC for Ca2+. In contrast, the TnT mutation ΔK210 may increase the ability of TnI to bind to actin-Tm and decrease the apparent sensitivity of TnC for Ca2+. Because the disease-related protein modifications and the engineered TnC constructs altered thin filament Ca2+ sensitivity through potentially different molecular mechanisms, they exerted an additive effect on thin filament Ca2+ binding when combined.
The qualitative similarity of the results obtained from the reconstituted thin filaments and skinned trabeculae suggests that the thin filament is a reliable model system to study thin filament Ca2+ sensitivity. Thus, the thin filament Ca2+ binding and skinned trabecula force-pCa assays are efficient platforms to rapidly screen different thin filament modifications and test the efficiency of engineered TnC constructs. Although numerous disease-related protein modifications have been identified and studied, approximately half of the diagnosed cardiomyopathies remain idiopathic. Even with an unknown genetic background, directly targeting TnC could provide a way to reset the contractile performance back to normal. Ultimately, gene therapy approaches could introduce the correcting TnCs into diseased hearts to evaluate in vivo cardiac function. On the other hand, structural studies on the engineered TnC constructs could facilitate more specific pharmaceutical drug design targeted to TnC. The strategies utilized to modify TnC can also be applied to engineer additional EF-hand proteins such as parvalbumin and calmodulin to design potentially more therapeutic proteins for the heart or other organs (57). Thus, in addition to a potential new avenue to correct aberrant cardiac disease-related Ca2+ binding, the current study provides a novel perspective for engineering EF-hand Ca2+-binding proteins that are universally involved in cellular signaling cascades. These protein engineering approaches in combination with other therapies may one day improve the function of the diseased heart.
Acknowledgments
We thank Sean Little, Elizabeth Brundage, and Kristopher Kline for technical assistance. We also thank Drs. Jianchao Zhang and Jack Rall for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL091986 (to J. P. D.), AR020792 (to J. A. Rall), HL091056 (to B. J. B.), and HL087462 (to S. B. T.). This work was also supported by the American Heart Association (to B. L. and J. P. D.).
The mutant designations used are: TnCT53C, Cys-less human cardiac TnC with T53C mutation; TnCIAANST53C, TnCT53C labeled with IAANS; TnIAANST53C, TnT53C labeled with IAANS; TnI R192H, TnI mutation with Arg-192 replaced with His; TnT ΔK210, TnT mutation with Lys-210 deleted; TnI-(1–192), truncated TnI with residues 193–210 removed; TnC M45Q, TnC mutation with Met-45 replaced with Gln; TnC S69D, TnC mutation with Ser-69 replaced with Asp; TnC S69D/D73N, TnC mutation with Ser-69 replaced with Asp and Asp-73 replaced with Asn; TnC M45Q/S69D, TnC mutation with Met-45 replaced with Gln and Ser-69 replaced with Asp.
- Tn
- troponin
- TnC
- troponin C
- TnI
- troponin I
- TnT
- troponin T
- DCM
- dilated cardiomyopathy
- RCM
- restrictive cardiomyopathy
- IAANS
- 2-(4′-(iodoacetamido)anilino)naphthalene-6-sulfonic acid, sodium salt
- Tm
- tropomyosin.
REFERENCES
- 1. Nakayama S., Moncrief N. D., Kretsinger R. H. (1992) Evolution of EF-hand calcium-modulated proteins. II. Domains of several subfamilies have diverse evolutionary histories. J. Mol. Evol. 34, 416–448 [DOI] [PubMed] [Google Scholar]
- 2. Yang K. (2001) Prokaryotic calmodulins: recent developments and evolutionary implications. J. Mol. Microbiol. Biotechnol. 3, 457–459 [PubMed] [Google Scholar]
- 3. Zhou Y., Frey T. K., Yang J. J. (2009) Viral calciomics: interplays between Ca2+ and virus. Cell Calcium 46, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kawasaki H., Nakayama S., Kretsinger R. H. (1998) Classification and evolution of EF-hand proteins. Biometals 11, 277–295 [DOI] [PubMed] [Google Scholar]
- 5. Davis J. P., Tikunova S. B. (2008) Ca2+ exchange with troponin C and cardiac muscle dynamics. Cardiovasc. Res. 77, 619–626 [DOI] [PubMed] [Google Scholar]
- 6. Schwaller B. (2009) The continuing disappearance of “pure” Ca2+ buffers. Cell. Mol. Life Sci. 66, 275–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berridge M. J., Bootman M. D., Roderick H. L. (2003) Calcium signaling: dynamics, homeostasis, and remodeling. Nat. Rev. Mol. Cell Biol. 4, 517–529 [DOI] [PubMed] [Google Scholar]
- 8. Haiech J., Audran E., Fève M., Ranjeva R., Kilhoffer M. C. (2011) Revisiting intracellular calcium signaling semantics. Biochimie 93, 2029–2037 [DOI] [PubMed] [Google Scholar]
- 9. Yasuda S., Coutu P., Sadayappan S., Robbins J., Metzger J. M. (2007) Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circ. Res. 101, 377–386 [DOI] [PubMed] [Google Scholar]
- 10. Zhang R., Zhao J., Mandveno A., Potter J. D. (1995) Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ. Res. 76, 1028–1035 [DOI] [PubMed] [Google Scholar]
- 11. Schiaffino S., Gorza L., Ausoni S. (1993) Troponin isoform switching in the developing heart and its functional consequences. Trends Cardiovasc Med. 3, 12–17 [DOI] [PubMed] [Google Scholar]
- 12. Wei B., Jin J. P. (2011) Troponin T isoforms and posttranscriptional modifications: evolution, regulation and function. Arch. Biochem. Biophys. 505, 144–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Westfall M. V., Rust E. M., Metzger J. M. (1997) Slow skeletal troponin I gene transfer, expression, and myofilament incorporation enhances adult cardiac myocyte contractile function. Proc. Natl. Acad. Sci. U.S.A. 94, 5444–5449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Metzger J. M., Westfall M. V. (2004) Covalent and noncovalent modification of thin filament action: the essential role of troponin in cardiac muscle regulation. Circ. Res. 94, 146–158 [DOI] [PubMed] [Google Scholar]
- 15. Gomes A. V., Venkatraman G., Davis J. P., Tikunova S. B., Engel P., Solaro R. J., Potter J. D. (2004) Cardiac troponin T isoforms affect the Ca2+ sensitivity of force development in the presence of slow skeletal troponin I: insights into the role of troponin T isoforms in the fetal heart. J. Biol. Chem. 279, 49579–49587 [DOI] [PubMed] [Google Scholar]
- 16. Davis J. P., Rall J. A., Alionte C., Tikunova S. B. (2004) Mutations of hydrophobic residues in the N-terminal domain of troponin C affect calcium binding and exchange with the troponin C-troponin I96–148 complex and muscle force production. J. Biol. Chem. 279, 17348–17360 [DOI] [PubMed] [Google Scholar]
- 17. Davis J. P., Norman C., Kobayashi T., Solaro R. J., Swartz D. R., Tikunova S. B. (2007) Effects of thin and thick filament proteins on calcium binding and exchange with cardiac troponin C. Biophys. J. 92, 3195–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kobayashi T., Solaro R. J. (2006) Increased Ca2+ affinity of cardiac thin filaments reconstituted with cardiomyopathy-related mutant cardiac troponin I. J. Biol. Chem. 281, 13471–13477 [DOI] [PubMed] [Google Scholar]
- 19. Lu Q. W., Hinken A. C., Patrick S. E., Solaro R. J., Kobayashi T. (2010) Phosphorylation of cardiac troponin I at protein kinase C site threonine 144 depresses cooperative activation of thin filaments. J. Biol. Chem. 285, 11810–11817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robinson P., Griffiths P. J., Watkins H., Redwood C. S. (2007) Dilated and hypertrophic cardiomyopathy mutations in troponin and α-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 101, 1266–1273 [DOI] [PubMed] [Google Scholar]
- 21. Tachampa K., Kobayashi T., Wang H., Martin A. F., Biesiadecki B. J., Solaro R. J., de Tombe P. P. (2008) Increased cross-bridge cycling kinetics after exchange of C-terminal truncated troponin I in skinned rat cardiac muscle. J. Biol. Chem. 283, 15114–15121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Willott R. H., Gomes A. V., Chang A. N., Parvatiyar M. S., Pinto J. R., Potter J. D. (2010) Mutations in troponin that cause HCM, DCM, AND RCM: what can we learn about thin filament function? J. Mol. Cell Cardiol. 48, 882–892 [DOI] [PubMed] [Google Scholar]
- 23. Sumandea M. P., Burkart E. M., Kobayashi T., De Tombe P. P., Solaro R. J. (2004) Molecular and integrated biology of thin filament protein phosphorylation in heart muscle. Ann. N.Y. Acad. Sci. 1015, 39–52 [DOI] [PubMed] [Google Scholar]
- 24. Gomes A. V., Liang J., Potter J. D. (2005) Mutations in human cardiac troponin I that are associated with restrictive cardiomyopathy affect basal ATPase activity and the calcium sensitivity of force development. J. Biol. Chem. 280, 30909–30915 [DOI] [PubMed] [Google Scholar]
- 25. Mirza M., Marston S., Willott R., Ashley C., Mogensen J., McKenna W., Robinson P., Redwood C., Watkins H. (2005) Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J. Biol. Chem. 280, 28498–28506 [DOI] [PubMed] [Google Scholar]
- 26. Tardiff J. C. (2011) Thin filament mutations: developing an integrative approach to a complex disorder. Circ. Res. 108, 765–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Du C. K., Morimoto S., Nishii K., Minakami R., Ohta M., Tadano N., Lu Q. W., Wang Y. Y., Zhan D. Y., Mochizuki M., Kita S., Miwa Y., Takahashi-Yanaga F., Iwamoto T., Ohtsuki I., Sasaguri T. (2007) Knock-in mouse model of dilated cardiomyopathy caused by troponin mutation. Circ. Res. 101, 185–194 [DOI] [PubMed] [Google Scholar]
- 28. Jagatheesan G., Rajan S., Petrashevskaya N., Schwartz A., Boivin G., Arteaga G. M., Solaro R. J., Liggett S. B., Wieczorek D. F. (2007) Rescue of tropomyosin-induced familial hypertrophic cardiomyopathy mice by transgenesis. Am. J. Physiol. Heart Circ. Physiol. 293, H949–H958 [DOI] [PubMed] [Google Scholar]
- 29. Li Y., Charles P. Y., Nan C., Pinto J. R., Wang Y., Liang J., Wu G., Tian J., Feng H. Z., Potter J. D., Jin J. P., Huang X. (2010) Correcting diastolic dysfunction by Ca2+-desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J. Mol. Cell Cardiol. 49, 402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pinto J. R., Yang S. W., Hitz M. P., Parvatiyar M. S., Jones M. A., Liang J., Kokta V., Talajic M., Tremblay N., Jaeggi M., Andelfinger G., Potter J. D. (2011) Fetal cardiac troponin isoforms rescue the increased Ca2+ sensitivity produced by a novel double deletion in cardiac troponin T linked to restrictive cardiomyopathy: a clinical, genetic, and functional approach. J. Biol. Chem. 286, 20901–20912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kass D. A., Solaro R. J. (2006) Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation 113, 305–315 [DOI] [PubMed] [Google Scholar]
- 32. Tikunova S. B., Liu B., Swindle N., Little S. C., Gomes A. V., Swartz D. R., Davis J. P. (2010) Effect of calcium-sensitizing mutations on calcium binding and exchange with troponin C in increasingly complex biochemical systems. Biochemistry 49, 1975–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tikunova S. B., Davis J. P. (2004) Designing calcium-sensitizing mutations in the regulatory domain of cardiac troponin C. J. Biol. Chem. 279, 35341–35352 [DOI] [PubMed] [Google Scholar]
- 34. Pardee J. D., Spudich J. A. (1982) Purification of muscle actin. Methods Enzymol. 85, 164–181 [DOI] [PubMed] [Google Scholar]
- 35. Smillie L. B. (1982) Preparation and identification of α- and β-tropomyosins. Methods Enzymol. 85, 234–241 [DOI] [PubMed] [Google Scholar]
- 36. Tobacman L. S., Adelstein R. S. (1984) Enzymatic comparisons between light chain isozymes of human cardiac myosin subfragment-1. J. Biol. Chem. 259, 11226–11230 [PubMed] [Google Scholar]
- 37. Kodama T., Fukui K., Kometani K. (1986) The initial phosphate burst in ATP hydrolysis by myosin and subfragment-1 as studied by a modified malachite green method for determination of inorganic phosphate. J. Biochem. 99, 1465–1472 [DOI] [PubMed] [Google Scholar]
- 38. Davis J. P., Rall J. A., Reiser P. J., Smillie L. B., Tikunova S. B. (2002) Engineering competitive magnesium binding into the first EF-hand of skeletal troponin C. J. Biol. Chem. 277, 49716–49726 [DOI] [PubMed] [Google Scholar]
- 39. Norman C., Rall J. A., Tikunova S. B., Davis J. P. (2007) Modulation of the rate of cardiac muscle contraction by troponin C constructs with various calcium binding affinities. Am. J. Physiol. Heart Circ. Physiol. 293, H2580–H2587 [DOI] [PubMed] [Google Scholar]
- 40. Biesiadecki B. J., Tachampa K., Yuan C., Jin J. P., de Tombe P. P., Solaro R. J. (2010) Removal of the cardiac troponin I N-terminal extension improves cardiac function in aged mice. J. Biol. Chem. 285, 19688–19698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Black D. J., Tikunova S. B., Johnson J. D., Davis J. P. (2000) Acid pairs increase the N-terminal Ca2+ affinity of CaM by increasing the rate of Ca2+ association. Biochemistry 39, 13831–13837 [DOI] [PubMed] [Google Scholar]
- 42. Narolska N. A., Piroddi N., Belus A., Boontje N. M., Scellini B., Deppermann S., Zaremba R., Musters R. J., dos Remedios C., Jaquet K., Foster D. B., Murphy A. M., van Eyk J. E., Tesi C., Poggesi C., van der Velden J., Stienen G. J. (2006) Impaired diastolic function after exchange of endogenous troponin I with C-terminal truncated troponin I in human cardiac muscle. Circ. Res. 99, 1012–1020 [DOI] [PubMed] [Google Scholar]
- 43. Foster D. B., Noguchi T., VanBuren P., Murphy A. M., Van Eyk J. E. (2003) C-terminal truncation of cardiac troponin I causes divergent effects on ATPase and force: implications for the pathophysiology of myocardial stunning. Circ. Res. 93, 917–924 [DOI] [PubMed] [Google Scholar]
- 44. Kruger M., Zittrich S., Redwood C., Blaudeck N., James J., Robbins J., Pfitzer G., Stehle R. (2005) Effects of the mutation R145G in human cardiac troponin I on the kinetics of the contraction-relaxation cycle in isolated cardiac myofibrils. J. Physiol. 564, 347–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dong W. J., Xing J., Ouyang Y., An J., Cheung H. C. (2008) Structural kinetics of cardiac troponin C mutants linked to familial hypertrophic and dilated cardiomyopathy in troponin complexes. J. Biol. Chem. 283, 3424–3432 [DOI] [PubMed] [Google Scholar]
- 46. Iorga B., Blaudeck N., Solzin J., Neulen A., Stehle I., Lopez Davila A. J., Pfitzer G., Stehle R. (2008) Lys-184 deletion in troponin I impairs relaxation kinetics and induces hypercontractility in murine cardiac myofibrils. Cardiovasc Res. 77, 676–686 [DOI] [PubMed] [Google Scholar]
- 47. Du J., Liu J., Feng H. Z., Hossain M. M., Gobara N., Zhang C., Li Y., Jean-Charles P. Y., Jin J. P., Huang X. P. (2008) Impaired relaxation is the main manifestation in transgenic mice expressing a restrictive cardiomyopathy mutation, R193H, in cardiac TnI. Am. J. Physiol. Heart Circ. Physiol. 294, H2604–H2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Murphy A. M., Kögler H., Georgakopoulos D., McDonough J. L., Kass D. A., Van Eyk J. E., Marbán E. (2000) Transgenic mouse model of stunned myocardium. Science 287, 488–491 [DOI] [PubMed] [Google Scholar]
- 49. Li Y., Charles P. Y., Nan C., Pinto J. R., Wang Y., Liang J., Wu G., Tian J., Feng H. Z., Potter J. D., Jin J. P., Huang X. (2010) Correcting diastolic dysfunction by Ca2+-desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J. Mol. Cell Cardiol. 49, 402–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Negele J. C., Dotson D. G., Liu W., Sweeney H. L., Putkey J. A. (1992) Mutation of the high affinity calcium-binding sites in cardiac troponin C. J. Biol. Chem. 267, 825–831 [PubMed] [Google Scholar]
- 51. Putkey J. A., Sweeney H. L., Campbell S. T. (1989) Site-directed mutation of the trigger calcium-binding sites in cardiac troponin C. J. Biol. Chem. 264, 12370–12378 [PubMed] [Google Scholar]
- 52. George S. E., Su Z., Fan D., Means A. R. (1993) Calmodulin-cardiac troponin C chimeras: effects of domain exchange on calcium binding and enzyme activation. J. Biol. Chem. 268, 25213–25220 [PubMed] [Google Scholar]
- 53. Putkey J. A., Liu W., Lin X., Ahmed S., Zhang M., Potter J. D., Kerrick W. G. (1997) Fluorescent probes attached to Cys-35 or Cys-84 in cardiac troponin C are differentially sensitive to Ca2+-dependent events in vitro and in situ. Biochemistry 36, 970–978 [DOI] [PubMed] [Google Scholar]
- 54. Gillis T. E., Moyes C. D., Tibbits G. F. (2003) Sequence mutations in teleost cardiac troponin C that are permissive of high Ca2+ affinity of site II. Am. J. Physiol. Cell Physiol. 284, C1176–C1184 [DOI] [PubMed] [Google Scholar]
- 55. Maune J. F., Klee C. B., Beckingham K. (1992) Ca2+ binding and conformational change in two series of point mutations to the individual Ca2+-binding sites of calmodulin. J. Biol. Chem. 267, 5286–5295 [PubMed] [Google Scholar]
- 56. Li M. X., Wang X., Sykes B. D. (2004) Structural based insights into the role of troponin in cardiac muscle pathophysiology. J. Muscle Res. Cell Motil. 25, 559–579 [DOI] [PubMed] [Google Scholar]
- 57. Zhang J., Shettigar V., Zhang G. C., Kindell D. G., Liu X., López J. J., Yerrimuni V., Davis G. A., Davis J. P. (2011) Engineering parvalbumin for the heart: optimizing the Mg2+ binding properties of Rat β-parvalbumin. Front. Physiol. 2, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]