

Background: FoxO1 regulates expression of lipogenic genes including srebp1.
Results: FoxO1 inhibits transcription of SREBP-1c via coordinated effects on key regulatory factors including Sp1 and SREBP-1c itself.
Conclusion: FoxO1 acts at multiple levels to prevent assembly of the transcriptional complex on the srebp1 gene.
Significance: FoxO1 effectively inhibits SREBP-1c gene expression, a major regulator of hepatic lipogenesis.
Keywords: Gene Regulation, Insulin, Lipids, Liver, Transcription Factors, FoxO1, SREBP-1c, Sp1
Abstract
Induction of lipogenesis in response to insulin is critically dependent on the transcription factor, sterol regulatory element-binding protein-1c (SREBP-1c). FoxO1, a forkhead box class-O transcription factor, is an important mediator of insulin action, but its role in the regulation of lipid metabolism has not been clearly defined. We examined the effects of FoxO1 on srebp1 gene expression in vivo and in vitro. In vivo studies showed that constitutively active (CA) FoxO1 (CA-FoxO1) reduced basal expression of SREBP-1c mRNA in liver by ∼60% and blunted induction of SREBP-1c in response to feeding. In liver-specific FoxO knock-out mice, SREBP-1c expression was increased ∼2-fold. Similarly, in primary hepatocytes, CA-FoxO1 suppressed SREBP1-c expression and inhibited basal and insulin-induced SREBP-1c promoter activity. SREBP-1c gene expression is induced by the liver X receptor (LXR), but CA-FoxO1 did not block the activation of SREBP-1c by the LXR agonist TO9. Insulin stimulates SREBP-1c transcription through Sp1 and via “feed forward” regulation by newly synthesized SREBP-1c. CA-FoxO1 inhibited SREBP-1c by reducing the transactivational capacity of both Sp1 and SREBP-1c. In addition, chromatin immunoprecipitation assays indicate that FoxO1 can associate with the proximal promoter region of the srebp1 gene and disrupt the assembly of key components of the transcriptional complex of the SREBP-1c promoter. We conclude that FoxO1 inhibits SREBP-1c transcription via combined actions on multiple transcription factors and that this effect is exerted at least in part through reduced transcriptional activity of Sp1 and SREBP-1c and disrupted assembly of the transcriptional initiation complex on the SREBP-1c promoter.
Introduction
FoxO5 transcription factors are important mediators of insulin and growth factor action (1), particularly in the liver where they have emerged as important regulators of hepatic gluconeogenesis (2). In contrast, hepatic lipid synthesis is regulated by the lipogenic transcription factor sterol regulatory element-binding protein 1c (SREBP-1c) (3). In the postprandial state, insulin suppresses the expression of gluconeogenic enzymes at least in part by promoting nuclear export of FoxO1 via AKT/PKB-mediated phosphorylation (4) while coordinately inducing hepatic lipid synthesis via SREBP-1c (5). In addition to its role in regulation of gluconeogenic gene expression, recent evidence suggests that FoxO1 contributes to the regulation of hepatic lipid synthesis by modulating expression of SREBP-1c (6, 7). We have previously shown that in livers of transgenic mice expressing a constitutively active FoxO1 mutant (CA-FoxO1), FoxO1 inhibits both SREBP-1c expression and de novo hepatic lipogenesis (6). This suggests that FoxO1 may play a role in maintaining low levels of SREBP-1c expression during fasting by inhibiting SREBP-1c transcription. Conversely, insulin-mediated export of FoxO1 from the nucleus may contribute to the induction of SREBP-1c expression and lipogenesis in response to feeding. Given the pivotal role of SREBP-1c in mediating the induction of hepatic lipogenesis in hyperinsulinemic states such as obesity and type II diabetes (5), it is important to identify and characterize factors that oppose the effects of insulin on SREBP-1c. Therefore, the current studies were undertaken to better define the role of FoxO1 in regulation of SREBP-1c expression and to identify the mechanism(s) by which FoxO1 represses srebp1 gene transcription.
The primary mechanism by which insulin regulates FoxO1 is via phosphorylation and translocation of FoxO1 from the nucleus to the cytoplasm (8, 9). Human FoxO1 contains three consensus Akt/PKB phosphorylation sites including Thr-24, Ser-256, and Ser-319 (1). Phosphorylation of Ser-256 is required for subsequent phosphorylation of Thr-24 and Ser-319. Phosphorylation of Ser-319 in turn is followed by phosphorylation of Ser-322 and Ser-325 by casein kinase 1 and Ser-329 by the dual-specificity kinase 1 (DYRK1), respectively (10). The sequential phosphorylation of FoxO1 leads to enhanced formation of a complex with the nuclear export proteins Ran and Crm-1 thereby promoting nuclear exclusion of FoxO1 (11). Furthermore, phosphorylation of Thr-24 is required for interaction with 14-3-3 proteins that promote cytoplasmic sequestration of FoxO proteins (12). Thus each of the three AKT phosphorylation sites on FoxO1 participate in insulin-mediated inhibition of FoxO1 action (13), and FoxO1 is excluded from the nucleus in response to activation of the AKT/PKB signaling pathway by insulin in the postprandial state. To examine the effect of FoxO1 on srebp1 gene expression independent of this mechanism, we used a constitutively active form of FoxO1 (CA-FoxO1) in which all three Akt/PKB phosphorylation sites (Thr-24, Ser-256, and Ser-319) are rendered inactive through mutation to alanine. These alterations allow the CA-FoxO1 to remain in the nucleus and exert its transcriptional effects in the presence of insulin. Using a combined strategy of overexpression of CA-FoxO1 and depletion of endogenous FoxO proteins, we conclude that FoxO1 inhibits SREBP-1c transcription. We also demonstrate that FoxO1 directly associates with the SREBP-1c promoter and negatively regulates srebp1 gene expression via multiple mechanisms including effects on promoter binding and transactivating capacity of key transcriptional activators including Sp1, LXRα, and SREBP-1c itself.
EXPERIMENTAL PROCEDURES
Transgenic and Knock-out Mice
Transgenic mice expressing constitutively active (T24A, S256A, S319A) FoxO1 mutant in liver, directed by the human A-antitrypsin gene promoter were generated as reported previously (6). For fasting and refeeding studies, 8-week-old male transgenic mice or wild-type littermate controls were fasted overnight for 18 h and sacrificed 6 h later with or without refeeding with high carbohydrate/low polyunsaturated fatty acid chow (TD03303, Harlan, Teklad, Madison, WI) (6). For studies with the LXR agonist TO901317 (TO9) (Sigma), freely feeding male CA-FoxO1 transgenic mice and wild-type littermate controls were given TO9 (40 mg/kg) or vehicle alone (1% Tween 80 in 1% critical micellar concentration and 7% dimethyl sulfoxide) at 10 a.m. on two consecutive days, and mice were sacrificed 4 h after the last dose. Liver-specific FoxO knock-out mice were created by crossing mice with “floxed” alleles for FoxO1, FoxO3a, and FoxO4 (provided by R. Depinho) (14) with mice expressing Cre recombinase in the liver under the control of the albumin promoter (Jackson Laboratories). Disruption of FoxO genes was confirmed by monitoring FoxO mRNA and protein levels by qRT-PCR and Western blotting. Eight-week-old knock-out and Cre-negative littermate controls were fasted for 18 h prior to sacrifice.
Plasmids
The rat SREBP-1c promoter-luciferase construct pSREBP(−1516/+40)-luc and the truncated and site-specific mutant constructs were described previously (15). The pCA-FoxO1 expression vector that expresses constitutively nuclear and active FoxO1 was created by inserting into pFlag-CMV5a vector the cDNA sequence of human FoxO1 in which the three phosphorylation target sites responsible for nuclear exclusion, Thr-24, Ser-256, and Ser-319, were each replaced by an alanine. The Gal4-SREBP1c expression plasmids were created by inserting the cDNA sequences for the transactivation domain of rat SREBP-1c into the pm expression vector (Clontech, Mountain View, CA). The Gal4-LXRα plasmid containing the Gal4 DNA-binding domain in-frame with the human LXRα ligand-binding domain and the Gal4 luciferase reporter plasmid have been described previously (15). The TK luciferase reporter construct containing 6 Sp1-binding sites (pGCx6-luc) was described previously (17).
Small Interference RNA for FoxO1 (RNAi-FoxO1)
Oligonucleotides for RNAi-FoxO1 and scrambled RNAi oligonucleotides (RNAi-Scr) were synthesized by IDT (Coralville, IA). The sequences of RNAi-FoxO1 are 5′-gugugcccuacuucaaggatt-3′ (forward) and 5′-uccuugaaguagggcacactt-3′ (reverse), respectively, targeting the rat FoxO1 coding sequence (GI: 300797650). The scrambled RNAi (RNAi-Scr) sequences were generated by randomly rearranging the bases for RNAi-FoxO1 and the sequences were 5′-gcugcuacgccuugaugaatt-3′ (forward) and 5′-uucaucaaggcguagcagctt-3′ (reverse). Single-stranded siRNAs were dissolved in the duplex buffer (100 mm potassium acetate, 30 mm HEPES, pH 7.5) and annealed by incubating at 90 °C for 2 min followed by cooling to room temperature.
Primary Hepatocytes and Transient Transfection
The isolation of rat primary hepatocytes was described previously (15). Rat hepatocytes were transfected with plasmid vectors for luciferase reporter genes or transcription factors using the Lipofectin reagent (Invitrogen). Hepatocytes were co-transfected with the phRL-null vector (Promega) as a transfection control. Eighteen hours after transfection, cells were incubated in RPMI medium containing 20 mm glucose alone or with the appropriate treatments as indicated for 24 h. Cells were lysed and luciferase activity was quantified fluorimetrically using the Dual Luciferase Reporter Assay System (Promega).
Adenovirus Preparation
The creation of recombinant adenoviruses expressing the constitutively active human FoxO1 (Ad-CAFoxO1) has been described previously (6). For studies in isolated hepatocytes, adenoviral vectors were re-amplified in HEK293 cells, and plaque forming units were titrated by Vector Biolabs (Philadelphia, PA) per service agreement.
Real-time PCR
RNA was prepared from liver or rat hepatocytes using RNeasy kits (Qiagen, Valencia CA), and cDNA was synthesized using the Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Sciences) and analyzed by real time PCR (RT-PCR). The amplification of target cDNA was detected by SYBR Green and analyzed by the ΔΔCt method. Cyclophilin D or ribosomal protein L32 mRNA were used as a reference. The primers used for amplifying the genes studied are as follows: mouse SREBP-1c forward primer, 5′-cggaagctgtcggggtag-3′ and reverse primer, 5′-gttgttgatgagctggagca-3′; mouse L32 forward primer, 5′-acatttgccctgaatgtggt-3′ and reverse primer, 5′-atcctcttgccctgatcctt-3′; rat SREBP-1c forward primer, 5′-catggattgcacatttgaagac-3′ and reverse primer, 5′-gcaggagaagagaagctctcag-3′; rat fatty acid synthase forward primer, 5′-ggccacctcagtcctgttat-3′ and reverse primer, 5′-agggtccagctagagggtaca-3′; rat insulin-like growth factor binding protein-1 (IGFBP-1) forward primer, 5′-gctaggcctttgatttctccct-3′ and reverse primer, 5′-cctatgtttgtcctgttgtgatgg-3′; rat cyclophilin D forward primer, 5′-gtgaagatgtcccacccatc-3′ and reverse primer, 5′-caattctaaaacaattcgtccaac-3′.
Western Blotting Analysis
Whole cell lysates or nuclear extracts from rat hepatocytes were prepared as described previously (15, 16). One hundred micrograms of proteins were separated by SDS-PAGE, transferred onto nitrocellulose membranes, and exposed to the indicted antibodies, and visualized by SuperSignal West Femto maximum sensitivity substrate (Pierce) using the Bio-Rad image system. The antibodies used for immunoblot analyses were LXRα (RLD-1) (SC-1206, Santa Cruz Biotechnology), SREBP-1 (557036, BD Biosciences), FoxO1 (C29H4, Cell Signaling Technology), β-actin (A1978, Sigma), and Histone H3 (3638, Cell Signaling Technology).
Chromatin Immunoprecipitation Assays
ChIP assays (17) were performed on liver samples from wild-type and transgenic mice expressing CA-FoxO1 in liver 6 h after re-feeding or primary rat hepatocytes infected with adenovirus expressing CA-FoxO1. For in vivo studies, minced liver tissue was cross-linked in 1% formaldehyde at room temperature for 10 min with constant rotation and cross-linking was stopped with 125 mm glycine, then pelleted by centrifugation and washed with ice-cold PBS. The washed pellet was resuspended in hypotonic solution (10 mm HEPES, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 1 mm EDTA, 1 mm dithiothreitol, 0.15 mm spermine, 0.5 mm spermidine, 5% sucrose, and protease inhibitors), then disrupted with a Dounce homogenizer and layered onto a cushion buffer (10 mm Tris-HCl, pH 7.5, 15 mm NaCl, 60 mm KCl, 1 mm EDTA, 0.15 mm spermine, 0.5 mm spermidine, 10% sucrose, and protease inhibitors) for centrifugation. The pellet was resuspended for chromatin fragmentation and immunoprecipitation using the SimpleChIPTM Enzymatic Chromatin IP kit (Cell Signaling) according to the manufacturer's directions.
ChIP assays in hepatocytes were performed as described previously (15). Hepatocytes were infected with adenovirus expressing GFP- or CA-FoxO1 for 24 h. Then cells were incubated in control or insulin-containing medium (100 nm) for 6 h. Subsequently, cells were cross-linked with 1% formaldehyde for 10 min. Chromatin was fragmented by sonication and immunoprecipitated with specific antibodies or nonimmune IgG. The antibodies used for ChIP are the same as the aforementioned Western blotting analysis except with an additional antibody for RNA polymerase II from Cell Signaling Technology (catalog number 2629). Protein-bound DNA was released by incubation at 65 °C for 6 h, purified, and amplified by PCR.
RESULTS
CA-FoxO1 Suppresses SREBP-1c Expression in Vivo
To elucidate the role of FoxO1 in the regulation of SREBP-1c we first examined the effect of FoxO1 on hepatic expression of SREBP-1c in response to changes in nutritional status in vivo using transgenic mice with liver-specific expression of the constitutively active form of human FoxO1 (CA-FoxO1) (6). We have previously reported that the hepatic expression of this FoxO1 transgene under control of the α1-antitrypsin promoter is at levels equal to ∼60% that of the endogenous FoxO1 gene (6). The transgene expresses CA-FoxO1 protein at levels comparable with that of endogenous FoxO1 (data not shown). In contrast to endogenous FoxO1 protein, CA-FoxO1 retains its nuclear localization and transcriptional activity in the presence of insulin. As shown in Fig. 1A, SREBP-1c mRNA levels are significantly reduced in livers of transgenic mice expressing CA-FoxO1 under both fasting and re-fed conditions compared with wild-type littermate controls (Fig. 1A, left panel). Although SREBP-1c expression was increased in both WT and CA-FoxO1 mice following re-feeding, the magnitude of this induction was significantly reduced in FoxO1 transgenic mice compared with wild-type controls (Fig. 1A, right panel). These findings indicate that FoxO1 suppresses the expression of SREBP-1c in the liver, and that nuclear export of FoxO proteins in response to insulin is important to allow full stimulation of SREBP-1c expression after re-feeding.
FIGURE 1.

FoxO1 reduces hepatic expression of SREBP-1c in transgenic mice. A, effect of constitutively active FoxO1 (CA-FoxO1) on hepatic SREBP-1c gene expression in response to fasting and refeeding. Wild-type (WT, open bars) and transgenic mice expressing constitutively active FoxO1 (CA-FoxO1, solid bars) in the liver were fasted for 18 h and sacrificed 6 h after refeeding or continued fasting. Total RNA was prepared and mRNA levels of SREBP-1c were analyzed by qRT-PCR and results were expressed relative to levels in fasting WT mice (left panel). The right panel shows the relative abundance of SREBP-1c mRNA following refeeding versus fasting levels (refed-fasting) for each genotype. *, signifies p < 0.05, CA-FoxO1 versus WT. B, effect of liver-specific FoxO knock-out on hepatic SREBP-1c gene expression. Alleles for FoxO1, FoxO3a, and FoxO4 were disrupted in the liver using Cre-lox technology (FoxO KO, black bar), and Cre-negative littermates where floxed alleles were not disrupted served as controls (WT, open bar). Left panel, total hepatic RNA was prepared after an 18-h fast and SREBP-1c transcripts were quantified by qRT-PCR. Results were expressed relative to SREBP-1c mRNA levels in fasting or wild-type mice. *, signifies p < 0.05, CA-FoxO1 versus WT. Right panel, total cell lysates were prepared from FoxO knock-out mice and littermates and analyzed by Western blotting with the high-mobility group protein-1 (HMGB1) as loading control.
As shown in Fig. 1B, we also used liver-specific FoxO knock-out mice to determine whether endogenous FoxO proteins regulate SREBP-1c. Because there can be significant redundancy in the effects of different FoxO isoforms (1, 18, 19), we targeted the disruption of the FoxO1, FoxO3a, and FoxO4 alleles in the liver using the Cre-lox system and examined SREBP-1c mRNA levels in the liver. We confirmed by Western blotting that FoxO1 and FoxO3 proteins are completely eliminated from the FoxO knock-out mice (Fig. 1B). FoxO4 is expressed at very low levels in liver and was not detected in the liver of wild-type or knock-out mice by Western blotting. The SREBP-1c mRNA level was increased 2-fold in liver-specific FoxO knock-out mice compared with floxed littermate controls at the end of an 18-h fast, supporting the concept that FoxO proteins contribute to the suppression of hepatic SREBP-1c expression in vivo under fasting conditions.
To determine whether aberrant regulation of SREBP-1c in FoxO knock-out mice and CA-FoxO1 transgenic mice results from direct actions of FoxO1, we determined the effect of FoxO1 on the expression of SREBP-1c in primary rat hepatocyte cultures. We infected rat hepatocytes with adenovirus expressing constitutively active FoxO1 (Ad-CAFoxO1) plus GFP or a control virus expressing GFP alone (Ad-GFP). Cells were maintained in medium containing insulin (100 nm) to suppress the function of endogenous FoxO proteins and enhance the expression of SREBP-1c. After 36 h of incubation, RNA was prepared and quantified by qRT-PCR. As shown in Fig. 2A, expression of SREBP-1c and its downstream lipogenic enzyme target fatty acid synthase were reduced by over 50% in hepatocytes infected with Ad-CAFoxO1. These inhibitory effects were specific insofar as the expression of IGFBP-1, a gene that is positively regulated by FoxO1 (20), was markedly increased in Ad-CAFoxO1-infected hepatocytes (Fig. 2A, right panel). Western blot analysis confirmed that the adenovirus-mediated expression of CA-FoxO1 increased the level of FoxO1 in the nucleus 4-fold compared with control (Fig. 2B). This was accompanied by a marked reduction in levels of both the precursor (∼125 kDa) and nuclear (∼56 kDa) forms of SREBP-1c proteins (Fig. 2C). These data indicate that FoxO1 directly inhibits transcription of the SREBP-1c gene and reduces SREBP-1c protein levels.
FIGURE 2.

CA-FoxO1 inhibits SREBP-1c expression in rat hepatocytes. A, effect of CA-FoxO1 on mRNA levels of SREBP-1c and fatty acid synthase. Rat hepatocytes were infected with adenovirus expressing GFP alone (Ad-GFP) or a constitutively active form of FoxO1 (Ad-CAFoxO1) and then treated with 100 nm insulin for 24 h. Total RNA was prepared and the abundance of transcripts for SREBP-1c, fatty acid (FA) synthase, and IGFBP-1 was analyzed by qRT-PCR. IGFBP-1 was included as a positive control for CA-FoxO1 action. Data for RT-PCR are the mean ± S.E. of three to six independent experiments. B, Ad-CAFoxO1 increased nuclear content of FoxO1. Rat hepatocytes were infected with Ad-GFP or Ad-CAFoxO1. Nuclear extracts were analyzed for FoxO1 protein by Western blot analysis. C, Ad-CAFoxO1-reduced SREBP-1c protein. Rat hepatocytes were treated in the same manner as described in B, and cytoplasmic and nuclear proteins were prepared for Western blot analysis of SREBP-1 protein levels. Levels of both the full-length precursor and N-terminal fragment of SREBP-1c (nuclear SREBP-1c) were measured in the cytoplasmic and nuclear fractions, respectively. Western blots are representative of three independent experiments and the numbers above the bands are average levels of relative expression of the proteins quantified by densitometry.
CA-FoxO1 Inhibits Activation of SREBP-1c Transcription by Insulin
Because insulin strongly activates transcription of the srebp1 gene (15, 21) and FoxO1 is a major target of insulin action (1), we next examined the effect of CA-FoxO1 on the response of the rat SREBP-1c promoter to insulin. Primary cultures of rat hepatocytes were co-transfected with pSREBP-1c (−1516/+40)-luc and the pCMV plasmid vector expressing either CA-FoxO1 or empty vector and then incubated in the absence or presence of insulin for 24 h prior to analysis of luciferase activity. As shown in Fig. 3A, insulin treatment increased SREBP-1c promoter activity 3-fold in cells transfected with empty vector. In contrast, co-expression of CA-FoxO1 reduced basal SREBP-1c promoter activity by 50% and completely abolished the stimulatory effect of insulin. Insulin treatment reduced the level of endogenous FoxO1 protein in the nucleus of cells transfected with the empty vector. In contrast, the level of nuclear FoxO1 in hepatocytes expressing the constitutively nuclear mutant CA-FoxO1 was unaltered following insulin treatment consistent with the loss of insulin regulation in the mutant FoxO1 (Fig. 3B). This suggests that the ability of insulin to stimulate srebp1 gene transcription is, in part, due to phosphorylation and nuclear exclusion of FoxO1. To test this hypothesis, we used small interference RNA targeted against FoxO1 (RNAi-FoxO1) to further evaluate the role of endogenous FoxO1 in the regulation of SREBP-1c promoter activity in hepatocytes. Suppression of the FoxO1 protein in hepatocytes using RNAi strongly enhanced the ability of insulin to stimulate SREBP-1c promoter activity (Fig. 3C). Transfection of RNAi-FoxO1 at a final concentration of 50 nm in the growth medium reduced expression of the FoxO1 protein in rat hepatocytes (Fig. 3D). These findings indicate that endogenous FoxO1 modulates the induction of SREBP-1c by insulin.
FIGURE 3.

FoxO1 reduces the stimulation of the SREBP-1c promoter activity by insulin. A, CA-FoxO1 reduces activation of the SREBP-1c promoter by insulin. Rat hepatocytes were co-transfected with the SREBP-1c reporter plasmid pSREBP-1c (−1516/+40)-luc and either the CA-FoxO1 expression plasmid or empty vector 24 h prior to treatment with (solid bars) or without (open bars) insulin (100 nm). Cell lysates were prepared 24 h later for analysis of luciferase activity. Cells were transfected with Renilla luciferase to control for transfection efficiency. B, nuclear content of endogenous FoxO1 but not CA-FoxO1 is regulated by insulin. Hepatocytes were co-transfected with the CA-FoxO1 expression plasmid or empty vector 24 h before treatment with or without insulin (100 nm), and nuclear extracts were prepared 12 h later for analysis of FoxO1 protein by Western blot. C, SREBP-1c promoter response to insulin is enhanced by suppression of endogenous FoxO1. Hepatocytes were co-transfected overnight with pSREBP-1c (−1516/+40)-luc and either scrambled or FoxO1-specific small RNA oligos (RNAi-Sc and RNAi-FoxO1, respectively) and then treated with or without insulin for 24 h. Cell extracts were assayed for luciferase activities. D, RNAi-FoxO1 reduced FoxO1 protein expression. Hepatocytes were treated in the same manner as that for C. Whole cell lysates were prepared and analyzed by Western blotting. A representative blot of three independent experiments is shown. *, signifies p < 0.05 insulin-treated cells, RNAi-Sc versus RNAi-FoxO1. Data in panels A and C are presented as the mean ± S.E. from three hepatocyte preparations each with triplicate treatments. Numbers above the bands in B and D are average relative levels of proteins quantified by densitometry.
CA-FoxO1 Reduced Recruitment of SREBP-1c to SREBP-1c Promoter by Insulin
Because FoxO1 reduced srebp1 gene expression in the livers of mice and rat primary hepatocytes, we conducted chromatin immunoprecipitation (17) assays to determine whether this effect was achieved by reducing binding of known regulators of SREBP-1c transcription, LXR, or SREBP-1c itself to the SREBP-1c promoter. Liver samples from transgenic CA-FoxO1 mice and wild-type littermates were cross-linked and subjected to ChIP as previously described (17). The results showed that the binding of SREBP-1 and LXR to the SREBP-1c promoter was reduced by ∼60 and 30%, respectively, in the transgenic CA-FoxO1 mice as compared with the wild-type littermates (Fig. 4A). In contrast, binding of the transcriptional co-activator CBP (p300) was not affected (Fig. 4A).
FIGURE 4.

CA-FoxO1 is associated with the SREBP-1c promoter and reduces promoter occupancy by SREBP-1c, and RNA-polymerase II. A, reduced binding of SREBP-1 to the proximal SREBP-1c promoter in liver of CA-FoxO1 overexpressing transgenic mice as determined by ChIP assay. Freshly isolated nuclei from livers of wild type (WT) and transgenic (TGN) mice were cross-linked with formaldehyde, and sheared chromatin was immunoprecipitated with specific antibodies against SREBP-1c, LXRα, CBP, or IgG. The occupancy of the SREBP-1c promoter by nuclear proteins was demonstrated by the amplification with primers targeting the proximal SREBP-1c promoter. B, CA-FoxO1 reduces binding of SREBP-1c and LXRα to the proximal SREBP-1c promoter as determined by ChIP assay. Rat hepatocytes were infected with adenovirus expressing constitutively active FoxO1 (Ad-CAFoxO1) or control adenovirus (Ad-GFP). After incubation for 24 h, hepatocytes were treated with or without insulin (100 nm) for 6 h. Chromatin was prepared and immunoprecipitated with specific antibodies against SREBP-1c, LXRα, RNA polymerase, histone 3, or IgG. Occupancy of the SREBP-1c promoter by nuclear proteins was determined by amplification with primers targeting the proximal SREBP-1c promoter. C, effect of CA-FoxO1 and insulin on SREBP-1c, LXRα, and RNA polymerase II protein levels in primary hepatocyte cultures. Hepatocytes were infected with adenoviral vectors and treated with/without insulin as above before preparation of nuclear protein extracts for analysis by Western blotting. One hundred micrograms of nuclear protein was resolved by SDS-PAGE and transferred for Western blot. Representative blots from three independent experiments are shown. D, recruitment of endogenous FoxO1 and CA-FoxO1 to the SREBP-1c and IGFBP-1 promoters. Hepatocytes were infected with Ad-CA-FoxO1 or control adenovirus and treated with or without insulin as described in panel A and chromatin was precipitated using FoxO1 specific antibody or IgG. Precipitated DNA fragments were PCR amplified as described under “Experimental Procedures” using specific primers directed against the proximal IGFBP-1 or SREBP-1c promoter as indicated. Numbers above bands in A, B, and D are the average relative signal intensities, and the numbers in C represent the average relative levels of proteins, all quantified by densitometry.
We also performed ChIP assays in rat primary hepatocytes. Hepatocytes were infected with Ad-CAFoxO1 or control Ad-vector for 24 h and then further incubated with or without insulin for 6 additional hours. Following cross-linking, DNA fragments were immunoprecipitated with antibodies against LXRα, SREBP-1c, or FoxO1. The DNA fragments were amplified by PCR using primers targeting the proximal region of the SREBP-1c promoter (−162 to −25). As shown in Fig. 4B, insulin strongly enhanced recruitment of SREBP-1c to the proximal SREBP-1c promoter, consistent with our previous report (15), but this effect was abolished by adenovirus-mediated expression of CA-FoxO1. LXRα was associated with the SREBP-1c promoter both in the presence and absence of insulin, and CA-FoxO1 attenuated LXRα promoter occupancy (Fig. 4B). Furthermore, we observed that insulin strongly enhanced recruitment of RNA polymerase II to the SREBP-1c promoter, but this effect was prevented by CA-FoxO1 (Fig. 4B) indicating that CA-FoxO1 interferes with assembly of the transcriptional initiation complex on the SREBP-1c promoter.
To determine the effect of CA-FoxO1 on abundance of these transcription factors, we performed Western blot analysis of nuclear proteins from hepatocytes infected with Ad-CAFoxO1 or Ad-GFP. CA-FoxO1 blunted the induction of the SREBP-1 nuclear protein by insulin (Fig. 4C). Thus the ability of CA-FoxO1 to prevent recruitment of SREBP-1 to the promoter in response to insulin treatment in hepatocytes transfected with CA-FoxO1 is due, at least in part, to the decreased availability of the newly synthesized SREBP-1 protein. In contrast, nuclear abundance of LXRα and RNA polymerase were not significantly altered (Fig. 4C). Thus reduced promoter occupancy by LXRα and RNA polymerase II did not result from reduced nuclear levels of these proteins in hepatocytes.
FoxO1 Is Recruited to SREBP-1c Promoter in an Insulin-regulated Fashion
Although the proximal SREBP-1c promoter does not include a consensus binding sequence for FoxO1 ((C/G)(T/A)AAA(C/T)), our findings that CA-FoxO1 reduced binding of SREBP-1c and LXRα to the SREBP-1c promoter suggested the possibility that that FoxO1 might interfere with transcription factor binding and/or co-activator recruitment via direct association with the SREBP-1c promoter. We therefore determined whether FoxO1 is associated with the SREBP-1c promoter. As a control, we examined binding of FoxO1 to the promoter region of IGFBP1, which has a known FoxO1 DNA-binding site (1). As expected, both endogenous FoxO1 and exogenous CA-FoxO1 were associated with the IGFBP1 promoter, and insulin treatment disrupted the association of endogenous FoxO1 but not CA-FoxO1 (Fig. 4D, lower panel). Similarly, we observed that endogenous FoxO1 was also associated with the SREBP-1c promoter and this association was attenuated by insulin treatment. Adenoviral expressed CA-FoxO1 protein was also associated with the SREBP-1c promoter, but consistent with the inability of insulin to promote nuclear exclusion of phosphorylation-defective CA-FoxO1, this association was not affected by insulin (Fig. 4D, upper panel). Taken together, these results are consistent with the conclusion that FoxO1 inhibits SREBP-1c gene expression, at least in part, by direct interaction with the promoter, most likely via protein-protein interactions.
FoxO1 Suppresses SREBP-1c Promoter Activity
The SREBP-1c promoter contains tandem LXR response elements (5, 21, 22) and LXR plays a key role in the activation of SREBP-1c transcription by insulin (15, 23). N-3 polyunsaturated fatty acids repress SREBP-1c transcription via an inhibition of LXRα (24). Chromatin immunoprecipitation studies indicated that CA-FoxO1 reduced recruitment of LXRα to the SREBP-1c promoter (above). We therefore determined if CA-FoxO1 alters the transcriptional activity of LXR. Consistent with the role of LXR in activation of the SREBP-1c promoter, the synthetic LXR agonist T0901317 (TO9) strongly stimulates SREBP-1c transcription (15). To determine whether FoxO1 might suppress SREBP-1c transcription by interfering with ligand-dependent activation of LXR, we examined the ability of oral administration of TO9 to induce hepatic SREBP-1c mRNA levels in vivo in wild-type and CA-FoxO1 transgenic mice. As shown in the left panel of Fig. 5A, treatment with TO9 stimulated SREBP-1c expression 3-fold in WT mice. Although basal expression of SREBP-1c mRNA was markedly reduced in CA-FoxO1 mice compared WT controls, induction of SREBP-1c in response to the LXR agonist TO9 was retained (6-fold induction) indicating intact activation of LXR in CA-FoxO1 transgenic mice (Fig. 5A, left panel). As a control, we assessed IGFBP-1 expression and observed that the basal and CA-FoxO1-induced expression of IGFBP-1 was unaltered by TO9 treatment (Fig. 5A, right panel). We tested for the physical interaction of LXRα and FoxO1 using co-immunoprecipitation of overexpressed proteins. However, we were unable to observe an interaction of FoxO1 and LXRα further suggesting that inhibition of LXR is not a mechanism for the inhibition of SREBP-1c gene expression (data not shown).
FIGURE 5.

CA-FoxO1 does not alter response of SREBP-1c to the LXR agonist TO901317. A, effect of T0901317 on SREBP-1c expression in transgenic mice expressing CA-FoxO1. Wild-type (WT) and transgenic mice expressing CA-FoxO1 in the liver were treated with 40 mg/kg of LXR agonist (T090137) or vehicle (control) by gavage every 24 h for 2 days and sacrificed 4 h after the second dose in the nonfasted state. SREBP-1c (left panel) and IGFBP-1 (right panel) mRNA levels in livers were determined by qRT-PCR and expressed relative to levels in vehicle-treated WT mice. Results shown are mean ± S.E. from 4 animals in each group. B, CA-FoxO1 suppresses SREBP-1c promoter activity. Hepatocytes were co-transfected with luciferase reporter constructs containing either the wild-type (−1516/+40) SREBP-1c promoter (pSREBP-1cWT-luc) or the corresponding construct where both LXREs in the proximal SREBP-1c promoter were mutated (pSREBP-1cMutLXRE-luc), together with the CA-FoxO1 expression vector or empty vector. Cells were treated with 10 μm TO901317 or carrier alone for 24 h before cell lysates were prepared for analysis of luciferase activity. Data are presented as mean ± S.E. from three independent experiments each performed in triplicate.
To further evaluate the role of LXR in FoxO1 inhibition of SREBP-1c expression, we examined the inhibitory effects of FoxO1 on basal and TO9-stimulated SREBP-1c promoter activity in rat hepatocytes transfected with luciferase reporter gene constructs containing either the wild-type (WT) SREBP-1c promoter or a promoter construct in which both LXR response elements were disrupted (pSREBP-1c Mut-LXRE-luc). After incubation with or without 10 μm TO9 for 24 h, cell extracts were assayed for luciferase activity. As shown in Fig. 5B, TO9 strongly activated the wild-type SREBP-1c promoter. Consistent with the in vivo response observed in Fig. 5A, although co-expression of CA-FoxO1 markedly reduced basal promoter activity, the ability of TO9 to stimulate WT-SREBP-1c promoter activity was preserved (7.5-fold) (Fig. 5B). The response to TO9 in the LXRE-mutant SREBP-1c promoter although attenuated, was still present (Fig. 5B). This residual response was most likely the result of indirect “feed forward” activation of the sterol response element (SRE) of the SREBP-1c promoter via induction of endogenous SREBP-1c (15, 22). Importantly, FoxO1 effectively suppressed both basal and TO9 induced activity of the LXRE mutant promoter, indicating that CA-FoxO1 inhibits promoter activity independent of LXR (Fig. 5B). Although LXR clearly plays an important role in SREBP-1c transcription and overall promoter activity, these observations suggested that CA-FoxO1 inhibits SREBP-1c promoter activity through other factors. We next examined the role of two other factors known to mediate induction of SREBP-1c by insulin, Sp1 and SREBP-1c itself.
CA-FoxO1 Inhibits srebp1 Gene Transcription via Multiple Transcription Factors
The proximal region of the rat SREBP-1c promoter contains cis-regulatory elements for multiple transcription factors including two LXR-binding sites, a binding site for SREBP proteins themselves, and several Sp1-binding sites that contribute to basal and insulin-stimulated transcription (16). We tested by luciferase reporter assays the effects of CA-FoxO1 on the activities of the 1.5-kb wild-type SREBP-1c promoter and two truncated promoter constructs, 400 and 150 bp, the latter lacking the two LXR-binding sites (Fig. 6A). The results showed CA-FoxO1 exerted similar degrees of inhibitory effects on the activities of these three constructs (Fig. 6A), providing further support for the concept that interaction with LXR is not required for FoxO1 suppression of SREBP-1c promoter activity.
FIGURE 6.
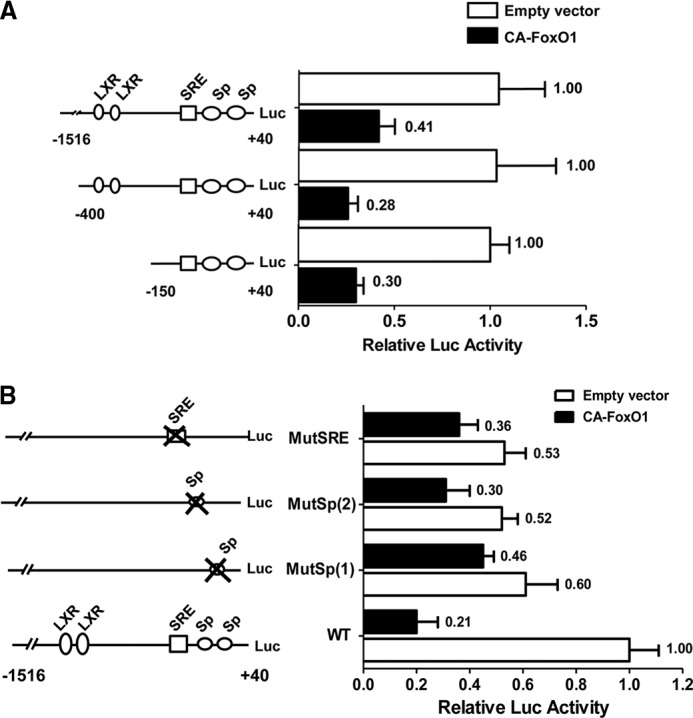
FoxO1 inhibits SREBP-1c through Sp1 and SREBP-1c. A, CA-FoxO1 inhibits full-length and truncated SREBP-1c promoter. Hepatocytes were transfected with serial deletions of the SREBP-1c promoter and empty pFlagCMV2 vector or a plasmid expressing CA-FoxO1. Thirty-six hours post-transfection, cell lysates were assayed for luciferase expression. B, CA-FoxO1 suppresses wild-type and mutant SREBP-1c promoters. Rat hepatocytes were transfected with the wild-type (WT) SREBP-1c promoter construct (pSREBP-1c (−1516/+40)-luc) or with mutant constructs in which the indicated transcription factor binding sites were disrupted, with or without plasmid expressing CA-FoxO1. Luciferase analyses were performed as described in A. The data are presented as percent inhibition of SREBP-c promoter activity by CA-FoxO1.
To further examine the roles of SREBP, and Sp1-binding sites in the inhibition of SREBP-1c by CA-FoxO1, we tested the effects of CA-FoxO1 on the 1.5-kb wild-type SREBP-1c promoter, or analogous promoter constructs in which the binding sites for SREBP and Sp1 were individually mutated. The results revealed that whereas ectopic expression of CA-FoxO1 inhibited the wild-type SREBP-1c promoter by 79%, its inhibitory effect was markedly diminished when each of these binding sites were individually mutated. The most striking effect was observed when the first proximal Sp1 site was mutated as CA-FoxO1 inhibited the promoter activity by only 24%, with luciferase activity reduced from 0.6 to 0.46 relative to that of the wild-type promoter in the absence CA-FoxO1 (Fig. 6B). These results showed that CA-FoxO1 exerts its inhibitory actions on srebp1 gene transcription via a combined effect on these multiple transcription factors.
CA-FoxO1 Reduces Transactivating Capacity of Sp1 and SREBP-1c
As the mutagenesis studies described above suggested a role of Sp1 and SREBP-1c in mediating the inhibitory effect of CA-FoxO1 on SREBP-1c promoter activity, we conducted experiments to determine the effects of FoxO1 on the transactivating capacity of Sp1 and SREBP-1c. We first tested whether CA-FoxO1 affects the transactivating capacity of LXRα independent of its interaction with an LXRE using a Gal4-LXRα construct expressing the DNA-binding domain of Gal4 in-frame with the ligand-binding and activation domains of LXRα. As shown in Fig. 7A, the activity of the Gal4X4-luc reporter construct was stimulated ∼6-fold by TO9 in the presence of ectopically expressed Gal4-LXRα. Although co-expression of CA-FoxO1 reduced the basal response of the Gal4-luciferase reporter to Gal4-LXRα in the absence of ligand, Gal4-LXRα activity was strongly (5-fold) induced by TO9 (Fig. 7A), indicating preservation of TO9 responsiveness in the presence of CA-FoxO1. The Gal4 reporter gene (Gal4X4-TK-Luc) control did not respond to either TO9 or FoxO1 alone.
FIGURE 7.

Inhibitory effects of FoxO1 on Sp1 and SREBP-1c. A, CA-FoxO1 does not reduce the response of Gal4-LXR to the LXR agonist TO9. Hepatocytes were co-transfected with pGal4-luc composed of four Gal4-binding sites fused to a minimal TK promoter along with plasmid expressing the Gal4 DNA binding alone (Gal4DBD) or a chimeric protein containing the Gal4 DNA-binding domain in-frame with the ligand binding and transactivation domains of human LXRα (Gal4LXRα), plus either the CA-FoxO1 expression vector or empty vector. Luciferase activity was quantified in cell extracts following 24 h treatment with/without TO901317. Data are presented as mean ± S.E. from three independent experiments each performed in triplicate. B, CA-FoxO1 inhibits the ability of Sp1 to activate a GC box-driven promoter. Hepatocytes were co-transfected with a luciferase reporter construct driven by six GC-box elements and an Sp1-expressing plasmid, in the presence or absence of CA-FoxO1. Cell extracts were assayed for luciferase expression 36 h post-transfection. C, CA-FoxO1 reduces transactivating capacity of Gal4-SREBP-1c. Hepatocytes were co-transfected with the pGal4-luc reporter gene construct, composed of four Gal4-binding sites fused to a minimal TK promoter (inset) together with plasmid vectors expressing the Gal4 DNA binding alone (Gal4DBD) or a chimeric protein containing the Gal4 DNA-binding domain in-frame with the transactivation domain of rat SREBP-1c (amino acids 1–300) (Gal4SREBP1c), plus either the CA-FoxO1 expression vector or empty vector. Luciferase activity was quantified in cell extracts following 36 h post-transfection. Data in A–C are presented as mean ± S.E. from 3–7 independent experiments done in triplicate.
To examine the effect of FoxO1 on the transactivating capacity of Sp1, we tested the ability of CA-FoxO1 to suppress activation of a Sp1 reporter construct containing 6 GC boxes linked to a firefly luciferase reporter gene in response to Sp1 overexpression. As shown in Fig. 7B, overexpression of Sp1 increased the promoter activity 3-fold, and this response was completely abolished by co-expressed CA-FoxO1 indicating that CA-FoxO1 repressed Sp1. This finding combined with previous observations that CA-FoxO1 reduced basal activity of the SREBP-1c promoter indicates an important role of Sp1 in mediating the inhibitory effect of CA-FoxO1.
The SRE site in the proximal SREBP-1c promoter for the binding of SREBP-1c itself plays an important role in the feed forward loop of SREBP-1c regulation. Although CA-FoxO1 attenuated recruitment of SREBP-1c to its promoter by insulin, SREBP-1c binding was not completely disrupted. Therefore, we tested the possibility that CA-FoxO1 may further disrupt feed forward stimulation of the promoter by reducing the transactivating capacity of SREBP-1c (Fig. 7C). A plasmid expressing chimeric proteins containing the activation domain of SREBP-1c coupled to the Gal4 DNA-binding domain (Gal4-SREBP1c) and a Gal4-luciferase reporter gene were co-transfected into the rat hepatoma McA cells. The activity of Gal4-luciferase was activated 7-fold by Gal4-SREBP-1c, but the activation was strongly attenuated by co-expression of CA-FoxO1 (Fig. 7C). CA-FoxO1 had no effect on the activity of the Gal4 reporter with expression of the Gal4 DNA-binding domain alone indicating that the effect of CA-FoxO1 was specific for SREBP-1c. CA-FoxO1 had no effect on expressed levels of the Gal4-SREBP-1c (data not shown). Thus CA-FoxO1 reduced the transactivating capacity of SREBP-1c independent of its effect on DNA binding.
DISCUSSION
In the current study, we employed a combination of in vivo and in vitro approaches to determine that FoxO1 inhibits SREBP-1c expression. This conclusion is based upon the following observations. 1) Transgenic mice expressing constitutively active FoxO1 (CA-FoxO1) in the liver had significantly lower SREBP-1c expression in both fasted and re-fed states than did wild-type mice (25). This inhibition of SREBP-1c expression was also evident in primary cultures of rat hepatocytes following adenovirus-mediated expression of CA-FoxO1 (3). SREBP-1c expression was increased in livers of transgenic mice with liver-specific triple deletion of FoxO1, FoxO3, and FoxO4 (4). In primary hepatocyte cultures, knockdown of FoxO1 by siRNA increased both basal and insulin-stimulated SREBP-1c promoter activity (5). CA-FoxO1 directly prevented induction of the SREBP-1c promoter by insulin. Thus our studies point to a direct inhibitory effect of FoxO1 on SREBP-1c gene expression and furthermore, support a physiological role of FoxO1 in suppression of SREBP-1c and de novo lipogenesis during fasting.
Although the current studies were being conducted, Liu et al. (26) proposed that the inhibitory effect of FoxO1 on SREBP-1c was the result of reduced binding and activity of LXR. Although LXRα is clearly important for SREBP-1c transcription, our results suggest that other mechanisms also contribute to mediating inhibitory effects of FoxO1. We also observed that FoxO1 reduces the binding of LXRα to the SREBP-1c promoter. However, we found that CA-FoxO1 inhibits SREBP-1c promoter activity even after LXR-binding sites are disrupted by either truncation or deletion and SREBP-1c expression and promoter activity remained fully responsive to the LXR agonist TO9, indicating an intact LXR signaling system. These observations indicated that the inhibitory effect of FoxO1 is not limited to impaired activation of LXR, and that other mechanisms also are involved. Thus, our findings do not support a primary role for LXR in the inhibitory effect of FoxO1. We have identified inhibition of Sp1 and SREBP-1c as additional novel mechanisms by which FoxO1 inhibits srebp1 gene expression.
The finding that both endogenous and CA FoxO1 are associated with the SREBP-1c promoter is particularly intriguing. The PCR primers used in the ChIP assays amplified the proximal portion of the SREBP-1c promoter. Although a potential FoxO1-binding site (TGTTTT) is present in the distal promoter (−1209/−1214), the proximal promoter has no known FoxO1-binding site. Furthermore, the suppressive effect of FoxO1 is equally evident in truncated SREBP-1c promoter constructs that do not contain a putative FoxO1-binding site suggesting that FoxO1 associates with the proximal SREBP-1c promoter, most likely via protein-protein interactions. Interestingly, a recent microarray analysis of a FoxO1 knock-out mouse model Gene Set Enrichment Analysis revealed that the promoter regions of most genes regulated by FoxO1 did not contain a FOXO recognition site (27).
Induction of SREBP-1c transcription is dependent upon the coordinated assembly of a transcriptional complex that includes LXRα, Sp1, and SREBP-1c itself (15, 16). We found that inhibition of srebp1 gene expression by CA-FoxO1 is mediated by its effects on Sp1 and SREBP-1c. CA-FoxO1 inhibits srebp1 gene expression both by reducing the transcriptional activity of Sp1 and SREBP-1c and by decreasing the feed forward activation of the srebp1 gene by newly synthesized SREBP-1c. We further demonstrated that CA-FoxO1 effectively prevents the recruitment of RNA polymerase to the promoter in response to insulin treatment and that FoxO1 itself is directly associated with the SREBP-1c promoter in vivo. These findings indicate that FoxO1 inhibits SREBP-1c gene transcription by preventing the assembly of the transcriptional complex on the SREBP-1c promoter. Although the exact mechanism remains undefined, our findings indicate that FoxO1 prevents assembly of the transcriptional complex via its effects on Sp1 and SREBP-1c. In this regard, it is important to note that FoxO proteins have been shown to suppress expression of other genes by sequestering transcription factors and co-activators (28, 29). In addition, FoxO proteins also can repress promoter activity by recruiting co-repressor proteins (30–32).
FoxO1 exerts both positive and negative effects on gene transcription. Our finding that FoxO1 inhibits Sp1 transactivating capacity may appear counterintuitive in this regard. However, it is apparent that the transcriptional effect of FoxO1 is specific to the promoter context. In contrast to the effect of FoxO1 on the SREBP-1c gene that lacks a functional FoxO1-binding site, FoxO1 directly induces transcription of genes such as IGFBP1 via a FKH factor site in the promoter (33). Regarding the roles of Sp1, on the basis of the data reported here and earlier published observations, we posit that a putative role of Sp1 in the regulatory mechanisms of FoxO1 appears to be highly promoter context-specific. For example, the gluconeogenic gene G6Pase, which is activated by FoxO1, has two bona fide Sp1 motifs in the proximal promoter that were required for basal expression of the G6Pase gene (34) and yet activation of G6Pase by FoxO1 is dependent on two FoxO1-binding sites (35). Similarly, activation of the IGFBP1 gene by FoxO1 is also mediated via two FoxO1-binding elements. However, unlike the promoters of G6Pase and the IGFBP1 genes that contain the TATA box element (34, 36), the SREBP-1c promoter lacks a canonical TATA element, and the Sp1 motif near the transcription start site plays a critical role in promoter activity (16, 21). In TATA-less promoters Sp1 may act as a key transcription factor where it tethers the transcription preinitiation complex to the promoter with a heat-labile complex of TFIID (37). Based on a large body of data it may be surmised that Sp1 is a specific transcriptional activator of many genes, whereas its function is dispensable for the activation of other genes (38). Our observation that CA-FoxO1 reduced the transactivating potential of Sp1 and SREBP-1c as well as the occupancy of RNA polymerase II on SREBP-1c promoter (Fig. 4C) is entirely consistent with this interpretation.
FoxO1 regulates SREBP-1c expression by complex mechanisms that likely reflect the sum of both direct inhibitory effects as observed in the present study and other indirect opposing actions of FoxO1 on insulin signaling, glucose metabolism, and gene expression (1, 39). Although we and others have observed negative regulation of SREBP-1c by FoxO1 both in vitro and in vivo (6, 26, 31), others have reported that the constitutively active FoxO1 mutants induce SREBP-1c in vivo (7, 40). These seemingly opposing effects of overexpressed constitutively active FoxO1 mutants may result from differences in genetic background of the animal models used, duration of treatment, levels of FoxO1 expressed, nutritional state, or differing characteristics of the FoxO1 mutants employed (6, 7, 40). In the current study, we used a mutant in which all three AKT phosphorylation sites (Thr-23, Ser-253, and Ser-319) were mutated to a nonphosphorylated amino acid (alanine) (FoxO1-AAA) (9). Other studies used a mutant construct in which serine 253 was mutated to the phosphomimetic amino acid aspartic acid instead of alanine (FoxO1-ADA) (41). As noted by Naimi et al. (42), the FoxO1-ADA mutant behaves as if it were phosphorylated on Ser-253. Thus the effects of these mutants may differ as each phosphorylation site has its own unique role in regulating nuclear export, DNA binding, protein-protein interactions, and transactivating capacity of FoxO1 (43). For example, we have shown previously that replacing Ser-256 with the phosphomimetic amino acid aspartate reduces the DNA binding and transactivating capacity of FoxO1 (44).
The effect of constitutively active FoxO1 mutants on SREBP-1c may also reflect indirect effects on insulin signaling pathways. Matsumoto et al. (7) noted that induction of SREBP-1c by FoxO1-ADA was associated with enhanced activation of the Akt/PKB pathway by insulin, an effect that was independent of DNA binding. Because the Akt/PKB pathway mediates induction of SREBP-1c by insulin (45), activation of this pathway by constitutively active FoxO mutants may oppose any direct inhibitory effects of FoxO1 on SREBP-1c in vivo. The complexity of the signaling pathways involved in induction of SREBP-1c by insulin (46) and the divergent effects of FoxO on various components of the insulin signaling pathway (47) further add to the potential for varying effects of FoxO1 under differing experimental conditions.
It is important to note that constitutively active phosphorylation defective FoxO1 mutants such as CA-FoxO1 are not regulated in a physiologic fashion by insulin. Thus, although they provide useful insights into FoxO1 function independent of insulin effects, they do not necessarily reflect regulation of endogenous FoxO1 under physiologic conditions. However, our evidence indicates that FoxO proteins participate in regulation of SREBP-1c in a physiologic manner. We found that reducing endogenous FoxO1 using RNAi in primary hepatocytes and in FoxO knock-out mice, respectively, resulted in enhanced SREBP-1c gene expression. These results are consistent with other studies where disrupting the expression of FoxO1 in the liver is associated with an increase in SREBP-1c expression in mice with insulin deficiency or disrupted insulin signaling (27, 48). Together, these studies provide strong genetic evidence that FoxO proteins negatively regulate the expression of SREBP-1c in the liver. In addition, our ChIP studies demonstrate that FoxO1 directly associates with the SREBP-1c promoter and that this association is regulated in a physiologic manner by insulin. Therefore, these studies support the hypothesis that FoxO1 contributes to the repression of SREBP-1c gene expression during fasting and/or insulin-deficient states (6, 26, 31). On the other hand, in conditions such as the metabolic syndrome, impaired suppression of FoxO1 may trigger an alternative activation of Akt signaling that can lead to increased hepatic lipid synthesis in the face of insulin resistance (7). Therefore, FoxO1 may have differing effects on SREBP-1c regulation in normal versus insulin-resistant states.
In summary, we have shown that FoxO1 represses SREBP-1c gene expression and that this inhibitory effect of FoxO1 is mediated at least in part via reduced trans activating capacity of Sp1 and SREBP-1c and by reduced feed forward activation of the SREBP-1c promoter via newly synthesized SREBP-1c. This results in failure to assemble the transcriptional complex as evidenced by lack of recruitment of polymerase II to the promoter in response to insulin. The inhibition by FoxO1 involves direct association of FoxO1 with the SREBP-1c promoter. Further delineation of the mechanism(s) by which FoxO1 and other regulatory factors suppress srebp1 gene expression may identify novel molecular targets to improve lipid metabolism in insulin-resistant and diabetic states.
This work was supported, in whole or in part, by National Institutes of Health Grants DK075505 (to M. B. E.), DK0059368 (to E. A. P.), and DK41430 (to T. G. U.) and a grant from the Department of Veterans Affairs Merit Review Program (to X. D., T. G. U., R. R., and M. B. E).
- FoxO
- Forkhead box class O transcription factor
- SREBP-1c
- sterol-regulatory element-binding protein-1c
- LXR
- liver X receptor
- CA-FoxO1
- constitutively active FoxO1 (FoxO1-AAA)
- IGFBP-1
- insulin growth factor-binding protein-1
- qRT
- quantitative real-time
- Ad
- adenovirus.
REFERENCES
- 1. Barthel A., Schmoll D., Unterman T. G. (2005) FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 16, 183–189 [DOI] [PubMed] [Google Scholar]
- 2. Haeusler R. A., Accili D. (2008) The double life of IRS. Cell Metab. 8, 7–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Horton J. D., Goldstein J. L., Brown M. S. (2002) SREBPs. Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Puigserver P., Rhee J., Donovan J., Walkey C. J., Yoon J. C., Oriente F., Kitamura Y., Altomonte J., Dong H., Accili D., Spiegelman B. M. (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature 423, 550–555 [DOI] [PubMed] [Google Scholar]
- 5. Raghow R., Yellaturu C., Deng X., Park E. A., Elam M. B. (2008) SREBPs. The cross-roads of physiological and pathological lipid homeostasis. Trends Endocrinol. Metab. 19, 65–73 [DOI] [PubMed] [Google Scholar]
- 6. Zhang W., Patil S., Chauhan B., Guo S., Powell D. R., Le J., Klotsas A., Matika R., Xiao X., Franks R., Heidenreich K. A., Sajan M. P., Farese R. V., Stolz D. B., Tso P., Koo S. H., Montminy M., Unterman T. G. (2006) FoxO1 regulates multiple metabolic pathways in the liver. Effects on gluconeogenic, glycolytic, and lipogenic gene expression. J. Biol. Chem. 281, 10105–10117 [DOI] [PubMed] [Google Scholar]
- 7. Matsumoto M., Han S., Kitamura T., Accili D. (2006) Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J. Clin. Invest. 116, 2464–2472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., Greenberg M. E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 [DOI] [PubMed] [Google Scholar]
- 9. Scheimann A. O., Durham S. K., Suwanichkul A., Snuggs M. B., Powell D. R. (2001) Role of three FKHR phosphorylation sites in insulin inhibition of FKHR action in hepatocytes. Horm. Metab. Res. 33, 631–638 [DOI] [PubMed] [Google Scholar]
- 10. Woods Y. L., Rena G., Morrice N., Barthel A., Becker W., Guo S., Unterman T. G., Cohen P. (2001) The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser-329 in vitro, a novel in vivo phosphorylation site. Biochem. J. 355, 597–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rena G., Guo S., Cichy S. C., Unterman T. G., Cohen P. (1999) Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 274, 17179–17183 [DOI] [PubMed] [Google Scholar]
- 12. Rena G., Prescott A. R., Guo S., Cohen P., Unterman T. G. (2001) Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in regulating 14-3-3 binding, transactivation, and nuclear targetting. Biochem. J. 354, 605–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao X., Gan L., Pan H., Kan D., Majeski M., Adam S. A., Unterman T. G. (2004) Multiple elements regulate nuclear/cytoplasmic shuttling of FOXO1. Characterization of phosphorylation- and 14-3-3-dependent and -independent mechanisms. Biochem. J. 378, 839–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paik J. H., Kollipara R., Chu G., Ji H., Xiao Y., Ding Z., Miao L., Tothova Z., Horner J. W., Carrasco D. R., Jiang S., Gilliland D. G., Chin L., Wong W. H., Castrillon D. H., DePinho R. A. (2007) FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 128, 309–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cagen L. M., Deng X., Wilcox H. G., Park E. A., Raghow R., Elam M. B. (2005) Insulin activates the rat sterol regulatory element-binding protein 1c (SREBP-1c) promoter through the combinatorial actions of SREBP, LXR, Sp-1, and NF-Y cis-acting elements. Biochem. J. 385, 207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deng X., Yellaturu C., Cagen L., Wilcox H. G., Park E. A., Raghow R., Elam M. B. (2007) Expression of the rat sterol regulatory element-binding protein-1c gene in response to insulin is mediated by increased transactivating capacity of specificity protein 1 (Sp1). J. Biol. Chem. 282, 17517–17529 [DOI] [PubMed] [Google Scholar]
- 17. Thijssen-Timmer D. C., Schiphorst M. P., Kwakkel J., Emter R., Kralli A., Wiersinga W. M., Bakker O. (2006) PGC-1α regulates the isoform mRNA ratio of the alternatively spliced thyroid hormone receptor α transcript. J. Mol. Endocrinol. 37, 251–257 [DOI] [PubMed] [Google Scholar]
- 18. Haeusler R. A., Kaestner K. H., Accili D. (2010) FoxOs function synergistically to promote glucose production. J. Biol. Chem. 285, 35245–35248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tothova Z., Kollipara R., Huntly B. J., Lee B. H., Castrillon D. H., Cullen D. E., McDowell E. P., Lazo-Kallanian S., Williams I. R., Sears C., Armstrong S. A., Passegué E., DePinho R. A., Gilliland D. G. (2007) FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128, 325–339 [DOI] [PubMed] [Google Scholar]
- 20. Guo S., Rena G., Cichy S., He X., Cohen P., Unterman T. (1999) Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J. Biol. Chem. 274, 17184–17192 [DOI] [PubMed] [Google Scholar]
- 21. Deng X., Cagen L. M., Wilcox H. G., Park E. A., Raghow R., Elam M. B. (2002) Regulation of the rat SREBP-1c promoter in primary rat hepatocytes. Biochem. Biophys. Res. Commun. 290, 256–262 [DOI] [PubMed] [Google Scholar]
- 22. Amemiya-Kudo M., Shimano H., Yoshikawa T., Yahagi N., Hasty A. H., Okazaki H., Tamura Y., Shionoiri F., Iizuka Y., Ohashi K., Osuga J., Harada K., Gotoda T., Sato R., Kimura S., Ishibashi S., Yamada N. (2000) Promoter analysis of the mouse sterol regulatory element-binding protein-1c gene. J. Biol. Chem. 275, 31078–31085 [DOI] [PubMed] [Google Scholar]
- 23. Chen G., Liang G., Ou J., Goldstein J. L., Brown M. S. (2004) Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. U.S.A. 101, 11245–11250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Howell G., 3rd, Deng X., Yellaturu C., Park E. A., Wilcox H. G., Raghow R., Elam M. B. (2009) N-3 polyunsaturated fatty acids suppress insulin-induced SREBP-1c transcription via reduced trans-activating capacity of LXRα. Biochim. Biophys. Acta 1791, 1190–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yellaturu C. R., Deng X., Cagen L. M., Wilcox H. G., Mansbach C. M., 2nd, Siddiqi S. A., Park E. A., Raghow R., Elam M. B. (2009) Insulin enhances post-translational processing of nascent SREBP-1c by promoting its phosphorylation and association with COPII vesicles. J. Biol. Chem. 284, 7518–7532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu X., Qiao A., Ke Y., Kong X., Liang J., Wang R., Ouyang X., Zuo J., Chang Y., Fang F. (2010) FoxO1 represses LXRα-mediated transcriptional activity of SREBP-1c promoter in HepG2 cells. FEBS Lett. 584, 4330–4334 [DOI] [PubMed] [Google Scholar]
- 27. Dong X. C., Copps K. D., Guo S., Li Y., Kollipara R., DePinho R. A., White M. F. (2008) Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 8, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hirota K., Daitoku H., Matsuzaki H., Araya N., Yamagata K., Asada S., Sugaya T., Fukamizu A. (2003) Hepatocyte nuclear factor-4 is a novel downstream target of insulin via FKHR as a signal-regulated transcriptional inhibitor. J. Biol. Chem. 278, 13056–13060 [DOI] [PubMed] [Google Scholar]
- 29. Dowell P., Otto T. C., Adi S., Lane M. D. (2003) Convergence of peroxisome proliferator-activated receptor γ and FoxO1 signaling pathways. J. Biol. Chem. 278, 45485–45491 [DOI] [PubMed] [Google Scholar]
- 30. Liu Z. P., Wang Z., Yanagisawa H., Olson E. N. (2005) Phenotypic modulation of smooth muscle cells through interaction of FoxO4 and myocardin. Dev Cell 9, 261–270 [DOI] [PubMed] [Google Scholar]
- 31. Kamei Y., Miura S., Suganami T., Akaike F., Kanai S., Sugita S., Katsumata A., Aburatani H., Unterman T. G., Ezaki O., Ogawa Y. (2008) Regulation of SREBP1c gene expression in skeletal muscle. Role of retinoid X receptor/liver X receptor and forkhead-O1 transcription factor. Endocrinology 149, 2293–2305 [DOI] [PubMed] [Google Scholar]
- 32. Liu P., Li S., Gan L., Kao T. P., Huang H. (2008) A transcription-independent function of FOXO1 in inhibition of androgen-independent activation of the androgen receptor in prostate cancer cells. Cancer Res. 68, 10290–10299 [DOI] [PubMed] [Google Scholar]
- 33. Nasrin N., Ogg S., Cahill C. M., Biggs W., Nui S., Dore J., Calvo D., Shi Y., Ruvkun G., Alexander-Bridges M. C. (2000) DAF-16 recruits the CREB-binding protein coactivator complex to the insulin-like growth factor-binding protein 1 promoter in HepG2 cells. Proc. Natl. Acad. Sci. U.S.A. 97, 10412–10417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wasner C., Grempler R., Walther R., Schmoll D. (2001) Basal level of glucose-6-phosphatase gene transcription requires binding site for Sp family proteins within the gene promoter. Biochim. Biophys. 1521, 126–129 [DOI] [PubMed] [Google Scholar]
- 35. Nakae J., Kitamura T., Ogawa W., Kasuga M., Accili D. (2001) Insulin regulation of gene expression through the forkhead transcription factor Foxo1 (Fkhr) requires kinases distinct from Akt. Biochemistry 40, 11768–11776 [DOI] [PubMed] [Google Scholar]
- 36. Unterman T. G., Lacson R. G., McGary E., Whalen C., Purple C., Goswami R. G. (1992) Cloning of the rat insulin-like growth factor binding protein-1 gene and analysis of its 5′ promoter region. Biochem. Biophys. Res. Commun. 185, 993–999 [DOI] [PubMed] [Google Scholar]
- 37. Pugh B. F., Tjian R. (1991) Transcription from a TATA-less promoter requires a multisubunit TFIID complex. Genes Dev. 5, 1935–1945 [DOI] [PubMed] [Google Scholar]
- 38. Solomon S. S., Majumdar G., Martinez-Hernandez A., Raghow R. (2008) A critical role of Sp1 transcription factor in regulating gene expression in response to insulin and other hormones. Life Sci. 83, 305–312 [DOI] [PubMed] [Google Scholar]
- 39. Tzivion G., Dobson M., Ramakrishnan G. (2011) FoxO transcription factors: Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta, 1813, 1938–1945 [DOI] [PubMed] [Google Scholar]
- 40. Qu S., Altomonte J., Perdomo G., He J., Fan Y., Kamagate A., Meseck M., Dong H. H. (2006) Aberrant Forkhead box O1 function is associated with impaired hepatic metabolism. Endocrinology 147, 5641–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakae J., Barr V., Accili D. (2000) Differential regulation of gene expression by insulin and IGF-1 receptors correlates with phosphorylation of a single amino acid residue in the forkhead transcription factor FKHR. EMBO J. 19, 989–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Naïmi M., Gautier N., Chaussade C., Valverde A. M., Accili D., Van Obberghen E. (2007) Nuclear forkhead box O1 controls and integrates key signaling pathways in hepatocytes. Endocrinology 148, 2424–2434 [DOI] [PubMed] [Google Scholar]
- 43. Rena G., Woods Y. L., Prescott A. R., Peggie M., Unterman T. G., Williams M. R., Cohen P. (2002) Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J. 21, 2263–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang X., Gan L., Pan H., Guo S., He X., Olson S. T., Mesecar A., Adam S., Unterman T. G. (2002) Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J. Biol. Chem. 277, 45276–45284 [DOI] [PubMed] [Google Scholar]
- 45. Fleischmann M., Iynedjian P. B. (2000) Regulation of sterol regulatory element-binding protein 1 gene expression in liver. Role of insulin and protein kinase B/cAkt. Biochem. J. 349, 13–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yecies J. L., Zhang H. H., Menon S., Liu S., Yecies D., Lipovsky A. I., Gorgun C., Kwiatkowski D. J., Hotamisligil G. S., Lee C. H., Manning B. D. (2011) Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and -independent pathways. Cell Metab. 14, 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen C. C., Jeon S. M., Bhaskar P. T., Nogueira V., Sundararajan D., Tonic I., Park Y., Hay N. (2010) FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev. Cell 18, 592–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Haeusler R. A., Han S., Accili D. (2010) Hepatic FoxO1 ablation exacerbates lipid abnormalities during hyperglycemia. J. Biol. Chem. 285, 26861–26868 [DOI] [PMC free article] [PubMed] [Google Scholar]