

Background: Neuropeptide Y and nitric oxide are key regulators of adult hippocampal neurogenesis.
Results: Pharmacological inhibition of intracellular NO signaling pathways abolished neuropeptide Y-mediated neuroproliferation.
Conclusion: An intracellular NO-cGMP-PKG and ERK pathway mediates the neuropeptide Y neuroproliferative effect.
Significance: This work unites two significant modulators of hippocampal neurogenesis and provides a mechanism for the independent extra- and intracellular regulation of postnatal neural precursors by NO.
Keywords: Cell Culture, Neurogenesis, Neuropeptide, Nitric Oxide, Signal Transduction, Neuronal Precursor Cells
Abstract
Neuropeptide Y (NPY) is widely expressed in the central and peripheral nervous systems and is proliferative for a range of cells types in vitro. NPY plays a key role in regulating adult hippocampal neurogenesis in vivo under both basal and pathological conditions, although the underlying mechanisms are largely unknown. We have investigated the role of nitric oxide (NO) on the neurogenic effects of NPY. Using postnatal rat hippocampal cultures, we show that the proliferative effect of NPY on nestin+ precursor cells is NO-dependent. As well as the involvement of neuronal nitric-oxide synthase, the proliferative effect is mediated via an NO/cyclic guanosine monophosphate (cGMP)/cGMP-dependent protein kinase (PKG) and extracellular signal-regulated kinase (ERK) 1/2 signaling pathway. We show that NPY-mediated intracellular NO signaling results in an increase in neuroproliferation. By contrast, extracellular NO had an opposite, inhibitory effect on proliferation. The importance of the NO-cGMP-PKG signaling pathway in ERK1/2 activation was confirmed using Western blotting. This work unites two significant modulators of hippocampal neurogenesis within a common signaling framework and provides a mechanism for the independent extra- and intracellular regulation of postnatal neural precursors by NO.
Introduction
Neuropeptide Y (NPY)2 is a highly conserved 36-amino acid polypeptide widely expressed in neural tissue of the central and peripheral nervous systems (1). Following its isolation from the porcine hypothalamus in 1982 (2), and determination of its amino acid sequence (1) and its distribution in the rat brain (3), NPY has been implicated in the regulation of a range of physiological processes, including circadian rhythms (4), feeding behavior (5), memory processing (6), affective disorders (7), and seizure control (8, 9). In the hippocampus, NPY is expressed by GABAergic interneurons of the dentate hilus and in the hippocampus proper (10, 11) and acts on G-protein coupled receptors (GPCR) in the rhodopsin-like GPCR family. Out of the five subtypes identified in mammals, four (Y1, Y2, Y4, and Y5) are functional in humans (12) and show tissue-specific expression patterns (13).
Hansel et al. (14) first demonstrated the proliferative effects of NPY on neuronal precursor cells derived from the postnatal rat olfactory epithelium, which appear to be mediated via the NPY Y1 receptor. Since then, we and others have shown that NPY is able to stimulate the proliferation of a range of cell types including neuronal precursor cells from the hippocampal subgranular zone (SGZ) (15), retinal glial (Muller) cells (16), endothelial cells (17), and precursor cells from the subventricular zone (SVZ) (18). Adult neurogenesis is well established in mammalian brains, including in humans (19), and NPY plays a key role in regulating adult hippocampal neurogenesis under both normal and pathological conditions (20, 21). It has been proposed that NPY-releasing interneurons in the dentate hilus release NPY onto progenitors within the SGZ to regulate their proliferation and adult hippocampal neurogenesis (22). The signaling mechanisms underlying the proliferative effect of NPY are not fully understood, although the extracellular signal-regulated kinases (ERK) 1/2 of the mitogen-activated protein kinases (MAPK) signaling pathway have been shown to play a major role (14, 20, 23).
Nitric oxide (NO) is a ubiquitous gaseous signaling molecule involved in mediating a vast range of intra- and intercellular signaling cascades and regulation of physiological processes (24). NO is synthesized by the enzyme nitric-oxide synthase (NOS), which converts the amino acid l-arginine into NO and citrulline through sequential oxygenation reactions (25). There are three isoforms of NOS, neuronal NOS (nNOS or NOS-I), inducible NOS (iNOS or NOS-II), and endothelial NOS (eNOS or NOS-III) (26). Since NO was first shown to act as an intercellular messenger in the brain (27), its role as a neurotransmitter/neuromodulator has been extensively studied (28).
NO has been implicated in the mechanisms underlying both the early stages (neurogenesis) and the advanced stages (synaptogenesis and neural map formation) of neuronal differentiation and also in developmental neurogenesis (28). In addition, NOS is localized to neurogenic sites in the dentate gyrus of the hippocampus and SGZ (29) and the SVZ, olfactory, and rostral migratory stream (30). NO has also been shown to exert a dual role on cell proliferation by mediating both antiproliferative and proliferative effects (25). Although NO exerts an antiproliferative effect on cells such as vascular smooth muscle cells (31) and endothelial cells (32), it mediates a proliferative effect on fibroblasts (33) and myoblasts (34). Given the importance of NPY in regulating adult hippocampal neurogenesis, the localization of NOS to NPY-responsive neurogenic areas, and the bifunctional effects of NO on cell proliferation, it is therefore possible that NO acts as a mediator of NPY signaling. In fact, a link between NO and the physiological effects of NPY was suggested by Morley et al. (35), who showed that a low dose of NPY was able to increase NOS expression in the hypothalamus by 147% and that an NPY-induced increase in food intake was mediated via NOS. More recently, Alvaro et al. (23) showed that NOS was involved in mediating the NPY-induced proliferation of retinal neural cells, further supporting our hypothesis. Through the use of pharmacological, immunocytochemical, and live cell imaging techniques, we describe the involvement of NO signaling in mediating the neuroproliferative effect of NPY on cultures derived from the postnatal rat hippocampus.
EXPERIMENTAL PROCEDURES
Generation of Hippocampal Cultures from Postnatal Rat
Primary hippocampal cell cultures were generated from 7–10-day-old postnatal Wistar rats as described previously by Howell et al. (15) and described in full in the supplemental Experimental Procedures. Cells were plated at 100,000 viable cells/ml directly onto poly-l-lysine (Sigma-Aldrich) coated 6- or 24-well plates for cell counting or poly-l-lysine-coated sterile borosilicate glass coverslips for confocal imaging (13-mm diameter; VWR International). The wells were rinsed and replenished with fresh Neurobasal A® medium supplemented with 2% B27 supplement (Invitrogen, Paisley, UK) and 0.5 mm glutamine (Sigma-Aldrich) (NBA/B27/Glu) with 1% antibiotic/antimycotic (penicillin/streptomycin and Fungizone, Invitrogen) 2 h after plating to remove nonadhered cells and debris (control conditions). Pharmacological reagents under study were also added at this time or cultured for 3 days in vitro (DIV) under control conditions until exposure for 6 h on the final day.
Exposure to Pharmacological Agonists and Antagonists and BrdU Incorporation
The concentrations of agonists or antagonists were chosen based on published literature and included NPY (1 μm; Sigma-Aldrich) (15), the nonselective NOS inhibitor N(ω)-nitro-l-arginine methyl ester (l-NAME) (500 μm; Sigma-Aldrich) (23), the inactive enantiomer N(ω)-nitro-d-arginine methyl ester (d-NAME) (500 μm; Sigma), the selective Y1 agonist [F7, P34]NPY (1 μm; courtesy of Prof. Dr. A. G. Beck-Sickinger, Universität Leipzig, Germany) (15), the NOS substrate l-arginine (500 μm; Sigma-Aldrich), the inactive enantiomer d-arginine (500 μm; Sigma-Aldrich), the cGMP analog 8-Br-cGMP (20 μm; Tocris Bioscience) (23), the selective soluble guanylate cyclase (sGC) inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 50 μm; Ascent Scientific) (23), the MEK 1/2 inhibitor U0126 (1 μm; Ascent Scientific) (15), the cGMP-dependent protein kinase (PKG) inhibitor KT5823 (1 μm; Tocris Bioscience), the selective nNOS inhibitor N-(4S)-4-amino-5-[aminoethyl]aminopentyl-N′-nitroguanidine (AAAN, 200 nm; Sigma-Aldrich), the selective iNOS inhibitor EIT hydrobromide (EITH, 100 nm; Tocris Bioscience), the NO donor DETA/NONOate (NOC-18, 50/100/200 μm; Calbiochem, Merck Biosciences) (42), and the NO scavenger carboxy-PTIO (CPTIO, 10 μm; Tocris Bioscience) (42, 43). Agonists or antagonists were added to cultures for 3 or 5 DIV or for 6 h on the final day before cells were rinsed once in 0.1 m phosphate-buffered saline (PBS; Sigma-Aldrich) and fixed in 4% paraformaldehyde (Sigma-Aldrich) for 20 min. Cell proliferation was assessed through incorporation of the thymidine analog 5-bromo-2-deoxyuridine (BrdU; Sigma-Aldrich). Cultures were exposed to 20 μm BrdU for 6 h before cell fixation.
Immunocytochemistry
Fixed cultures were washed in PBS before BrdU-treated cultures were incubated in 2 m HCl for 30 min at 37 °C. Cells were then washed in PBS before the nonspecific antibody-binding sites were blocked by incubation in 5% donkey blocking serum (Sigma-Aldrich) in PBS-0.1% Triton (PBS-T) for 30 min at 25 °C. Cells were then incubated in appropriate dilutions of primary antibodies in PBS-T, 5% donkey blocking serum overnight at 4 °C. Primary antibodies were used at the following concentrations: rat anti-BrdU (1:500; AbD Serotec), mouse anti-rat nestin (1:200; BD Biosciences), mouse anti-class III β-tubulin (1:500; Cambridge Bioscience), rabbit anti-class III β-tubulin (1:500; Cambridge Bioscience), and rabbit anti-nNOS (1:500, 1:1000; Millipore). Cells were washed once in PBS before incubation with secondary antibodies in PBS-T for 2 h at 25 °C. Secondary antibodies were used at the following concentrations: Alexa Fluor donkey anti-rat 488 (1:1000; Molecular Probes, Invitrogen), Alexa Fluor donkey anti-mouse 488 (1:500; Molecular Probes, Invitrogen), Alexa Fluor donkey anti-rabbit 488 (1:500; Molecular Probes, Invitrogen), Alexa Fluor donkey anti-mouse 555 (1:500; Molecular Probes, Invitrogen), and Alexa Fluor donkey anti-rabbit 555 (1:500; Molecular Probes, Invitrogen). Cells were rinsed once with PBS before counterstaining with 4′,6-diamidino-2-phenylindole (DAPI, 20 μg/ml; Sigma) for 6 min. Samples were rinsed in PBS and maintained in distilled water for imaging. Cells cultured on coverslips underwent an additional mounting step after immunostaining. Coverslips were transferred face down onto mounting medium Mowiol aqueous mount.
Quantification and Statistical Analysis
An inverted Leica DM IRB microscope (Leica Microsystems UK Ltd., Milton Keynes, UK) was used to capture fluorescent images for cell counting. Cell quantification was performed on six random 20× fields/well using Volocity version 4.2.0 (Improvision Inc., Lexington, KY). The area of a 20× field was previously measured using a 255-μm graticule slide. Raw data counts were averaged and expressed as the mean number of cells/mm2/well and plotted ± S.E. Plots were based on the mean of a sample of at least four wells per condition per experiment from a minimum of three independent experiments, with each experiment consisting of 16 hippocampi pooled from eight animals. The mitotic index (in percentage), which reflects the proliferation status of a cell population, was quantified by dividing the numbers of BrdU+ cells (cells in S-phase) by the total cell count. Data were plotted using GraphPad Prism data analysis software (GraphPad Software Inc., San Diego, CA) and the appropriate statistical test (ANOVA with Dunnett's multiple comparison post hoc test, Bonferroni's post hoc test, or the unpaired Student's t test) applied to the means. Confocal images were taken on a Nikon C1 Digital Eclipse modular confocal microscope system (Nikon UK Ltd.) using the Nikon EZ-C1 Gold v3.70 software (Nikon Corp.).
Western Blot Analysis
Western blotting was modified from a method previously described by Howell et al. (20) and described in full in the supplemental Experimental Procedures. Hippocampal cells derived from primary cell culture generation were plated at a density of 106 cells/ml in 6-well plates and cultured for 3 DIV under control conditions (NBA/B27/Glu and 1% antibiotic/antimycotic) before treatment with 1 μm NPY, 1 μm NPY and 500 μm l-NAME, or control (NBA/B27/Glu) for 8 min at 37 °C. The wells treated with l-NAME were pretreated with 1 mm l-NAME for 10 min prior to ensure adequate NOS inhibition (36). After treatment, the cells were lysed before Western blotting was carried out on the cell lysate against phosphorylated and total ERK1/2, which was visualized using the enhanced chemiluminescence (ECL) system (LumiGLO reagent and peroxide; Cell Signaling Technology). The two bands of ERK1/2 were analyzed separately, as they may have different phosphorylation profiles in response to NPY exposure, using the ImageJ analysis software (National Institutes of Health). Data were plotted using GraphPad Prism (GraphPad Software Inc.), and ANOVA with Dunnett's multiple comparison post hoc test or Bonferroni's post hoc test was applied on the means.
Diaminofluorescein Diacetate Live Cell Imaging
The fluorescent probe for the detection of NO, diaminofluorescein diacetate (DAF-FM DA; Sigma-Aldrich) (37), was utilized in two separate methods on hippocampal cultures. The first involved pretreating cultures under control or NPY conditions for 3 DIV before loading with DAF-FM DA and live imaging to determine differences in DAF-FM DA fluorescence due to NPY treatment. In the second method, cells were cultured under control conditions for 3 DIV before loading with DAF-FM DA and using time-lapse imaging to directly investigate the cellular response to an NPY pulse. Please refer to the supplemental Experimental Procedures for a full description of DAF-FM DA imaging. To confirm that DAF-FM DA fluorescence reflected NO levels, the NO donor (S)-nitroso-N-acetylpenicillamine (100 μm; Tocris Bioscience) was used as a positive control. Cells were fixed in paraformaldehyde, and the phenotype of cells (nestin+ or class III β-tubulin+) was identified via immunocytochemistry. The fluorescence intensity of cells was analyzed using ImageJ analysis software whereby the average background intensity was subtracted from the cell intensity within each image. Time-lapse data of individual time points were normalized across experiments by plotting data relative to the cell intensity at time point 0 (t0).
RESULTS
NPY Is Proliferative for Postnatal Hippocampal Cultures and Progenitor Cells
The proliferation status of primary hippocampal cultures was significantly increased over 5 DIV in the presence of 1 μm NPY. NPY exposure statistically significantly enhanced the mitotic index (supplemental Fig. 1A). In addition, NPY also statistically significantly increased the mitotic index of the nestin+ precursor cell population (supplemental Fig. 1B). This confirmed the neuroproliferative effects of NPY we had previously shown (15, 20) before the intracellular mechanisms involved were explored.
Proliferative Effect of NPY Is Mediated via NOS
To investigate the role of NOS and NO in NPY-mediated proliferation, cultures were exposed for 3 DIV to 1 μm NPY and/or 500 μm of the (non-subtype-selective) nitric-oxide synthase inhibitor l-NAME or inactive enantiomer d-NAME. NPY induced a statistically significant increase in the numbers of BrdU+ cells (Fig. 1A) and the mitotic index (Fig. 1C), which was reduced to control levels in the presence of the active NOS inhibitor l-NAME (Fig. 1, A and C). Similarly, the statistically significant increase in nestin+ BrdU+ precursors and the mitotic index of nestin+ cells by NPY was reduced to control levels by the presence of l-NAME (Fig. 1, B and D). In contrast, d-NAME had no effect on NPY-induced BrdU incorporation in nestin+ precursors, suggesting that the inactive enantiomer had no effect on NPY action (Fig. 1, A and B). Although d-NAME appeared to mediate a slight inhibitory effect on the mitotic indices (Fig. 1, C and D), this is most likely due to d-NAME impurity and the presence of minute amounts of active l-NAME. Nevertheless, the mitotic indices of the NPY + d-NAME condition were not statistically significantly different from the NPY condition.
FIGURE 1.
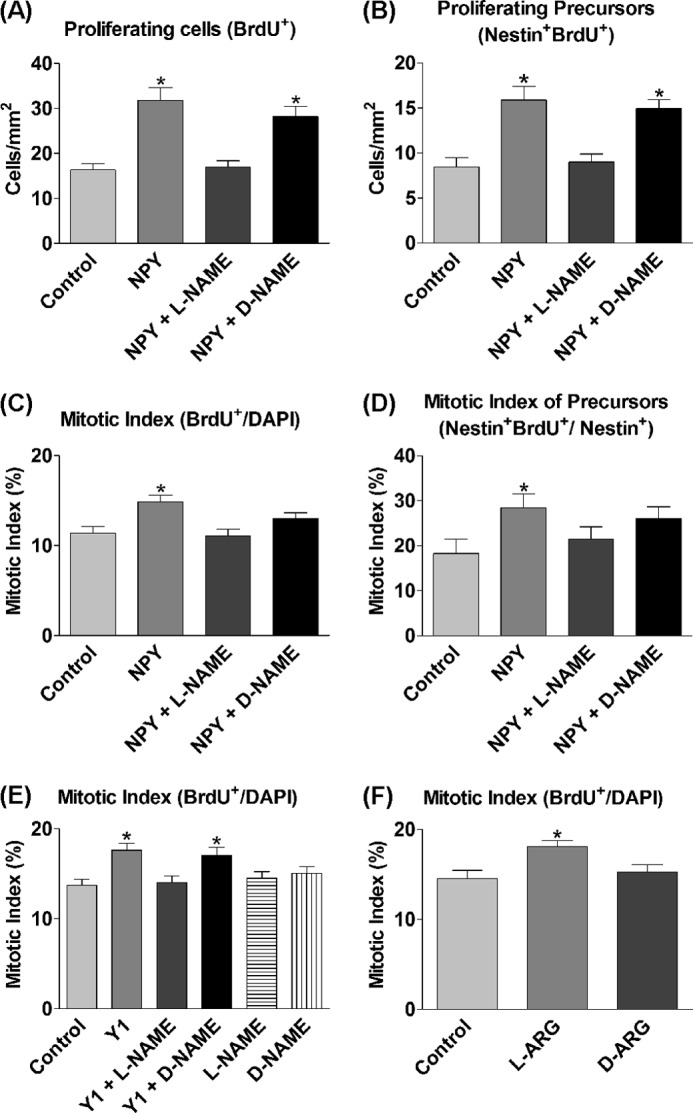
Proliferative effect of NPY is mediated via NOS. A–F, cells were grown under control conditions (NBA/B27/Glu) and 1 μm NPY and/or 500 μm l-NAME or d-NAME or 1 μm Y1 agonist [F7, P34]NPY (Y1) (A–D) and/or 500 μm l-NAME or d-NAME (E) or 500 μm l-arginine (l-ARG) and 500 μm d-arginine (d-ARG) (F) for 3 DIV. BrdU was added for the final 6 h. NPY induced a statistically significant increase in BrdU+ cells (A), nestin+ BrdU+ precursors (B), the overall mitotic index (C), and the mitotic index of precursor cells (D), which was reduced by l-NAME back down to control levels, but unaffected in general by the inactive enantiomer d-NAME. E, the Y1 agonist statistically significantly increased the mitotic index, which was reduced back down to control levels by the addition of l-NAME. The inactive enantiomer d-NAME had no effect on Y1 agonist action. l-NAME and d-NAME had no effects on basal proliferation rates on their own. F, l-arginine statistically significantly increased the mitotic index of cells, whereas the enantiomer d-arginine had no effect. Data represent mean ± S.E. based on a sample that represents at least 12 wells/condition from at least three separate experiments. One-way ANOVA with Dunnett's multiple comparison test as compared with control condition was performed. *, p < 0.05.
The proliferative effect of NPY is mediated via its Y1 receptor (15), and Kopp et al. (38) have shown that there are high levels of Y1 receptor localization within the neurogenic SGZ of the dentate. To investigate the involvement of NOS in mediating the proliferative effects of NPY through the Y1 receptor, primary hippocampal cells were cultured under control and in the presence of the Y1 agonist [F7, P34]NPY and/or l-NAME or d-NAME for 3 DIV. The proliferative effect, as demonstrated by a statistically significant increase in the mitotic index induced by the Y1 agonist, was reduced to control levels by the presence of l-NAME, but was unaffected by the presence of d-NAME (Fig. 1E). l-NAME and d-NAME alone had no effect on the mitotic index (Fig. 1E).
To further determine the involvement of NO in inducing proliferative effects, l-arginine, the substrate for NOS in the synthesis of NO, was added to increase the substrate for NO synthesis. d-Arginine, the inactive enantiomer of l-arginine, was used as an experimental control. Although d-arginine showed no effect on basal proliferation rates, l-arginine exposure to cells over 3 DIV statistically significantly enhanced the mitotic index of the cultures as compared with control conditions (Fig. 1F).
nNOS Is Likely to Mediate Proliferative Effect of NPY
We have shown that NOS appears to play an important role in mediating the neurogenic effect of NPY; however, the isoform mediating this process is unknown. The different isoforms of NOS show a variety of physiological roles. To identify the isoform involved in proliferation, our studies were restricted to commercially available and selective inhibitors of nNOS or iNOS, which was achieved through the use of 200 nm AAAN (39) or 100 nm EITH (40), respectively, along with 1 μm NPY on cells cultured for 3 DIV. NPY exerted a statistically significant proliferative effect on cultures, which was reduced to control levels by the nNOS inhibitor (Fig. 2A), but was unaffected by the iNOS inhibitor (Fig. 2B). The inhibitors had no effect on basal proliferation rates on their own. This result indicates a key involvement by nNOS in mediating the effect of NPY. Although the involvement of endothelial NOS could not be fully rejected, the complete inhibition of NPY-mediated proliferation by AAAN suggests the involvement of only nNOS. The immunocytochemical analysis of hippocampal cultures showed the localization of nNOS to nestin+ and class III β-tubulin+ cells (Fig. 2C), the same cell types that Howell et al. (15) had previously shown to be responsive to NPY.
FIGURE 2.

nNOS is likely to mediate proliferative effect of NPY. nNOS and iNOS were inhibited using subtype selective inhibitors. Hippocampal cells were cultured under control conditions (NBA/B27/glutamine) (A) or control conditions and 1 μm NPY and/or 100 nm EITH (iNOS inhibitor) (B). On the 3rd day, BrdU and 1 μm NPY and/or 200 nm AAAN (nNOS inhibitor) (A) or BrdU (B) was added for the final 6 h. NPY induced a statistically significant increase in the mitotic index, which was reduced back down to control levels in the presence of AAAN (A) and unaffected by the presence of EITH (B). AAAN and EITH had no effects on basal proliferation rates on their own. Data represent mean ± S.E. based on a sample that represents at least 12 wells/condition from three different experiments. One-way ANOVA with Dunnett's multiple comparison test as compared with control condition was performed. *, p < 0.05. ns, not significant. C, each set of images represents fluorescence images captured on a confocal microscope separated into filter sets of DAPI/nNOS, nNOS, nNOS/nestin, or nNOS/class III β-tubulin (TUJ1) with DAPI in blue, nNOS in green, and cell phenotype nestin or class III β-tubulin in red. nNOS was localized throughout the soma and cytoplasm of both cell types, although staining showed more intense immunostaining located in the soma of nestin+ cells and cytoplasm of class III β-tubulin+ cells (arrows). Scale bar, 25 μm.
Proliferative Effect of NPY Is Mediated via Intracellular NO Signaling, whereas Extracellular NO Is Antiproliferative
NO is a highly diffusible signaling molecule (41) that is able to mediate both intracellular and intercellular signaling through intracellular and/or extracellular release (24, 42). To determine whether intracellular or extracellular NO was involved in mediating the proliferative effect of NPY, hippocampal cultures were exposed to NPY in the presence of the cell impermeable NO scavenger, CPTIO (42, 43), for 3 DIV as a means to reduce any extracellular NPY. CPTIO failed to affect NPY-induced total cell (Fig. 3A) or nestin+ precursor cell (Fig. 3B) proliferation, arguing against the involvement of extracellular NO signaling pathways in mediating the proliferative effect of NPY.
FIGURE 3.
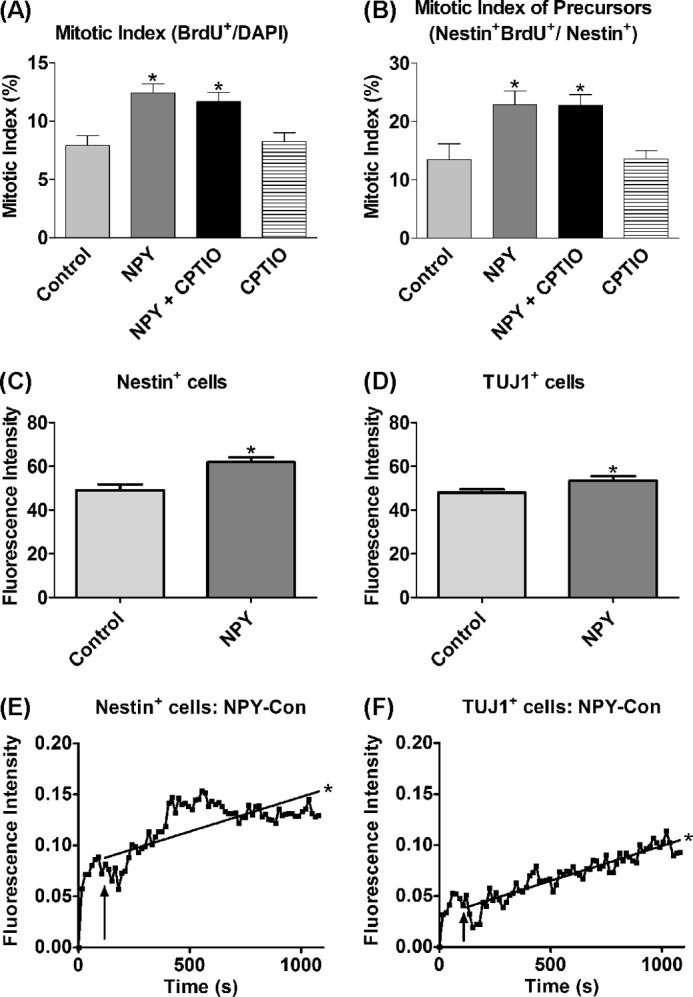
Proliferative effect of NPY is mediated via endogenous NO signaling. A and B, cells were cultured for 3 DIV under control (NBA/B27/Glu), 1 μm NPY, and/or 10 μm CPTIO (NO scavenger) conditions before BrdU was added for the final 6 h. A statistically significant increase was observed in the mitotic indices of the overall cell population (A) and the nestin+ cell population in response to NPY exposure (B), which was still significantly increased in the presence of CPTIO, suggesting an endogenous, rather than exogenous, signaling pathway in the NPY-bound cell. Data represent mean ± S.E. based on a sample that represents at least 12 wells/condition from at least three different experiments. One-way ANOVA with Dunnett's multiple comparison test as compared with control condition was performed. *, p < 0.05. C–F, DAF-FM DA fluorescence intensity in cells treated with NPY. Hippocampal cells were cultured under control conditions (NBA/B27/glutamine) or 1 μm NPY for 3 DIV before DAF-FM DA loading and imaging. The fluorescent intensity of cells treated with DAF-FM DA was assessed using two distinct methods. In the first approach, NPY-pretreated nestin+ (C) and class III β-tubulin+ (TUJ1+) (D) cell populations showed statistically significantly increased fluorescence intensity as compared with control-treated cell populations (Student's t test, p = 0.0003 and p = 0.0324 for nestin+ and class III β-tubulin+ cell populations, respectively). Data represent mean ± S.E. based on a sample that represents at least 50 individual cells/condition. E and F, Similarly, in the second approach, an NPY pulse statistically significantly enhanced the DAF-FM DA fluorescence intensity of nestin+ (E) and class III β-tubulin+ (F) cells even after subtraction of the respective control (Con) traces (PBS pulse), which demonstrates a direct involvement of NO and its production within the intracellular signaling pathway of NPY (linear regression analysis; *, p < 0.0001). Experimental settings and conditions were replicated across 4–5 independent experiments for both the pretreated cultures and the time-lapse investigations.
Live cell imaging using DAF-FM DA was used to assess endogenous NO activity in response to NPY exposure. The NO donor (S)-nitroso-N-acetylpenicillamine was used as a positive control for DAF-FM DA fluorescence (supplemental Fig. 2). The fluorescence intensity of cells treated with DAF-FM DA was assessed using two methods. First, cultures were pretreated under either control or NPY conditions before DAF-FM DA fluorescence intensity in the nestin+ and class III β-tubulin+ cell populations was assessed. In both nestin+ and class III β-tubulin+ populations, the NPY-pretreated group showed statistically significantly enhanced DAF-FM DA fluorescence as compared with the untreated control group (Fig. 3, C and D), suggesting increased NO activity in response to NPY exposure. Second, an NPY pulse statistically significantly enhanced the DAF-FM DA fluorescence intensity of individual nestin+ and class III β-tubulin+ cells over time (Fig. 3, E and F), demonstrating increased intracellular NO activity directly in response to NPY.
If the proliferative effect of NPY is mediated via intracellular NO production, what effects, if any, would extracellular NO confer? To further examine this effect, hippocampal cells were cultured in the presence of the NO donor, DETA/NONOate (NOC-18), for 3 DIV to determine the effects of extracellular NO on cell proliferation. An NOC-18 concentration range of 50–200 μm (42) was investigated to explore the dose response. Indeed, extracellular NO exerted a negative effect on proliferation rates as has been previously reported (42). Although NOC-18 at the lowest concentration of 50 μm had no effect on cell proliferation, 100 and 200 μm NOC-18 exerted an inhibitory effect, which statistically significantly decreased the mitotic index (Fig. 4A), as well as the number of nestin+ BrdU+ precursors (Fig. 4B), to below control levels. This antiproliferative effect on basal cell proliferation increased in response to NOC-18 concentration, with higher concentrations exerting a more prominent inhibitory effect. To exclude cell damage/death as a possible explanation, studies using propidium iodide staining showed no change in percentage of cell death as compared with control at the NOC-18 concentrations investigated (data not shown). Far from being a possible proliferative pathway for NPY, extracellular NO was antiproliferative for hippocampal cultures.
FIGURE 4.
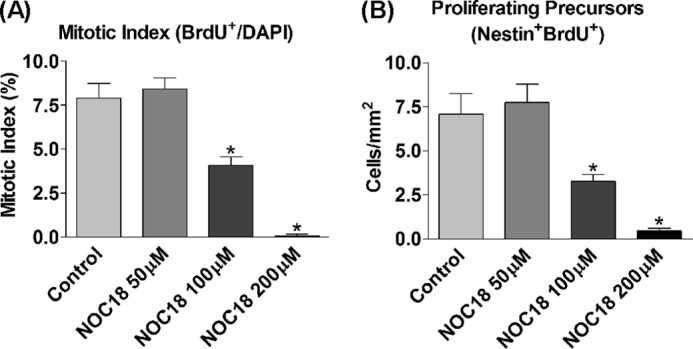
Exogenous NO is inhibitory for cell proliferation. A and B, Hippocampal cells were cultured under control conditions (NBA/B27/glutamine) or 50, 100, or 200 μm DETA/NONOate (NOC-18; NO donor) for 3 DIV before BrdU was added for the final 6 h. NOC-18 at 50 μm had no effect on basal proliferation rates, whereas 100 and 200 μm NOC-18 exerted a negative effect on basal proliferation rates, which statistically significantly reduced the mitotic index (A) and the number of nestin+ BrdU+ precursors to below control levels (B). Data represent mean ± S.E. based on a sample that represents at least 12 wells/condition from at least three different experiments. One-way ANOVA with Dunnett's multiple comparison test as compared with control condition was performed. *, p < 0.05.
NO-cGMP-PKG Pathway Mediates Proliferative Effect of NPY
sGC, which converts GTP into cGMP, is a major intracellular target of NO (44). The intracellular downstream targets of NO were analyzed using the cell-permeable cGMP analog, 8-Br-cGMP (45), and the potent sGC antagonist, ODQ (46). The NPY-induced statistically significant increase in proliferation was mimicked by 8-Br-cGMP (Fig. 5A), and there were no cumulative effects in response to combined exposure to NPY and 8-Br-cGMP, consistent with NPY signaling via cGMP. This was supported by ODQ reducing the proliferative effect of NPY to below control levels, which was also prevalent in ODQ-only controls, suggesting a role for sGC in also maintaining basal proliferation rates (Fig. 5A). In further verification of sGC involvement, cultures supplemented with l-arginine as substrate for NO synthesis showed statistically significantly increased proliferation rates, which were also reduced to below control levels by the presence of ODQ (Fig. 5B). Downstream of sGC is PKG, a common target of cGMP. PKG is potently and selectively inhibited by the compound KT5823 (47), which was exposed to cultures for 3 DIV in the presence of NPY and 8-Br-cGMP. The statistically significantly increased proliferation status induced by NPY or 8-Br-cGMP was reduced to, and in the case of 8-Br-cGMP, below control levels by the presence of KT5823 (Fig. 5C). There was a trend for the presence of KT5823 to have a negative effect on basal proliferation rates (Fig. 5C), which may be explained by the fact that PKG, like sGC, may normally be involved in maintaining basal proliferation rates in culture.
FIGURE 5.
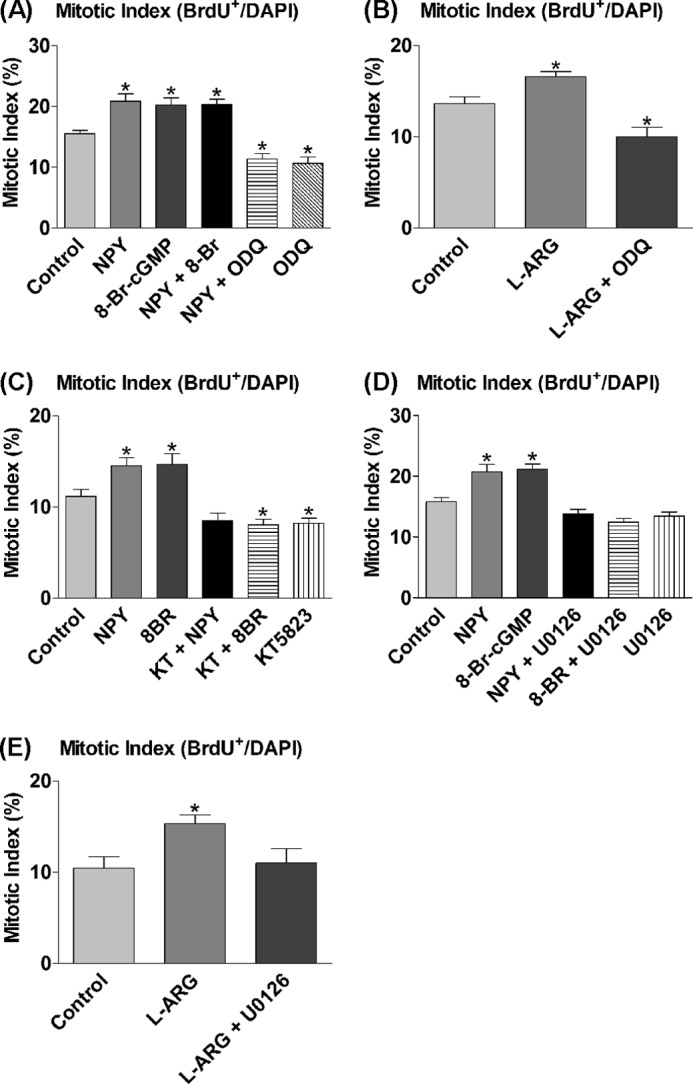
sGC, cGMP, and ERK1/2 mediate proliferative effect of NPY. A–E, cells were cultured under control conditions (NBA/B27/Glu) and 1 μm NPY, 20 μm 8-Br-cGMP (8-Br; cGMP analog), and/or 50 μm ODQ (sGC inhibitor) (A); 500 μm l-arginine (l-ARG) and/or 50 μm ODQ (B); 1 μm NPY or 20 μm 8-Br-cGMP in the presence of 1 μm KT5823 (KT; PKG inhibitor) (C); 1 μm NPY, 20 μm 8-Br-cGMP, and/or 1 μm U0126 (inhibitor of ERK1/2 activation by inhibiting MEK) (D); or 500 μm l-arginine and/or 1 μm ODQ (E) for 3 DIV. BrdU was added for the final 6 h. A, NPY or 8-Br-cGMP both induced a statistically significant increase in the mitotic index. There were no cumulative effects from combined NPY and 8-Br-cGMP treatment. ODQ reduced the effect of NPY down below control levels and had negative effects on basal proliferation rates on its own. B, l-arginine induced a statistically significant proliferative effect, which was also reduced to below control levels by the presence of ODQ. C, NPY or 8-Br-cGMP induced a statistically significant increase in the mitotic index, which was significantly reduced by inhibiting PKG with KT5823. Both treatment with KT5823 on its own and in combination with 8-Br-cGMP resulted in a decrease to below control levels. D, NPY and 8-Br-cGMP both induced a statistically significant increase in the mitotic index, which was reduced back down to control levels by U0126. E, l-arginine statistically significantly increased the mitotic index, which was reduced back down to control levels by the presence of U0126. Data represent mean ± S.E. based on a sample that represents at least 12 wells/condition from at least three independent experiments. One-way ANOVA with Dunnett's multiple comparison test as compared with control condition was performed. *, p < 0.05.
ERK1/2 Activation Is Required for NPY Action and Is a Downstream Target of NOS-NO Signaling Pathway
The involvement of the MAPKs, ERK1/2, in mediating the effect of NPY had been shown previously, among others by Hansel et al. (14) and Howell et al. (20), in postnatal precursor cells from the olfactory epithelium and the hippocampus, respectively. U0126, an inhibitor of MEK 1/2 (kinases upstream of ERK1/2), prevents the activation (phosphorylation) of ERK1/2 and was added to cultures for 3 DIV in the presence of NPY and 8-Br-cGMP to confirm its involvement and determine its role within the signaling cascade. NPY or 8-Br-cGMP again induced a statistically significant increase in proliferation, whereas the presence of U0126 completely reduced proliferation levels to control levels (Fig. 5D). This inhibition of 8-Br-cGMP activity suggests a role for ERK1/2 which is situated downstream of sGC and cGMP, and possibly after PKG. In addition, cultures were supplemented with l-arginine and U0126 for 3 DIV to demonstrate the link between NOS-NO and ERK1/2. The statistically significantly increased proliferation in response to l-arginine was reduced to control levels by the presence of U0126 (Fig. 5E), which reinforces the role of NO in the NPY signaling pathway.
The signaling mechanisms initiated by the NPY Y receptors are diverse, and activation of ERK1/2 may be through an NO-independent pathway (13, 23). The levels of ERK1/2 phosphorylation were assessed by Western blotting in response to NOS inhibition (l-NAME) to determine the importance of the NO-cGMP pathway in mediating ERK1/2 activation and the proliferative effects of NPY (Fig. 6A). ERK1/2 phosphorylation levels increased in response to NPY exposure, especially ERK2 (p42) (Fig. 6C), and were statistically significantly decreased to control levels by the presence of l-NAME (Fig. 6, B and C). l-NAME on its own had no effects on basal phosphorylation levels of ERK1/2 (data not shown). Thus ERK1/2 activation by NPY was mediated via an NOS-NO signaling pathway.
FIGURE 6.
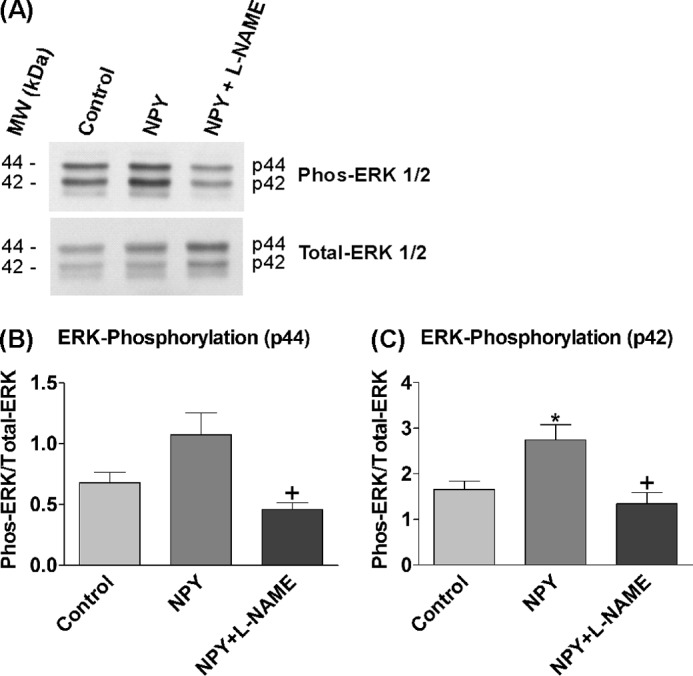
ERK1/2 activation by NPY is solely mediated via NOS-NO mechanisms. 3-day-old hippocampal cultures were treated with control medium (NBA/B27/Glu), 1 μm NPY, or 1 μm NPY in the presence of 500 μm l-NAME for 8 min. l-NAME treatment wells were pretreated with 1 mm l-NAME for 10 min beforehand to ensure adequate inhibition. A, immunoblots were probed with antibodies against phosphorylated (Phos-ERK 1/2) and total ERK (Total-ERK 1/2). ERK1 (p44) and ERK2 (p42) showed distinct bands. The addition of NPY increased the phosphorylation levels of ERK1/2, whereas the presence of l-NAME reduced levels. MW, molecular mass markers. B and C, ERK1 (p44) (B) and ERK2 (p42) (C) showed an increase in phosphorylation levels in response to NPY treatment, which was statistically significant with ERK2 (*), and both were statistically significantly reduced to control levels by the presence of l-NAME (+). Data represent mean ± S.E. based on three separate experiments. One-way ANOVA with Dunnett's (*) or Bonferroni's (+) multiple comparison test was performed. *, p < 0.05.
DISCUSSION
NPY has been shown to exert proliferative effects on a wide range of different cell types (14–16, 18, 23), but little was known about its intracellular signaling pathways. Recently, Lou et al. (42) have shown that the effects of NO on neural stem/precursor cells differ significantly depending on whether it was released extracellularly (antiproliferative) or intracellularly (proliferative). Although the involvement of NO mechanisms was thought to underlie the proliferative effect of NPY in retinal neural cells (23), our results are the first to demonstrate that intracellular NO produced by nNOS signals the complete Y1 receptor-mediated proliferative effect of NPY on hippocampal nestin+ precursors, whereas extracellular NO had the opposite antiproliferative effect. We also show that ERK1/2 activation by NPY is solely mediated via NOS-NO mechanisms.
We identified the involvement of NO in mediating the neuroproliferative effect of NPY via the NPY Y1 receptor by significantly reducing the effects of both NPY and a selective Y1 agonist by inhibiting NOS using l-NAME. Imaging studies using the NO probe DAF-FM DA before a NPY or PBS pulse showed that hippocampal cells had some endogenous NO activity. However, it is unlikely that this background level of NO was involved in mediating basal proliferative processes as pharmacological analysis using l-NAME showed that it had little effect on basal cell proliferation. On the contrary, both sGC and PKG were associated with maintaining basal proliferation rates, which may be due to NO-independent sGC activation (48, 49), which is less efficient than NO at promoting cGMP synthesis. Moreover, many of the physiological effects of NO are concentration-dependent, and an increase in endogenous (basal) NO levels, as a result of NPY stimulation or l-arginine supplementation, may initiate proliferative processes, which would otherwise not occur if endogenous NO levels were low.
Irrespective of NPY, however, NO itself has been shown to have both neuroinhibitory and neuroproliferative effects on adult neurogenesis (50). In vivo investigations by Packer et al. (51) showed that the chronic inhibition of NOS in rats with l-NAME increased levels of BrdU+ cells in neurogenic zones such as the SGZ (+68%) and SVZ (+58%). Similarly, Moreno-López et al. (52, 53) observed an increase in proliferating cells in mice in response to systemic l-NAME administration, although this was observed only in the SVZ and not the SGZ. The systemic administration of a different NOS inhibitor, 7-nitroindazole, induced a dose- and time-dependent increase in the number of BrdU+ cells, again in the mouse SVZ, but not in the dentate gyrus (53). This disparity in region response was probably due to sensitivity differences and the presence of specific cell types (52). Although these studies all support the idea of NO as a negative regulator of adult neurogenesis, it must also be noted that both l-NAME and 7-nitroindazole are relatively nonselective NOS inhibitors. Conversely, others have described NO as a positive regulator of adult neurogenesis. The administration of the NO donor, DETA/NONOate, to young adult rats increased cell proliferation in the SGZ and SVZ both under normal conditions and in response to stroke (54). Likewise, the prior administration of 7-nitroindazole and the iNOS inhibitor aminoguanidine significantly reduced the number of BrdU+ cells in the dentate gyrus after pentylenetrazol-induced seizures in the adult rat brain (55), supporting a role for NO in brain repair after brain injury or seizures. DETA/NONOate has also been shown to produce antidepressant effects by promoting hippocampal neurogenesis in young adult mice (56). The explanation for these paradoxical effects of NO lie in the existence and differential effects of the NOS isoforms and/or the opposing effects of intracellular and extracellular NO signaling on proliferation, which formed the basis of our investigations.
nNOS was identified as the main NOS isoform responsible for NPY-mediated proliferation, and the NPY-responsive nestin+ and class III β-tubulin+ precursor cells (15, 20) were immunocytochemically positive for nNOS. Indeed, nNOS immunostaining is localized to sites of neuronal proliferation and migration in the hippocampal dentate gyrus (29), and signaling between GPCRs, such as the Y1 receptor, and nNOS may be possible through GPCR-mediated mechanisms. In the cholecystokinin-mediated proliferation of Chinese hamster ovary cells, for example, the cholecystokinin A GPCR activated nNOS via the G-protein βγ subunit and the tyrosine phosphatase SHP-2 (57). In vivo studies using NOS isoform-specific knock-out animals suggest an inhibitory role for NO derived from nNOS (58), which also appears to mediate the suppressed neurogenesis seen in chronic, stress induced-depression (59). The cellular signaling pathways underlying these effects, however, are not known. Indeed, the cell type, cellular source, reactive status, timing of synthesis, and concentration are major determinants of NO effects (50, 60).
Our findings suggest that the NO released by nNOS in response to NPY acts intracellularly and mediates a proliferative effect. This finding is entirely consistent with the dual role of nNOS-derived NO recently demonstrated by Luo et al. (42), who found that intracellular NO produced by nNOS in neural stem cells was important in mediating their proliferation and neuronal differentiation, whereas nNOS-derived NO from adjacent neuronal cells was extracellularly released and exerted a negative control on neurogenesis. The addition of DETA/NONOate significantly decreased proliferation rates (42, 61, 62), which we also found when we exposed our hippocampal cultures to the donor. Indeed, our study significantly extends these findings by identifying NPY as a mediator of intracellular NO-induced proliferation. This dual role of NO may be policed by the differential subcellular compartmentalization of nNOS, for example, to the nuclei for neural stem cells or to the cytoplasm for neurons as proposed by Luo et al. (42), a trend also observed during the nNOS immunostaining of our own hippocampal cultures (Fig. 2C), or possibly, the existence of the nNOS splice variants, such as nNOSα, nNOSβ, and nNOSγ (26, 63). The mechanisms underlying the effects of NO on adult neurogenesis are highly complex in vivo and complicate further investigations, and the in vivo confirmation of our findings will be challenging given the difficulties in selectively targeting intracellular nNOS and Y1 receptors in neural stem cells.
A significant increase in intracellular NO activity occurs in response to NPY, which we visualized using DAF-FM DA, and the enzyme sGC and its product cGMP were identified as downstream targets of NPY/NO signaling. The cGMP analog, 8-Br-cGMP, initiated a proliferative effect similar to that of NPY, which has also been shown by Zhang et al. (64) to promote angiogenesis in the rat brain. PKG, a common target of cGMP, was implicated in the underlying signaling cascade of NPY after the effects of NPY were found to be significantly reduced following the inhibition of PKG with KT5823. Similarly, Hood and Granger (65) found that the VEGF-induced proliferation of human endothelial cells was mediated via an NO-cGMP-PKG signaling pathway, much like the pathway identified within this study.
Unlike the NO-cGMP-PKG pathway, the ERK1/2 of the MAPK signaling pathway has long been implicated in the processes underlying the proliferative effect of NPY (14, 20, 23). As well as confirming these previous results, we also investigated the links within this signaling pathway. Although it is still unclear how ERK1/2 may be activated from upstream NO-cGMP-PKG signaling, it is most likely due to kinase phosphorylation cascades. It is clear that the NPY-, l-arginine- (endogenous NO source), and 8-Br-cGMP-mediated proliferation of hippocampal cells is significantly reduced by inhibiting ERK1/2 activation (phosphorylation) with U0126. Upstream NO-cGMP-PKG signaling was directly linked to ERK1/2 activation, and all are involved in mediating the proliferative effect of NPY. Through assessing levels of ERK1/2 phosphorylation in response to NPY in the presence of l-NAME, we also confirmed that ERK1/2 activation was only mediated via NOS-NO mechanisms, which was uncertain before as ERK1/2 could have been activated via secondary signaling pathways. The activation of ERK1/2 by an NO-cGMP-PKG pathway has been shown to enhance declustering of the ionotropic glutamate receptor subunit 2/3 in rat cerebellar Purkinje cells (66) and is important in regulating synaptic plasticity and fear memory consolidation in the lateral amygdala of the rat (67). ERK1/2 themselves mediate further pathways involved in regulating the expression of genes controlling cell proliferation and differentiation through the phosphorylation of a variety of transcription factors (68, 69) (Fig. 7). The involvement of such transcription factors is a topic for further investigation.
FIGURE 7.

Intracellular mechanisms underlying NPY-mediated neuroproliferation. NPY released by interneurons of the dentate hilus acts on NPY Y1 receptors in the SGZ to initiate a neuroproliferative effect. The Y1 GPCR activates nNOS, which synthesizes NO from l-arginine. The target of NO is sGC, which converts GTP into cGMP. cGMP activates PKG, which in turn leads to the activation of a range of protein targets or kinase pathways, ultimately resulting in ERK1/2 activation (phosphorylation). ERK1/2 are involved in regulating the expression of genes involved in controlling cell proliferation and differentiation (adapted from Ref. 23).
NPY exerts a proliferative effect on postnatal rat hippocampal cultures, which was mediated via intracellular NO production and signaled via the nNOS-cGMP-PKG and ERK1/2 signaling pathway. We thus identify NPY as a selective agonist of intracellular NO signaling in postnatal hippocampal neural stem precursor cells. Indeed, NO has also been shown to be essential for the proliferation of embryonic hippocampal neural stem/progenitor cells (70). Although the literature on the effects of NO signaling on hippocampal neurogenesis is complex and conflicting, our study identifies the NPY Y1 receptor as a key target to selectively promote NO-mediated neural stem/precursor cell proliferation as a possible therapeutic intervention for promoting hippocampal neurogenesis.
Acknowledgments
We are grateful to Dr. Hansjürgen Schuppe and Dr. Andreas Wyttenbach for assistance and advice on diaminofluorescein diacetate imaging. The Nikon C1 confocal microscope system was purchased with funds from the Biotechnology and Biological Sciences Research Council (to P. L. Newland).
This work was supported by the Gerald Kerkut Charitable Trust.

This article contains supplemental Experimental Procedures and Figs. 1 and 2.
- NPY
- neuropeptide Y
- PKG
- cGMP-dependent protein kinase
- DAF-FM DA
- diaminofluorescein diacetate
- GPCR
- G-protein-coupled receptor
- SGZ
- subgranular zone
- SVZ
- subventricular zone
- iNOS
- inducible NOS
- nNOS
- neuronal NOS
- l-NAME
- N(ω)-nitro-l-arginine methyl ester
- d-NAME
- N(ω)-nitro-d-arginine methyl ester
- DIV
- days in vitro
- 8-Br-cGMP
- 8-bromo-cyclic GMP
- ODQ
- 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- EITH
- (S)-ethylisothiourea hydrobromide
- AAAN
- N-(4S)-4-amino-5-[aminoethyl]aminopentyl-N′-nitroguanidine
- DETA/NONOate
- diethylenetriamine NONOate
- CPTIO
- 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide
- ANOVA
- analysis of variance
- sGC
- soluble guanylate cyclase
- NBA
- neurobasal A.
REFERENCES
- 1. Lundberg J. M., Terenius L., Hökfelt T., Tatemoto K. (1984) Comparative immunohistochemical and biochemical analysis of pancreatic polypeptide-like peptides with special reference to presence of neuropeptide Y in central and peripheral neurons. J. Neurosci. 4, 2376–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tatemoto K. (1982) Neuropeptide Y: complete amino acid sequence of the brain peptide. Proc. Natl. Acad. Sci. U.S.A. 79, 5485–5489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Allen Y. S., Adrian T. E., Allen J. M., Tatemoto K., Crow T. J., Bloom S. R., Polak J. M. (1983) Neuropeptide Y distribution in the rat brain. Science 221, 877–879 [DOI] [PubMed] [Google Scholar]
- 4. Albers H. E., Ferris C. F. (1984) Neuropeptide Y: role in light-dark cycle entrainment of hamster circadian rhythms. Neurosci. Lett. 50, 163–168 [DOI] [PubMed] [Google Scholar]
- 5. Flood J. F., Morley J. E. (1989) Dissociation of the effects of neuropeptide Y on feeding and memory: evidence for pre- and postsynaptic mediation. Peptides 10, 963–966 [DOI] [PubMed] [Google Scholar]
- 6. Redrobe J. P., Dumont Y., St-Pierre J. A., Quirion R. (1999) Multiple receptors for neuropeptide Y in the hippocampus: putative roles in seizures and cognition. Brain. Res. 848, 153–166 [DOI] [PubMed] [Google Scholar]
- 7. Heilig M. (2004) The NPY system in stress, anxiety, and depression. Neuropeptides 38, 213–224 [DOI] [PubMed] [Google Scholar]
- 8. Erickson J. C., Clegg K. E., Palmiter R. D. (1996) Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature 381, 415–421 [DOI] [PubMed] [Google Scholar]
- 9. Vezzani A., Sperk G., Colmers W. F. (1999) Neuropeptide Y: emerging evidence for a functional role in seizure modulation. Trends. Neurosci. 22, 25–30 [DOI] [PubMed] [Google Scholar]
- 10. Freund T. F., Buzsáki G. (1996) Interneurons of the hippocampus. Hippocampus 6, 347–470 [DOI] [PubMed] [Google Scholar]
- 11. Köhler C., Eriksson L., Davies S., Chan-Palay V. (1986) Neuropeptide Y innervation of the hippocampal region in the rat and monkey brain. J. Comp. Neurol. 244, 384–400 [DOI] [PubMed] [Google Scholar]
- 12. Michel M. C., Beck-Sickinger A., Cox H., Doods H. N., Herzog H., Larhammar D., Quirion R., Schwartz T., Westfall T. (1998) XVI. International Union of Pharmacology recommendations for the nomenclature of neuropeptide Y, peptide YY, and pancreatic polypeptide receptors. Pharmacol. Rev. 50, 143–150 [PubMed] [Google Scholar]
- 13. Mullins D. E., Zhang X., Hawes B. E. (2002) Activation of extracellular signal regulated protein kinase by neuropeptide Y and pancreatic polypeptide in CHO cells expressing the NPY Y1, Y2, Y4, and Y5 receptor subtypes. Regul. Pept. 105, 65–73 [DOI] [PubMed] [Google Scholar]
- 14. Hansel D. E., Eipper B. A., Ronnett G. V. (2001) Neuropeptide Y functions as a neuroproliferative factor. Nature 410, 940–944 [DOI] [PubMed] [Google Scholar]
- 15. Howell O. W., Scharfman H. E., Herzog H., Sundstrom L. E., Beck-Sickinger A., Gray W. P. (2003) Neuropeptide Y is neuroproliferative for postnatal hippocampal precursor cells. J. Neurochem. 86, 646–659 [DOI] [PubMed] [Google Scholar]
- 16. Milenkovic I., Weick M., Wiedemann P., Reichenbach A., Bringmann A. (2004) Neuropeptide Y-evoked proliferation of retinal glial (Muller) cells. Graefes Arch. Clin. Exp. Ophthalmol. 242, 944–950 [DOI] [PubMed] [Google Scholar]
- 17. Movafagh S., Hobson J. P., Spiegel S., Kleinman H. K., Zukowska Z. (2006) Neuropeptide Y induces migration, proliferation, and tube formation of endothelial cells bimodally via Y1, Y2, and Y5 receptors. FASEB J. 20, 1924–1926 [DOI] [PubMed] [Google Scholar]
- 18. Agasse F., Bernardino L., Kristiansen H., Christiansen S. H., Ferreira R., Silva B., Grade S., Woldbye D. P., Malva J. O. (2008) Neuropeptide Y promotes neurogenesis in murine subventricular zone. Stem Cells 26, 1636–1645 [DOI] [PubMed] [Google Scholar]
- 19. Eriksson P. S., Perfilieva E., Björk-Eriksson T., Alborn A. M., Nordborg C., Peterson D. A., Gage F. H. (1998) Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317 [DOI] [PubMed] [Google Scholar]
- 20. Howell O. W., Doyle K., Goodman J. H., Scharfman H. E., Herzog H., Pringle A., Beck-Sickinger A. G., Gray W. P. (2005) Neuropeptide Y stimulates neuronal precursor proliferation in the postnatal and adult dentate gyrus. J. Neurochem. 93, 560–570 [DOI] [PubMed] [Google Scholar]
- 21. Howell O. W., Silva S., Scharfman H. E., Sosunov A. A., Zaben M., Shatya A., McKhann G., 2nd, Herzog H., Laskowski A., Gray W. P. (2007) Neuropeptide Y is important for basal and seizure-induced precursor cell proliferation in the hippocampus. Neurobiol. Dis. 26, 174–188 [DOI] [PubMed] [Google Scholar]
- 22. Gray W. P. (2008) Neuropeptide Y signaling on hippocampal stem cells in health and disease. Mol. Cell. Endocrinol. 288, 52–62 [DOI] [PubMed] [Google Scholar]
- 23. Alvaro A. R., Martins J., Araújo I. M., Rosmaninho-Salgado J., Ambrósio A. F., Cavadas C. (2008) Neuropeptide Y stimulates retinal neural cell proliferation: involvement of nitric oxide. J. Neurochem. 105, 2501–2510 [DOI] [PubMed] [Google Scholar]
- 24. Garthwaite J., Boulton C. L. (1995) Nitric oxide signaling in the central nervous system. Annu. Rev. Physiol. 57, 683–706 [DOI] [PubMed] [Google Scholar]
- 25. Villalobo A. (2006) Nitric oxide and cell proliferation. FEBS J. 273, 2329–2344 [DOI] [PubMed] [Google Scholar]
- 26. Alderton W. K., Cooper C. E., Knowles R. G. (2001) Nitric-oxide synthases: structure, function, and inhibition. Biochem. J. 357, 593–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garthwaite J., Charles S. L., Chess-Williams R. (1988) Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 336, 385–388 [DOI] [PubMed] [Google Scholar]
- 28. Contestabile A., Ciani E. (2004) Role of nitric oxide in the regulation of neuronal proliferation, survival, and differentiation. Neurochem. Int. 45, 903–914 [DOI] [PubMed] [Google Scholar]
- 29. Islam A. T., Kuraoka A., Kawabuchi M. (2003) Morphological basis of nitric oxide production and its correlation with the polysialylated precursor cells in the dentate gyrus of the adult guinea pig hippocampus. Anat. Sci. Int. 78, 98–103 [DOI] [PubMed] [Google Scholar]
- 30. Moreno-López B., Noval J. A., González-Bonet L. G., Estrada C. (2000) Morphological bases for a role of nitric oxide in adult neurogenesis. Brain Res. 869, 244–250 [DOI] [PubMed] [Google Scholar]
- 31. Garg U. C., Hassid A. (1989) Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J. Clin. Invest. 83, 1774–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. RayChaudhury A., Frischer H., Malik A. B. (1996) Inhibition of endothelial cell proliferation and bFGF-induced phenotypic modulation by nitric oxide. J. Cell. Biochem. 63, 125–134 [DOI] [PubMed] [Google Scholar]
- 33. Du M., Islam M. M., Lin L., Ohmura Y., Moriyama Y., Fujimura S. (1997) Promotion of proliferation of murine BALB/C3T3 fibroblasts mediated by nitric oxide at lower concentrations. Biochem. Mol. Biol. Int. 41, 625–631 [DOI] [PubMed] [Google Scholar]
- 34. Ulibarri J. A., Mozdziak P. E., Schultz E., Cook C., Best T. M. (1999) Nitric oxide donors, sodium nitroprusside and S-nitroso-N-acetylpencillamine, stimulate myoblast proliferation in vitro. In Vitro Cell Dev. Biol. Anim. 35, 215–218 [DOI] [PubMed] [Google Scholar]
- 35. Morley J. E., Alshaher M. M., Farr S. A., Flood J. F., Kumar V. B. (1999) Leptin and neuropeptide Y (NPY) modulate nitric-oxide synthase: further evidence for a role of nitric oxide in feeding. Peptides 20, 595–600 [DOI] [PubMed] [Google Scholar]
- 36. El Mabrouk M., Singh A., Touyz R. M., Schiffrin E. L. (2000) Antiproliferative effect of l-NAME on rat vascular smooth muscle cells. Life Sci. 67, 1613–1623 [DOI] [PubMed] [Google Scholar]
- 37. Kojima H., Nakatsubo N., Kikuchi K., Urano Y., Higuchi T., Tanaka J., Kudo Y., Nagano T. (1998) Direct evidence of NO production in rat hippocampus and cortex using a new fluorescent indicator: DAF-2 DA. Neuroreport 9, 3345–3348 [DOI] [PubMed] [Google Scholar]
- 38. Kopp J., Xu Z. Q., Zhang X., Pedrazzini T., Herzog H., Kresse A., Wong H., Walsh J. H., Hökfelt T. (2002) Expression of the neuropeptide Y Y1 receptor in the CNS of rat and of wild-type and Y1 receptor knock-out mice: focus on immunohistochemical localization. Neuroscience 111, 443–532 [DOI] [PubMed] [Google Scholar]
- 39. Hah J. M., Roman L. J., Martásek P., Silverman R. B. (2001) Reduced amide bond peptidomimetics. (4S)-N-(4-amino-5-[aminoakyl]aminopentyl)-N′-nitroguanidines, potent and highly selective inhibitors of neuronal nitric-oxide synthase. J. Med. Chem. 44, 2667–2670 [DOI] [PubMed] [Google Scholar]
- 40. Garvey E. P., Oplinger J. A., Tanoury G. J., Sherman P. A., Fowler M., Marshall S., Harmon M. F., Paith J. E., Furfine E. S. (1994) Potent and selective inhibition of human nitric-oxide synthases. Inhibition by non-amino acid isothioureas. J. Biol. Chem. 269, 26669–26676 [PubMed] [Google Scholar]
- 41. Lancaster J. R., Jr. (1997) A tutorial on the diffusibility and reactivity of free nitric oxide. Nitric Oxide 1, 18–30 [DOI] [PubMed] [Google Scholar]
- 42. Luo C. X., Jin X., Cao C. C., Zhu M. M., Wang B., Chang L., Zhou Q. G., Wu H. Y., Zhu D. Y. (2010) Bidirectional regulation of neurogenesis by neuronal nitric-oxide synthase derived from neurons and neural stem cells. Stem Cells 28, 2041–2052 [DOI] [PubMed] [Google Scholar]
- 43. Tolias C. M., McNeil C. J., Kazlauskaite J., Hillhouse E. W. (1999) Superoxide generation from constitutive nitric-oxide synthase in astrocytes in vitro regulates extracellular nitric oxide availability. Free Radic. Biol. Med. 26, 99–106 [DOI] [PubMed] [Google Scholar]
- 44. Denninger J. W., Marletta M. A. (1999) Guanylate cyclase and the •NO/cGMP signaling pathway. Biochim. Biophys. Acta 1411, 334–350 [DOI] [PubMed] [Google Scholar]
- 45. Francis S. H., Noblett B. D., Todd B. W., Wells J. N., Corbin J. D. (1988) Relaxation of vascular and tracheal smooth muscle by cyclic nucleotide analogs that preferentially activate purified cGMP-dependent protein kinase. Mol. Pharmacol. 34, 506–517 [PubMed] [Google Scholar]
- 46. Garthwaite J., Southam E., Boulton C. L., Nielsen E. B., Schmidt K., Mayer B. (1995) Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol. Pharmacol. 48, 184–188 [PubMed] [Google Scholar]
- 47. Francis S. H., Busch J. L., Corbin J. D. (2010) cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 62, 525–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ignarro L. J., Wood K. S., Wolin M. S. (1982) Activation of purified soluble guanylate cyclase by protoporphyrin IX. Proc. Natl. Acad. Sci. U.S.A. 79, 2870–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stasch J. P., Becker E. M., Alonso-Alija C., Apeler H., Dembowsky K., Feurer A., Gerzer R., Minuth T., Perzborn E., Pleiss U., Schröder H., Schroeder W., Stahl E., Steinke W., Straub A., Schramm M. (2001) NO-independent regulatory site on soluble guanylate cyclase. Nature 410, 212–215 [DOI] [PubMed] [Google Scholar]
- 50. Cárdenas A., Moro M. A., Hurtado O., Leza J. C., Lizasoain I. (2005) Dual role of nitric oxide in adult neurogenesis. Brain Res. Rev. 50, 1–6 [DOI] [PubMed] [Google Scholar]
- 51. Packer M. A., Stasiv Y., Benraiss A., Chmielnicki E., Grinberg A., Westphal H., Goldman S. A., Enikolopov G. (2003) Nitric oxide negatively regulates mammalian adult neurogenesis. Proc. Natl. Acad. Sci. U.S.A. 100, 9566–9571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Matarredona E. R., Murillo-Carretero M., Moreno-López B., Estrada C. (2005) Role of nitric oxide in subventricular zone neurogenesis. Brain Res. Rev. 49, 355–366 [DOI] [PubMed] [Google Scholar]
- 53. Moreno-López B., Romero-Grimaldi C., Noval J. A., Murillo-Carretero M., Matarredona E. R., Estrada C. (2004) Nitric oxide is a physiological inhibitor of neurogenesis in the adult mouse subventricular zone and olfactory bulb. J. Neurosci. 24, 85–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang R., Zhang L., Zhang Z., Wang Y., Lu M., Lapointe M., Chopp M. (2001) A nitric oxide donor induces neurogenesis and reduces functional deficits after stroke in rats. Ann. Neurol. 50, 602–611 [DOI] [PubMed] [Google Scholar]
- 55. Jiang W., Xiao L., Wang J. C., Huang Y. G., Zhang X. (2004) Effects of nitric oxide on dentate gyrus cell proliferation after seizures induced by pentylenetrazol in the adult rat brain. Neurosci. Lett. 367, 344–348 [DOI] [PubMed] [Google Scholar]
- 56. Hua Y., Huang X. Y., Zhou L., Zhou Q. G., Hu Y., Luo C. X., Li F., Zhu D. Y. (2008) DETA/NONOate, a nitric oxide donor, produces antidepressant effects by promoting hippocampal neurogenesis. Psychopharmacology 200, 231–242 [DOI] [PubMed] [Google Scholar]
- 57. Cordelier P., Estève J. P., Rivard N., Marletta M., Vaysse N., Susini C., Buscail L. (1999) The activation of neuronal NO synthase is mediated by G-protein βγ subunit and the tyrosine phosphatase SHP-2. FASEB J. 13, 2037–2050 [DOI] [PubMed] [Google Scholar]
- 58. Sun Y., Jin K., Childs J. T., Xie L., Mao X. O., Greenberg D. A. (2005) Neuronal nitric-oxide synthase and ischemia-induced neurogenesis. J. Cereb. Blood Flow Metab. 25, 485–492 [DOI] [PubMed] [Google Scholar]
- 59. Zhou Q. G., Hu Y., Hua Y., Hu M., Luo C. X., Han X., Zhu X. J., Wang B., Xu J. S., Zhu D. Y. (2007) Neuronal nitric-oxide synthase contributes to chronic stress-induced depression by suppressing hippocampal neurogenesis. J. Neurochem. 103, 1843–1854 [DOI] [PubMed] [Google Scholar]
- 60. Park C., Sohn Y., Shin K. S., Kim J., Ahn H., Huh Y. (2003) The chronic inhibition of nitric-oxide synthase enhances cell proliferation in the adult rat hippocampus. Neurosci. Lett. 339, 9–12 [DOI] [PubMed] [Google Scholar]
- 61. Covacu R., Danilov A. I., Rasmussen B. S., Hallén K., Moe M. C., Lobell A., Johansson C. B., Svensson M. A., Olsson T., Brundin L. (2006) Nitric oxide exposure diverts neural stem cell fate from neurogenesis towards astrogliogenesis. Stem Cells 24, 2792–2800 [DOI] [PubMed] [Google Scholar]
- 62. Carreira B. P., Morte M. I., Inácio A., Costa G., Rosmaninho-Salgado J., Agasse F., Carmo A., Couceiro P., Brundin P., Ambrósio A. F., Carvalho C. M., Araújo I. M. (2010) Nitric oxide stimulates the proliferation of neural stem cells bypassing the epidermal growth factor receptor. Stem Cells 28, 1219–1230 [DOI] [PubMed] [Google Scholar]
- 63. Corso-Díaz X., Krukoff T. L. (2010) nNOSα and nNOSβ localization to aggresome-like inclusions is dependent on HSP90 activity. J. Neurochem. 114, 864–872 [DOI] [PubMed] [Google Scholar]
- 64. Zhang R., Wang L., Zhang L., Chen J., Zhu Z., Zhang Z., Chopp M. (2003) Nitric oxide enhances angiogenesis via the synthesis of vascular endothelial growth factor and cGMP after stroke in the rat. Circ. Res. 92, 308–313 [DOI] [PubMed] [Google Scholar]
- 65. Hood J., Granger H. J. (1998) Protein kinase G mediates vascular endothelial growth factor-induced Raf-1 activation and proliferation in human endothelial cells. J. Biol. Chem. 273, 23504–23508 [DOI] [PubMed] [Google Scholar]
- 66. Endo S., Launey T. (2003) Nitric oxide activates extracellular signal-regulated kinase 1/2 and enhances declustering of ionotropic glutamate receptor subunit 2/3 in rat cerebellar Purkinje cells. Neurosci. Lett. 350, 122–126 [DOI] [PubMed] [Google Scholar]
- 67. Ota K. T., Pierre V. J., Ploski J. E., Queen K., Schafe G. E. (2008) The NO-cGMP-PKG signaling pathway regulates synaptic plasticity and fear memory consolidation in the lateral amygdala via activation of ERK/MAP kinase. Learn. Mem. 15, 792–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cano E., Mahadevan L. C. (1995) Parallel signal processing among mammalian MAPKs. Trends Biochem. Sci. 20, 117–122 [DOI] [PubMed] [Google Scholar]
- 69. Lopez-Ilasaca M. (1998) Signaling from G-protein-coupled receptors to mitogen-activated protein (MAP)-kinase cascades. Biochem. Pharmacol. 56, 269–277 [DOI] [PubMed] [Google Scholar]
- 70. Yoneyama M., Kawada K., Gotoh Y., Shiba T., Ogita K. (2010) Endogenous reactive oxygen species are essential for proliferation of neural stem/progenitor cells. Neurochem. Int. 56, 740–746 [DOI] [PubMed] [Google Scholar]