

Background: The mechanism of action of the potent antibiotic empedopeptin was not known.
Results: Empedopeptin forms calcium-dependent complexes with peptidoglycan precursors, particularly lipid II.
Conclusion: Bacterial cell wall synthesis is blocked by sequestration of the bactoprenol carrier.
Significance: This mechanism is proposed for a wider class of structurally related antibiotics including empedopeptin, tripropeptins, and plusbacins.
Keywords: Antibiotic Action, Cell Wall, Lipids, Peptidoglycan, Staphylococcus aureus, Empedopeptin, Lipid II, Plusbacin, Tripropeptin, Undecaprenyl Pyrophosphate
Abstract
Empedopeptin is a natural lipodepsipeptide antibiotic with potent antibacterial activity against multiresistant Gram-positive bacteria including methicillin-resistant Staphylococcus aureus and penicillin-resistant Streptococcus pneumoniae in vitro and in animal models of bacterial infection. Here, we describe its so far elusive mechanism of antibacterial action. Empedopeptin selectively interferes with late stages of cell wall biosynthesis in intact bacterial cells as demonstrated by inhibition of N-acetylglucosamine incorporation into polymeric cell wall and the accumulation of the ultimate soluble peptidoglycan precursor UDP-N-acetylmuramic acid-pentapeptide in the cytoplasm. Using membrane preparations and the complete cascade of purified, recombinant late stage peptidoglycan biosynthetic enzymes and their respective purified substrates, we show that empedopeptin forms complexes with undecaprenyl pyrophosphate containing peptidoglycan precursors. The primary physiological target of empedopeptin is undecaprenyl pyrophosphate-N-acetylmuramic acid(pentapeptide)-N-acetylglucosamine (lipid II), which is readily accessible at the outside of the cell and which forms a complex with the antibiotic in a 1:2 molar stoichiometry. Lipid II is bound in a region that involves at least the pyrophosphate group, the first sugar, and the proximal parts of stem peptide and undecaprenyl chain. Undecaprenyl pyrophosphate and also teichoic acid precursors are bound with lower affinity and constitute additional targets. Calcium ions are crucial for the antibacterial activity of empedopeptin as they promote stronger interaction with its targets and with negatively charged phospholipids in the membrane. Based on the high structural similarity of empedopeptin to the tripropeptins and plusbacins, we propose this mechanism of action for the whole compound class.
Introduction
Empedopeptin is an amphoteric, cyclic lipodepsipeptide produced by the Gram-negative soil bacterium Empedobacter haloabium ATCC 31962. Two accompanying publications in 1984 described its isolation, structure determination, and antibacterial activity in vitro and in vivo (1, 2). Empedopeptin showed potent antibacterial activity against a broad range of aerobic and anaerobic Gram-positive bacteria including the important pathogens Staphylococcus aureus, Streptococcus pneumoniae, and Clostridium difficile. Minimal inhibitory concentrations (MICs)2 in the low μg/ml range for susceptible as well as antibiotic-resistant isolates, substantial therapeutic efficacy in lethal bloodstream infections in mice, good pharmacokinetic parameters, and low acute toxicity form a promising profile for the compound (1) especially in light of rising vancomycin resistance among staphylococci and enterococci. Therefore, we revisited empedopeptin and elucidated its molecular mechanism of action.
The overall negatively charged, water-soluble empedopeptin consists of an octapeptide core cyclized by an ester bond and a C14-myristic acid tail (2) (Fig. 1A). The macrolactone core comprises alternating stretches of d- and l-configured amino acids (arginine, proline, and serine) as well as the non-proteinogenic amino acids hydroxyaspartic acid and hydroxyproline. Empedopeptin has considerable structural similarity to the lipodepsipeptides of the tripropeptin and plusbacin groups (3–7). The lower portion of the octapeptide core, which contains the three hydroxyamino acids plus arginine, is actually identical in empedopeptin, the tripropeptins (A–E and Z), and plusbacins (A1–B4) (4), whereas moderate variations occur in the amino acid composition of the upper ring segment as well as the fatty acid side chain (Fig. 1B). The lipid tails are branched or unbranched and are variable in length, the latter directly influencing antimicrobial activity. Although produced by different organisms, these lipodepsipeptides can basically be regarded as variants of the same structural scaffold.
FIGURE 1.

Structure of lipodepsipeptide antibiotic empedopeptin (A) and comparison with tripropeptins and plusbacins (B). Hy, hydroxy.
It has been demonstrated previously that plusbacin A3 interferes with bacterial peptidoglycan biosynthesis (8), although the specific enzymatic reaction or target structure had remained elusive. While work on this publication was in progress, it was reported that tripropeptin C can bind to undecaprenyl-pyrophosphate (C55-PP), thereby inhibiting the activity of undecaprenyl-pyrophosphate phosphatase (UppP) (9). Here, we show that empedopeptin exerts its antibacterial action by inhibition of peptidoglycan synthesis. However, using a comprehensive set of purified late stage peptidoglycan synthetic enzymes and the corresponding purified substrates, we demonstrate that this class of lipopeptide antibiotics sequesters further peptidoglycan precursors besides C55-PP and that the primary target of empedopeptin is lipid II, which is bound in a Ca2+-dependent fashion.
EXPERIMENTAL PROCEDURES
Production and Purification of Empedopeptin
Production of empedopeptin by E. haloabium ATCC 31962 on a 3-liter scale and extraction of the broth with butanol were performed as described (1). The crude butanol extract was subjected to reversed-phase vacuum liquid chromatography using stepwise gradient elution from H2O/MeOH (70:30) containing increasing proportions of MeOH followed by dichloromethane. LC-MS profiling indicated the pseudomolecular ion [M + H]+ and [M − H]− peaks at 1126.6 and 1124.6 m/z indicative of empedopeptin in the 100% MeOH fraction, which was further purified by reversed-phase HPLC using a linear gradient of 62:38–75:25 MeOH/H2O (2 mm NH4OAc) over a period of 30 min (Macherey-Nagel Nucleodur 100-5 C18, 250 × 8 mm, 5 μm; 1.8 ml/min flow rate; UV monitoring at 210 nm). Rechromatography of semipure empedopeptin by reversed-phase HPLC using two linear gradients of 60:40–65:35 MeOH/H2O (2 mm NH4OAc) over a period of 15 min and 65:35–90:10 over a 5-min period followed by isocratic elution at 90:10 for an additional 10 min (Macherey-Nagel Nucleosil 120-5 C18, 250 × 4 mm, 5 μm; 0.8 ml/min flow rate; UV monitoring at 210 nm) afforded 6.2 mg of pure empedopeptin.
Antibiotic Susceptibility Testing
MICs were determined by standard broth microdilution according to the Clinical and Laboratory Standards Institute guidelines (10) in polypropylene microtiter plates (Nunc brand) using cation-adjusted Mueller-Hinton broth (Oxoid). Experiments were conducted in the presence and absence of 1.25 mm Ca2+, which equates to the concentration of ionized calcium in human serum (11).
Precursor Incorporation Studies
To study the effect of empedopeptin on the synthesis of the major bacterial biopolymers, Bacillus subtilis was grown in Belitzky defined minimal medium (12), and the incorporation of tritium-labeled precursors (uridine, thymidine, phenylalanine, and N-acetylglucosamine (GlcNAc); 0.02 MBq/ml) into the acid-precipitable cell fraction was measured as described previously (13). Cultures were incubated with empedopeptin at a concentration of 8 μg/ml and with vancomycin at 0.25 μg/ml, both corresponding to 8× MIC in this medium. Belitzky medium contains 2 mm Ca2+.
Determination of Membrane Potential
The membrane potential was determined by a previously described method (14) using the lipophilic cation tetraphenylphosphonium (TPP+), which diffuses across the bacterial membrane in response to a trans-negative membrane potential (ΔΨ). 1 μCi/ml [3H]TPP+ (26 Ci/mmol) was added to a culture of S. aureus SG511 (A600 = 0.7) in half-concentrated Mueller-Hinton broth at 37 °C supplemented with 1.25 mm Ca2+, and cell-bound radioactivity was determined after filtration. For calculation of the membrane potential, cell-associated versus free TPP+ concentrations were applied to the Nernst equation. Culture aliquots were treated with empedopeptin (10 μg/ml, corresponding to 10-fold MIC) or the uncoupler carbonyl cyanide m-chlorophenylhydrazone (500 μm) as a control.
Quantification of Intracellular UDP-N-acetylmuramic Acid-pentapeptide (UDP-MurNAc-pp)
We followed the protocol of Kohlrausch and Höltje (15) to analyze the cytoplasmic nucleotide precursor pool. S. aureus ATCC 29213 was grown in 20 ml of half-concentrated Mueller-Hinton broth with 1.25 mm Ca2+ to an A600 of 0.6 and incubated with 130 μg/ml chloramphenicol for 15 min. Chloramphenicol is necessary to prevent (under the impact of the antibiotic under investigation) induction of autolytic processes and de novo synthesis of enzymes hydrolyzing the nucleotide-activated sugars, thereby interfering with determination of the soluble precursor (16). Empedopeptin was added at 40 μg/ml and vancomycin was added at 5 μg/ml (both 10× MIC) and incubated for another 45 min. Extraction of UDP-linked peptidoglycan precursors with boiling water and their analysis by HPLC was performed as described previously (17). Mass spectrometric confirmation of UDP-MurNAc-pp was conducted using a MALDI-TOF mass spectrometer (Bruker Biflex, Bruker Daltronics, Bremen, Germany) working in a linear negative mode. An aliquot of the HPLC fraction containing UDP-MurNAc-pp was mixed with 6-aza-2-thiothymine in 50% (v/v) ethanol, 20 mm ammonium citrate as matrix material and spotted onto a ground steel MALDI target plate. Mass spectra were recorded in the range of 1000–1500 Da and analyzed by Flexanalysis (Bruker Daltronics).
In Vitro Lipid II Synthesis with Membrane Preparations of Micrococcus luteus
In vitro lipid II synthesis was performed using membranes of M. luteus as described (18, 19). In short, for analytical assays, membrane preparations (200 μg of protein) were incubated in the presence of purified substrates (5 nmol undecaprenyl phosphate (C55-P), 50 nmol UDP-MurNAc-pp, and 50 nmol UDP-GlcNAc) in 60 mm Tris-HCl, 5 mm MgCl2, pH 7.5, 1.25 mm Ca2+, 0.5% (w/v) Triton X-100 in a total volume of 50 μl for 1 h at 30 °C. Incorporation of [14C]UDP-GlcNAc was used to quantify lipid II synthesized. Bactoprenol-containing products were extracted with an equal volume (50 μl) of butanol/pyridine acetate, pH 4.2 (2:1, v/v) and analyzed by thin layer chromatography (TLC) using chloroform/methanol/water/ammonia (88:48:10:1, v/v/v/v) as the solvent (20) and phosphomolybdic acid staining (21). Quantification was carried out by phosphorimaging in a StormTM imaging system (GE Healthcare). Empedopeptin (molecular mass, 1126.2 Da) was added in molar ratios of 0.5–2 with respect to C55-P. Synthesis and purification of lipid II on a preparative scale were performed as described (21, 22).
Synthesis of [14C]UDP-MurNAc-peptides by S. aureus MurA-F and DdlA Enzymes
[14C]UDP-MurNAc-pp was synthesized as described (23) with modifications (24). UDP-GlcNAc (100 nmol) was incubated with 15 μg of each of the recombinant histidine-tagged muropeptide synthetases MurA to MurF and d-alanyl-d-alanine ligase DdlA in 50 mm Bis-Tris propane, pH 8, 25 mm (NH4)2SO4, 5 mm MgCl2, 5 mm KCl, 0.5 mm DTT, 2 mm ATP, 2 mm phosphoenolpyruvate, 2 mm NADPH, 1 mm of each amino acid (14C-l-Ala, as well as unlabeled d-Glu, l-Lys, and d-Ala), 10% DMSO in a total volume of 125 μl for 60 min at 30 °C. 31.25 μl of the reaction mixture, corresponding to 25 nmol of [14C]UDP-MurNAc-pp, were used in the MraY synthesis assay without further purification. UDP-MurNAc-peptide variants with shortened stem peptide, i.e. UDP-MurNAc-dipeptide and -tripeptide, were synthesized in the presence of the corresponding subset of muropeptide synthetases and were used for the synthesis of lipid I(dipeptide) and lipid I(tripeptide), respectively.
In Vitro Peptidoglycan Synthesis Reactions Using Purified Proteins and Substrates
The MurG (UDP-N-acetylglucosamine:N-acetylmuramic acid(pentapeptide) undecaprenyl-pyrophosphate N-acetylglucosamine transferase) assay was performed in a final volume of 30 μl containing 2.5 nmol of purified lipid I (25) and 25 nmol of [14C]UDP-GlcNAc in 200 mm Tris-HCl, 5.7 mm MgCl2, pH 7.5, 0.8% Triton X-100 in the presence of 0.45 μg of purified MurG-His6 enzyme. The reaction mixture was incubated for 60 min at 30 °C. The assay for synthesis of lipid II-Gly1 catalyzed by FemX was performed as described previously (21). In vitro transglycosylation was determined by incubating 2 nmol of [14C]lipid II in 100 mm MES, 10 mm MgCl2, pH 5.5 in a total volume of 50 μl. The reaction was initiated by the addition of 7.5 μg of PBP2-His6 and incubated for 1.5 h at 30 °C (24). In all in vitro assays, empedopeptin was added in molar ratios ranging from 0.5 to 2 with respect to the amount of C55-P, lipid I, and lipid II, respectively. 1.25 mm Ca2+ was added as indicated. Synthesized lipid intermediates were extracted from the reaction mixtures with n-butanol/pyridine acetate, pH 4.2 (2:1, v/v), analyzed by TLC as described above, and quantified by phosphorimaging. In the PBP2 assay, reaction mixtures were applied directly onto the thin layer plates without extraction and were developed in solvent B (butanol/acetic acid/water/pyridine (15:3:12:10, v/v/v/v)). Experimental details related to assaying MraY, TagO, and UppP are provided in supplemental Figs. S1–S3.
Complex Formation of Empedopeptin with Lipid I/II and C55-PP
Binding of empedopeptin to C55-PP, lipid I, or lipid II was analyzed by incubating 2 nmol of each purified cell wall precursor with 1–8 nmol of empedopeptin in 50 mm Tris-HCl, pH 7.5 supplemented with 1.25 mm Ca2+ for 30 min. Analysis of complex formation between empedopeptin and the respective peptidoglycan precursor was carried out by applying reaction mixtures directly onto TLC plates and developing the plates in solvent B. Lipid-containing peptidoglycan precursors were detected by phosphomolybdic acid staining.
Antagonization Assays
Antagonization of the antibiotic activity of empedopeptin by potential target molecules was performed by an MIC-type assay setup in microtiter plates. Empedopeptin (8 μg/ml, corresponding to 8× MIC) in Mueller-Hinton broth with 1.25 mm Ca2+ was mixed with potential antagonists (UDP-GlcNAc, UDP-MurNAc-pp, C55-P, farnesyl-PP, C55-PP, lipid I, lipid II, undecaprenyl-PP-GlcNAc (lipid III), and lipid I variants with shortened stem peptides) in 0.156–10-fold molar excess with respect to the antibiotic. B. subtilis 168 (5 × 105cfu/ml) was added, and visible bacterial growth was recorded after overnight incubation. The concentration of purified peptidoglycan precursors was quantified on the basis of their phosphate content as described (26).
Quartz Crystal Microbalance
Quartz crystals were prepared and cleaned as described previously (27). Supported bilayers were completed by transferring a monolayer consisting of 90 mol % 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine and 10 mol % 1,2-dioleoyl-sn-glycero-3-phospho-(1′-racemic glycerol) onto a covalently fixed C16H33SH monolayer using the Langmuir-Blodgett technique. Fitting the quartz crystals into the flow chamber was performed under water to guarantee the integrity of the bilayer. Empedopeptin was investigated at 1 μm in the presence and absence of 1.25 mm CaCl2. A LiquiLab21 quartz crystal microbalance (ifake.V., Barleben, Germany), which enables the simultaneous detection of frequency and damping changes in real time, was used. Measurements and calculation of the kinetic binding constants were performed as described (28, 29).
RESULTS
Antimicrobial Activity of Empedopeptin
Empedopeptin exhibited potent antibacterial activity against multiresistant staphylococci and streptococci including methicillin-resistant S. aureus and penicillin-resistant S. pneumoniae (Table 1). The activity against enterococci was moderate in general but notably included vancomycin-resistant isolates. Escherichia coli was only marginally affected in accordance with the lack of activity reported earlier for other Gram-negative species (1), which probably reflects insufficient penetration of the rather large (1126.2 Da), net negatively charged compound through the outer membrane. Minimal inhibitory concentrations of empedopeptin were strongly influenced by the concentration of calcium ions in the culture medium. Addition of 1.25 mm Ca2+, equating to the Ca2+ concentration in human serum (11), to standard Mueller Hinton broth reduced the MIC values 2–16-fold depending on the test strain. It is important to note in this context that the calcium levels recommended by the Clinical Laboratory Standards Institute for cation-adjusted Mueller-Hinton broth (20–25 mg/liter Ca2+) (10) were not sufficient to achieve full empedopeptin activity.
TABLE 1.
Antibacterial activity of empedopeptin against representative bacterial strains in absence and presence of 1.25 mm Ca2+
MRSA, methicillin-resistant S. aureus; PRSP, penicillin-resistant S. pneumoniae; VRE, vancomycin-resistant Enterococcus.
| Bacterial strain | MIC |
|
|---|---|---|
| Without Ca2+ | +1.25 mm Ca2+ | |
| μg/ml | ||
| S. aureus ATCC 29213 | 32 | 4 |
| S. aureus N315 (MRSA) | 32 | 4 |
| S. aureus SG 511 | 8 | 1 |
| Staphylococcus simulans 22 | 16 | 1 |
| S. pneumoniae DSM 11865 (PRSP) | 1 | 0.25 |
| Streptococcus pyogenes ATCC 10389 | 1 | 0.25 |
| Enterococcus faecalis ATCC 29212 | 16 | 8 |
| Enterococcus faecium BM4147 (VRE) | 32 | 16 |
| B. subtilis 168 | 8 | 1 |
| M. luteus DSM 1790 | 0.125 | <0.062 |
| E. coli W3110 | >64 | >64 |
Metabolic Pathway Targeted by Empedopeptin
To study the impact of the antibiotic on the biosynthetic capacity of intact bacterial cells, we followed the incorporation of radiolabeled metabolic precursors into bacterial macromolecules. Empedopeptin at 8-fold the MIC rapidly and strongly interfered with the incorporation of [3H]GlcNAc into the cell wall of B. subtilis, whereas DNA and protein biosynthesis remained unaffected (Fig. 2). Incorporation of [3H]uridine into RNA was also diminished after prolonged exposure of the cells to the antibiotic (30 min). This can be explained by the fact that empedopeptin triggered a substantial accumulation of a soluble UDP-activated peptidoglycan precursor (UDP-N-acetylmuramic acid-pentapeptide) in the bacterial cytoplasm as outlined below, thereby depleting the uridine pool for RNA synthesis. Vancomycin, an established and specific inhibitor of peptidoglycan synthesis, induced a comparable effect at 8× MIC (Fig. 2).
FIGURE 2.
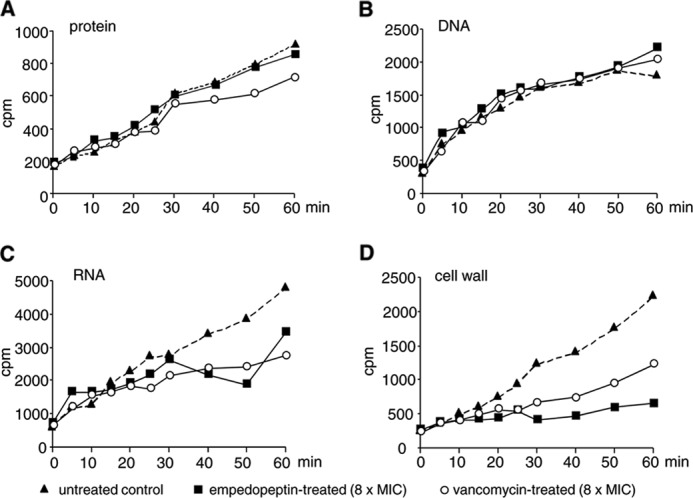
Impact of empedopeptin on macromolecular biosyntheses of B. subtilis 168. Untreated (triangles), empedopeptin-treated (squares), or vancomycin-treated (open circles) cells (both 8× MIC) were incubated with tritium-labeled precursors to monitor their incorporation into proteins (A), DNA (B), RNA (C), and cell wall (D). [3H]Phenylalanine was used to quantify protein synthesis, [3H]thymidine was used to quantify DNA synthesis, [3H]uridine was used to quantify RNA synthesis, and [3H]GlcNAc was used to quantify cell wall synthesis.
Peptidoglycan synthesis takes place in two different cellular compartments. At first, several cytoplasmic enzymes assemble the soluble, activated UDP-MurNAc-pp. The sugar-peptide moiety is then transferred to the membrane anchor C55-P to yield a membrane-standing monomer, which is further decorated, flipped across the membrane to the exterior, and incorporated into the three-dimensional peptidoglycan network. To distinguish whether empedopeptin interferes with peptidoglycan synthesis in the cytoplasmic versus the membrane-associated phase, we determined the cytoplasmic levels of UDP-MurNAc-pp. Antibiotics that interfere with the late stages of peptidoglycan synthesis, such as vancomycin, are known to trigger an accumulation of this ultimate soluble peptidoglycan precursor in the cytoplasm. Treatment of S. aureus with empedopeptin (10× MIC) led to a significant accumulation of UDP-MurNAc-pp similar to vancomycin-treated controls at corresponding multiples of the MIC (Fig. 3A).
FIGURE 3.
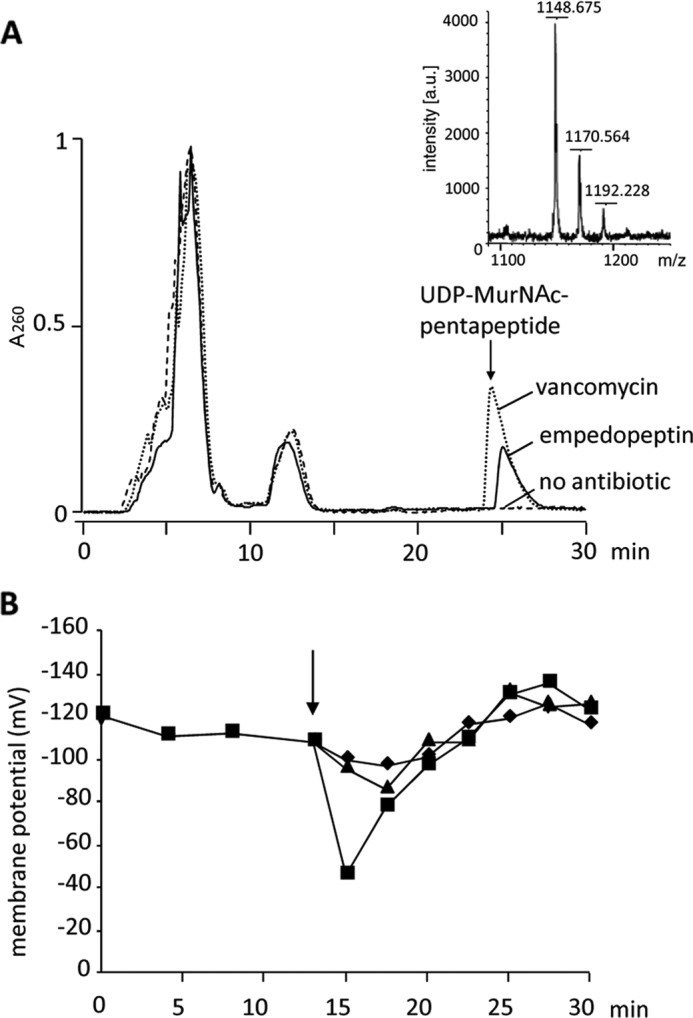
Impact of empedopeptin on the peptidoglycan precursor pool and membrane potential of staphylococci. Intracellular accumulation of the soluble cell wall precursor UDP-MurNAc-pentapeptide in S. aureus ATCC 29213 is shown (A). Cells were treated with empedopeptin (solid line), vancomycin (dotted line), or both at 10-fold MIC or left untreated (dashed line). The intracellular nucleotide pool was analyzed after extraction. HPLC analysis and subsequent mass spectrometry (small inset panel) confirmed the identity of UDP-MurNAc-pp. Its monoisotopic mass (m/z) is 1149.35 Da. In addition to the singly charged ion, the mono- and disodium salts were detected. The influence of empedopeptin on the membrane potential of S. aureus SG511 (B) is shown. ΔΨ was calculated from the distribution of the lipophilic cation TPP+ inside and outside the cells. The arrow indicates the time of antibiotic or carbonyl cyanide m-chlorophenylhydrazone addition, respectively. Triangles, empedopeptin (10× MIC); squares, carbonyl cyanide m-chlorophenylhydrazone (500 μm); diamonds, control.
The results with whole cells indicated peptidoglycan synthesis as the target pathway of empedopeptin. However, as the antibiotic structure contains a lipophilic tail of considerable length, we also investigated whether the compound might cause additional damage to the cytoplasmic membrane. Disruption of membrane integrity results in a strong decrease of ΔΨ. To determine ΔΨ in the presence of empedopeptin, we quantified the intracellular versus extracellular concentration of the tritium-labeled lipophilic cation TPP+, which diffuses freely across the bacterial membrane in response to a trans-negative ΔΨ. When growing cells of S. aureus were treated with the antibiotic, only a slight drop of membrane potential by 20 mV was observed followed by a rapid recovery (Fig. 3B). In contrast, the addition of the uncoupling ionophore carbonyl cyanide m-chlorophenylhydrazone resulted in a substantial ΔΨ decrease of about 70 mV (Fig. 3B).
Impact of Empedopeptin on in Vitro Peptidoglycan Biosynthesis
The biosynthesis of the bacterial cell wall requires the coordinated interplay of a number of enzymes and their respective substrates. (For an overview of the reactions involved, see Fig. 6.) After completion of UDP-MurNAc-pp synthesis in the cytoplasm, the membrane-bound enzyme MraY couples the sugar-peptide precursor to the lipid carrier C55-P yielding lipid I (30). The transferase MurG subsequently adds GlcNAc to the muramic acid moiety of lipid I yielding lipid II (31). This central cell wall building block is characteristically modified in different species. In staphylococci, the peptidyltransferases FemXAB catalyze the attachment of a specific pentaglycine interpeptide bridge to the stem peptide (21, 32) before the precursor is translocated across the cytoplasmic membrane (33) and incorporated into the peptidoglycan network. As our studies with intact bacterial cells had suggested an inhibitory activity of empedopeptin on membrane-associated stages of peptidoglycan synthesis, we next investigated the effect of the antibiotic in various in vitro systems capable of the respective reactions.
FIGURE 6.

Overview of peptidoglycan biosynthesis in S. aureus and schematic model for mechanism of action of empedopeptin. Starting from UDP-GlcNAc, the ultimate soluble precursor UDP-MurNAc is synthesized in the cytoplasm by the sequential action of MurA to MurF enzymes. Lipid I is formed in the first membrane-associated step at the inner face of the membrane by MraY, which transfers the soluble sugar-peptide moiety to the lipid carrier C55-P. The transferase MurG subsequently links a GlcNAc moiety to the muramoyl portion of lipid I yielding lipid II. In S. aureus, lipid II is further decorated by the addition of five glycine residues, catalyzed by FemXAB enzymes. Finally, lipid II is translocated across the membrane, and the peptidoglycan-monomer is incorporated into the growing peptidoglycan network by transglycosylation and transpeptidation. Once the lipid II monomer appears at the outside of the cytoplasmic membrane, empedopeptin binds to it in a Ca2+-dependent manner at a region involving the pyrophosphate group, the first sugar, and the proximal parts of the stem peptide and the undecaprenyl chain. Dimerization of empedopeptin may occur in the course of this process. C55-PP is also bound at the outer leaflet of the cytoplasmic membrane but with lower affinity than lipid II. Size, shape, and positioning of all components are schematic and not based on structural data. PEP, phosphoenolpyruvate.
Membrane preparations of M. luteus catalyze the membrane-associated synthesis of lipid II by MraY and MurG in vitro in the presence of defined amounts of the soluble precursors UDP-MurNAc-pp and UDP-GlcNAc as well as the bactoprenol carrier C55-P. In the control reaction without antibiotic, C55-P was completely converted to lipid II by the sequential activity of MraY and MurG, whereas addition of empedopeptin reduced the amount of lipid II (Fig. 4A). Quantitative analysis using [14C]UDP-GlcNAc to specifically label lipid II revealed a concentration-dependent reduction of the lipid II amount. Addition of equimolar concentrations of empedopeptin with respect to C55-P resulted in a 50% decrease in extracted lipid II, and a 2-fold molar excess of antibiotic led to an almost complete absence of lipid II (Fig. 4B).
FIGURE 4.

Effect of empedopeptin on membrane-associated stages of peptidoglycan synthesis in vitro. A and B, in vitro lipid II synthesis catalyzed by M. luteus membrane preparations. A, thin layer chromatography of n-butanol/pyridine acetate extracts and detection by phosphomolybdic acid staining. B, quantification of synthesized lipid II in n-butanol/pyridine acetate extracts of the membrane preparation via incorporated [14C]UDP-GlcNAc and phosphorimaging. C, D, E, and F, reactions catalyzed by purified enzymes from purified substrates. C, lipid II synthesis from lipid I by MurG quantified via incorporated [14C]UDP-GlcNAc. D and F, lipid II-Gly1 synthesis from lipid II by FemX quantified via incorporated [14C]glycine. E, the transglycosylation reaction catalyzed by PBP2 was quantified by the reduction of free [14C]lipid II. The inhibitory activity of empedopeptin in the absence (gray bars) or presence (white bars) of 1.25 mm Ca2+ (C, D, and E) or in the presence of a calcium gradient (F) is shown. The FemX reaction was chosen as an exemplary system to probe the effect of a calcium concentration range. For all reactions, the amount of products synthesized by control reactions in the absence of antibiotic was set to 100%. Empedopeptin was added at molar ratios of 0.5:1, 1:1, and 2:1 with respect to the lipid substrates as indicated. In F, the molar ratio of lipid II:empedopeptin was 1:2. The lantibiotic nisin in a 2-fold molar excess served as a control. Mean values from three independent experiments are shown. Error bars indicate the standard deviation. EMP, empedopeptin; LI, lipid I; LII, lipid II.
Target of Empedopeptin and Impact of Ca2+ on Target Interaction
To narrow down the molecular target of empedopeptin and to elucidate the role of Ca2+ in the inhibitory process, individual peptidoglycan biosynthesis reactions were analyzed in vitro using the purified recombinant proteins MraY, MurG, FemX, and PBP2 together with their respective purified substrates in the presence and absence of empedopeptin. Testing the MurG-catalyzed conversion of lipid I to lipid II as well as the incorporation of the first glycine residue into lipid II by recombinant FemX and the transglycosylation reaction by PBP2, we found all three reactions to be inhibited in a concentration-dependent manner (Fig. 4, C–F). The addition of Ca2+ considerably enhanced the effect of empedopeptin and led to a complete inhibition of the reactions at a 2:1 stoichiometry of empedopeptin to lipid precursor. Approximately physiological concentrations of calcium (1–1.25 mm) were required for empedopeptin to exert full inhibition as exemplified for the FemX-catalyzed glycine addition (Fig. 4F).
To investigate whether the stimulatory effect was specific for calcium or would likewise be triggered by other divalent cations, the influence of Mg2+, Mn2+, Fe2+, and Zn2+ was explored in several of the in vitro systems described above. For Mg2+, we can only draw limited conclusions because all in vitro systems depend on the presence of Mg2+, and omitting it from the assay mixtures prevented enzyme activity. Moreover, the presence of Zn2+ inhibited some enzymes, whereas others were more tolerant to the ion. Because of these experimental constraints, we measured all enzymes in the presence of 10 mm Mg2+ and determined the empedopeptin efficacy after further addition of a 1.25 mm concentration of the particular cation under investigation. Under these conditions, the cations stimulated the activity of empedopeptin in the following order: Ca2+ > Fe2+ > Zn2+ > Mn2+ > Mg2+. Calcium takes up a special position among all cations tested because it substantially increased the efficacy of empedopeptin even in the presence of 10 mm Mg2+. Taking into account the physiological concentrations of the cations in human blood where Fe2+, Zn2+, and Mn2+ are only available in traces, it is reasonable to presume that calcium has an important and prominent role in promoting the activity of empedopeptin against bacterial infections in the host.
It is important to note that empedopeptin inhibited the MurG, FemX, and PBP2 reactions at comparable concentrations, which made a specific interference with one of the enzymes highly unlikely and pointed to an interaction with the peptidoglycan precursors instead. The three enzymes discussed so far utilize undecaprenyl pyrophosphate-containing peptidoglycan precursors that also include a sugar moiety. In contrast, two enzymes that use the structurally simpler C55-P as a substrate were unaffected by empedopeptin when applied in the same molar ratio, namely MraY, which catalyzes the synthesis of lipid I (supplemental Fig. S1), and TagO, which catalyzes the formation of lipid III, the first membrane-bound lipid intermediate of teichoic acid biosynthesis (34, 35) (supplemental Fig. S2). Moreover, dephosphorylation of C55-PP by UppP (36) was also not markedly inhibited at a 2-fold molar excess of empedopeptin over C55-PP (supplemental Fig. S3).
Binding Site of Empedopeptin at Peptidoglycan Precursors
The interaction of empedopeptin with different undecaprenol-bound peptidoglycan precursors was directly demonstrated by incubating each lipid precursor with the antibiotic in buffer in various molar ratios followed by analysis via thin layer chromatography (Fig. 5). Free lipid I and lipid II were clearly visible at defined positions in the chromatogram, whereas free empedopeptin was not detectable by the phosphomolybdic acid staining method applied. In complex with empedopeptin, lipid I and lipid II migrated considerably more slowly and remained close to the application spot. In accordance with the results obtained in the enzyme reaction assays described above, a 2-fold molar excess of empedopeptin was sufficient for full sequestration of the peptidoglycan precursors lipid I and lipid II in a stable complex that even withstood solvent treatment during separation by thin layer chromatography. In contrast, even a 4-fold molar excess of the antibiotic could not trap undecaprenyl pyrophosphate in a stable complex under these conditions (Fig. 5). Notwithstanding, empedopeptin had some influence on the migration behavior of undecaprenyl pyrophosphate during thin layer chromatography because the lipid moved as a more focused band in the presence of the antibiotic (Fig. 5C, compare lane 1 with lanes 2–4).
FIGURE 5.
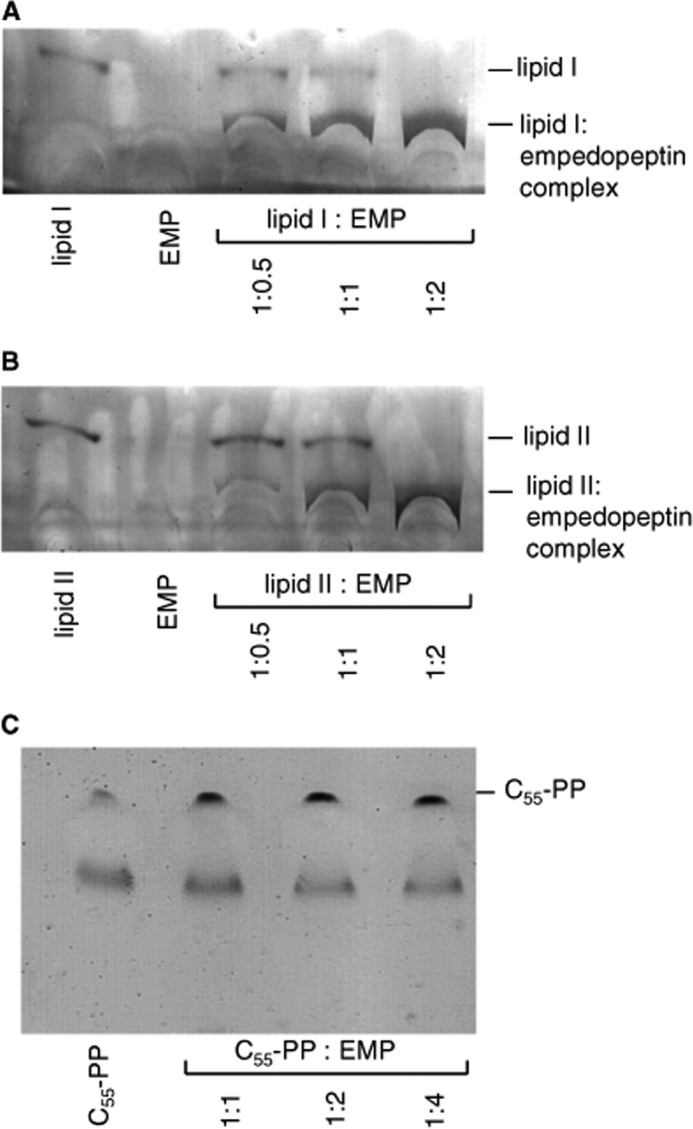
Complex formation of empedopeptin with peptidoglycan precursors lipid I, lipid II, and C55-PP and estimation of binding stoichiometry. Lipid I (A), lipid II (B), and C55-PP (C) were incubated with empedopeptin (EMP) at the indicated molar ratios in the presence of 1.25 mm calcium. The stable complex of empedopeptin with lipid I or lipid II remains close to the application spot, whereas free lipids migrate to the indicated positions. C55-PP was not trapped in a complex up to a 4-fold molar excess of the antibiotic. Rf(lipid I), 0.33; Rf(lipid I-EMP complex), 0.17; Rf(lipid II), 0.3; Rf(lipid II-EMP complex), 0.16; Rf(C55-PP), 0.37.
To narrow down the region of the lipid precursors that interacts with empedopeptin, we performed antagonization assays with diverse cell wall precursors and truncated variants thereof (Table 2). A concentration of empedopeptin corresponding to 8× MIC was preincubated with a 0.156–10-fold molar excess of the potentially antagonizing agents before being tested for growth inhibition of B. subtilis in Ca2+-supplemented (1.25 mm) culture broth. Lipid II and lipid I showed the strongest antagonizing effect on the antimicrobial activity of empedopeptin, confirming that the antibiotic is effectively trapped in a complex with the purified peptidoglycan precursors. Hence, it is no longer available for interaction with its membrane-standing targets when added to the cells. A lipid I congener with the peptide side chain shortened by the three C-terminal amino acids was only slightly less effective in antagonization. Also lipid III and C55-PP still retained substantial antagonizing potential, although their affinity for empedopeptin decreased in that order. In contrast, C55-P and the nucleotide-activated peptidoglycan sugars had no effect in the concentration range tested (Table 2).
TABLE 2.
Antagonistic effect of peptidoglycan and wall teichoic acid precursors and truncated variants thereof on antimicrobial activity of empedopeptin
Empedopeptin at 8× MIC was exposed to the potentially antagonizing agents in the indicated molar ratios prior to mixture with B. subtilis in culture broth containing 1.25 mm Ca2+. Results of two independent experiments are summarized. +, antagonization; −, no antagonization.
| Lipid intermediate | Molar ratio of precursor to empedopeptin |
||||||
|---|---|---|---|---|---|---|---|
| 10× | 5× | 2.5× | 1.25× | 0.625× | 0.3125× | 0.156× | |
| UDP-GlcNAc | − | − | − | − | − | − | − |
| UDP-MurNAc-pp | − | − | − | − | − | − | − |
| C55-P | − | − | − | − | − | − | − |
| C15-PP | + | + | + | − | − | − | − |
| C55-PP | + | + | + | − | − | − | − |
| Lipid I(dipeptide) | + | + | + | + | + | − | − |
| Lipid I(tripeptide) | + | + | + | + | + | − | − |
| Lipid I(pentapeptide) | + | + | + | + | + | + | − |
| Lipid II | + | + | + | + | + | + | − |
| Lipid III | + | + | + | + | − | − | − |
Interaction of Empedopeptin with Model Membranes
The target sites of empedopeptin are embedded among the phospholipid head groups of the cytoplasmic membrane. Bearing in mind the structure of empedopeptin with its hydrophobic C14-myristic acid tail, it is highly likely that the antibiotic establishes close contacts with the membrane that might also promote target binding. To determine the affinity of empedopeptin for phospholipids bilayers per se, we used the quartz crystal microbalance biosensor technique. The capacity of the antibiotic to interact with model membranes was investigated on the basis of the mass increase that occurs upon its binding to the lipid bilayer. A mixture of 90 mol % 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine and 10 mol % 1,2-dioleoyl-sn-glycero-3-phospho-(1′-racemic glycerol) served as a model system for the negatively charged bacterial membranes. Kinetic binding constants, such as ka, kd, and kD, were calculated from the quartz crystal microbalance frequency curves in the presence and absence of the physiological calcium concentration. Empedopeptin demonstrated a high membrane affinity in accordance with its overall hydrophobic nature (Table 3). Addition of calcium further increased membrane affinity by ∼2.5-fold, resulting from a higher association rate and a diminished dissociation rate. The rapid association kinetics and the kD values match those of the lantibiotic gallidermin (37). Cyclic voltammetry revealed membrane perturbation upon binding, suggesting a tight contact of the antibiotic with the lipid layer (data not shown).
TABLE 3.
Binding affinity of empedopeptin to phospholipid model membranes
Association rate (ka), dissociation rate (kd), and overall binding affinity (kD) of empedopeptin as determined by the quartz crystal microbalance technique either in the absence or presence of 1.25 mm CaCl2.
| ka | kd | kD | |
|---|---|---|---|
| m−1s−1 | s−1 | μm | |
| Without Ca2+ | 2835 ± 507 | 2.10 × 10−3 ± 0.86 × 10−3 | 0.77 ± 0.41 |
| 1.25 mm Ca2+ | 4736 ± 1262 | 1.47 × 10−3 ± 0.70 × 10−3 | 0.30 ± 0.11 |
DISCUSSION
The lipodepsipeptide empedopeptin inhibits bacterial growth by blocking peptidoglycan biosynthesis. It forms complexes with undecaprenyl pyrophosphate-containing cell wall precursors, thereby interrupting the lipid cycle and depleting the utilizing enzymes of their substrates. Empedopeptin displayed the highest affinity for both lipid I and lipid II. Several distinct assay systems indicated a 2:1 binding stoichiometry of antibiotic to lipid precursors, and the complex was remarkably stable even in the presence of the butanol/pyridine acetate used to develop the thin layer plates depicted in Fig. 5.
The presence of Ca2+ ions at the concentration at which they occur in human serum (11) was required for full inhibitory activity of empedopeptin in our in vitro peptidoglycan synthesis assays. As several of these assay systems contained purified enzymes and substrates, it can be excluded that this stimulating effect is merely due to facilitated interaction of the antibiotic with the bacterial membrane. Rather, our data suggest that calcium markedly promotes the direct interaction of empedopeptin with its target. Empedopeptin is an amphipathic molecule with a hydrophobic region containing the myristoyl side chain and two proline residues opposite to a decidedly hydrophilic, net negative region that includes the arginine as well as two aspartic acid residues. It is reasonable to conclude that positively charged calcium ions play a crucial role in bridging the antibiotic to the pyrophosphate region of the lipid precursors. Considering the 2:1 stoichiometry of the complex, Ca2+ ions might also facilitate the formation of empedopeptin dimers by bridging two aspartic acid moieties. In addition, our study with target-free artificial phospholipids layers (Table 3) demonstrates that the interaction with negatively charged membranes is also enhanced in the presence of calcium. The strong decrease of the MIC values in medium containing physiological calcium concentrations (Table 1) indicates that these ionic effects are critical for the antibacterial activity of empedopeptin.
Calcium ions are also involved in the interaction of other antibiotics with peptidoglycan precursors. Recently it was demonstrated that the antibiotic activity of friulimicin B is dependent on Ca2+-mediated complex formation with C55-P (38). Calcium was also shown to promote the interaction of particular lantibiotics carrying the mersacidin-type binding motif with their target lipid II, although the antibiotic activity was only slightly enhanced in these cases (39).
Our phospholipid binding study demonstrated high affinity of empedopeptin for negatively charged membranes per se. The electric currents detected by cyclic voltammetry on model membranes even pointed to membrane perturbations upon empedopeptin binding. The effects detected in this model membrane system correlated with a slight drop of the membrane potential of bacterial cells after empedopeptin addition. However, the effect on ΔΨ was only temporary, and there was no indication for a significant disturbance of membrane integrity. Rather, this effect suggests an intimate interaction of the lipophilic antibiotic with the cytoplasmic membrane in accordance with its membrane-standing targets. Along the same line, the continuation of DNA, RNA, and protein synthesis in the presence of empedopeptin precludes a gross depolarization of the cytoplasmic membrane. These biosynthetic processes are highly energy-consuming and require constant ATP replenishment by oxidative phosphorylation. Also, the fact that UDP-MurNAc-pp accumulated in the cytoplasm indicates a functional membrane barrier because the precursor was retained inside the cells.
In antagonization assays, very low amounts of externally added lipid I or lipid II were sufficient to fully complex free empedopeptin and to prevent its interaction with bacterial cells (Table 2). Among the different peptidoglycan precursors tested, lipid II and lipid I were equally well bound by the antibiotic, indicating that the second sugar GlcNAc is not crucial for the interaction. The C-terminal region of the stem peptide containing the l-lysyl-d-alanyl-d-alanine moiety also does not significantly contribute to binding as evident from the comparison of lipid I with lipid I(dipeptide). This result highlights that the target site of empedopeptin differs from that of the glycopeptide antibiotics (40) and is in accordance with the activity of empedopeptin against vancomycin-resistant bacterial isolates. However, if the cell wall precursor lacks the whole stem peptide and the lactyl moiety of MurNAc as it occurs in lipid III, its potential for complex formation is 4-fold lower than that of lipid I or lipid II. Undecaprenyl pyrophosphate as such is also recognized by empedopeptin, but the absence of the GlcNAc moiety further reduced the affinity by a factor of 2. It is also noteworthy that in a similar antagonization study with tripropeptin C20-PP showed stronger antagonizing potential than C15-PP (9), suggesting that the undecaprenyl chain also takes part in the interaction. In summary, the interaction of empedopeptin with lipid II appears to involve the pyrophosphate group, the first sugar, and the first one or two amino acids of the stem peptide as well as the proximal part of the undecaprenyl chain (Fig. 6).
Because of its comparably large size, empedopeptin will probably not enter the bacterial cell. Whether it can flip across the membrane to interact with lipid I that resides within the inner phospholipid leaflet is questionable and remains to be studied. Therefore, the surface-exposed lipid II appears to be the likely physiological target of empedopeptin. Because lipid II is bound with high affinity, its sequestration at low antibiotic concentrations and concomitant inhibition of peptidoglycan biosynthesis are probably the primary killing event. As shown for a variety of other antibacterial agents among them lipopeptides, lantibiotics, and defensins (25, 41), lipid II binding is sufficient to prevent the synthesis of novel peptidoglycan followed by bacterial cell lysis and death. Bacterial cells exposed to a lipid II binder do not merely suffer from inhibition of the transglycosylation step in peptidoglycan synthesis but they also suffer from interruption of the lipid cycle and general depletion of the undecaprenyl pool. Depletion of free bactoprenol inhibits several reactions in peptidoglycan synthesis and the membrane-associated stages of teichoic acid and capsule biosynthesis, which also rely on C55-P as a carrier.
However, empedopeptin differs from several other lipid II binders described to date (e.g. mersacidin and plectasin) (18, 25) by its affinity for additional bactoprenol-containing precursors that appear outside the cell. C55-PP can be sequestered as probably can the lipid III structural motif within extracellular teichoic acid precursors (e.g. within C55-PP-GlcNAc-N-acetylmannosamine- (glycerol phosphate)2-(ribitol phosphate)10–40 in S. aureus) (42). Among these structures, empedopeptin has the highest affinity for lipid II, which we consider its primary and main interaction site. However, if bacterial cells are exposed to sufficiently high empedopeptin concentrations, the antibiotic will probably recognize the other structures as well, thereby trapping the bactoprenol carrier at more than one stage in parallel. As interaction with multiple targets is beneficial in the light of reduced resistance development (43, 44), this characteristic is an advantage of empedopeptin.
While this work was in progress, a report on the mechanism of action of tripropeptin C was published, proposing C55-PP as the primary target and its dephosphorylation as the target reaction (9). The authors based their conclusion on biosynthesis reactions with bacterial membrane fractions and a MIC-type antagonization assay with externally added C55-PP. Similar to our results, they found that the activity of tripropeptin C was antagonized by a comparably high molar excess of C55-PP. However, neither purified lipid I, II, or III nor purified peptidoglycan biosynthetic enzymes were used in this study. As a consequence, the assay systems applied did not allow measurement of the affinity of tripropeptin C for the full spectrum of peptidoglycan precursors or investigation of individual enzyme reactions separately in the absence of other interfering precursors and enzymes. On the basis of the clear results obtained in our defined systems and in light of the strong structural similarity between empedopeptin, the tripropeptins, and the plusbacins (Fig. 1), we propose that complex formation with lipid II as the primary target and with C55-PP and the lipid III moiety as secondary targets represents the mechanism of antibacterial action for this entire class of compounds.
Acknowledgment
We gratefully acknowledge MALDI support by Michaele Josten, Bonn, Germany.
This work was supported by the German Research Organization (Grant FOR-854, Projects BR 3783/1-1, SA 292/15-1, SA 292/13-1, GR 2673/2-1, and BE 2242/3-1) and by the research fund of the Medical Faculty of the University of Bonn (BONFOR).

This article contains supplemental Figs. S1–S3.
- MIC
- minimal inhibitory concentration
- PP
- pyrophosphate
- C55-P
- undecaprenyl phosphate
- C55-PP
- undecaprenyl pyrophosphate
- GlcNAc
- N-acetylglucosamine
- lipid I
- undecaprenyl pyrophosphate-N-acetylmuramic acid(pentapeptide)
- lipid II
- undecaprenyl pyrophosphate-N-acetylmuramic acid(pentapeptide)-N-acetylglucosamine
- lipid II-Gly1
- undecaprenyl pyrophosphate-N-acetylmuramic acid(pentapeptide-glycine)-N-acetylglucosamine
- lipid III
- undecaprenyl-pyrophosphate-GlcNAc
- TPP+
- tetraphenylphosphonium
- UDP-MurNAc-pp
- UDP-N-acetylmuramic acid-pentapeptide
- UppP
- undecaprenyl-pyrophosphate phosphatase
- Bis-Tris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- PBP
- penicillin-binding protein
- ΔΨ
- membrane potential.
REFERENCES
- 1. Konishi M., Sugawara K., Hanada M., Tomita K., Tomatsu K., Miyaki T., Kawaguchi H., Buck R. E., More C., Rossomano V. Z. (1984) Empedopeptin (BMY-28117), a new depsipeptide antibiotic. I. Production, isolation and properties. J. Antibiot. 37, 949–957 [DOI] [PubMed] [Google Scholar]
- 2. Sugawara K., Numata K., Konishi M., Kawaguchi H. (1984) Empedopeptin (BMY-28117), a new depsipeptide antibiotic. II. Structure determination. J. Antibiot. 37, 958–964 [DOI] [PubMed] [Google Scholar]
- 3. Hashizume H., Igarashi M., Hattori S., Hori M., Hamada M., Takeuchi T. (2001) Tripropeptins, novel antimicrobial agents produced by Lysobacter sp. I. Taxonomy, isolation and biological activities. J. Antibiot. 54, 1054–1059 [DOI] [PubMed] [Google Scholar]
- 4. Hashizume H., Hattori S., Igarashi M., Akamatsu Y. (2004) Tripropeptin E, a new tripropeptin group antibiotic produced by Lysobacter sp. BMK333-48F3. J. Antibiot. 57, 394–399 [DOI] [PubMed] [Google Scholar]
- 5. Hashizume H., Hirosawa S., Sawa R., Muraoka Y., Ikeda D., Naganawa H., Igarashi M. (2004) Tripropeptins, novel antimicrobial agents produced by Lysobacter sp. J. Antibiot. 57, 52–58 [DOI] [PubMed] [Google Scholar]
- 6. Shoji J., Hinoo H., Katayama T., Nakagawa Y., Ikenishi Y., Iwatani K., Yoshida T. (1992) Structures of new peptide antibiotics, plusbacins A1-A4 and B1-B4. J. Antibiot. 45, 824–831 [DOI] [PubMed] [Google Scholar]
- 7. Shoji J., Hinoo H., Katayama T., Matsumoto K., Tanimoto T., Hattori T., Higashiyama I., Miwa H., Motokawa K., Yoshida T. (1992) Isolation and characterization of new peptide antibiotics, plusbacins A1-A4 and B1-B4. J. Antibiot. 45, 817–823 [DOI] [PubMed] [Google Scholar]
- 8. Maki H., Miura K., Yamano Y. (2001) Katanosin B and plusbacin A(3), inhibitors of peptidoglycan synthesis in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 45, 1823–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hashizume H., Sawa R., Harada S., Igarashi M., Adachi H., Nishimura Y., Nomoto A. (2011) Tripropeptin C blocks the lipid cycle of cell wall biosynthesis by complex formation with undecaprenyl pyrophosphate. Antimicrob. Agents Chemother. 55, 3821–3828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clinical and Laboratory Standards Institute (2008) Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard-Eighth Edition, M07-A8, Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 11. Barry A. L., Fuchs P. C., Brown S. D. (2001) In vitro activities of daptomycin against 2,789 clinical isolates from 11 North American medical centers. Antimicrob. Agents Chemother. 45, 1919–1922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stülke J., Hanschke R., Hecker M. (1993) Temporal activation of β-glucanase synthesis in Bacillus subtilis is mediated by the GTP pool. J. Gen. Microbiol. 139, 2041–2045 [DOI] [PubMed] [Google Scholar]
- 13. Freiberg C., Brunner N. A., Schiffer G., Lampe T., Pohlmann J., Brands M., Raabe M., Häbich D., Ziegelbauer K. (2004) Identification and characterization of the first class of potent bacterial acetyl-CoA carboxylase inhibitors with antibacterial activity. J. Biol. Chem. 279, 26066–26073 [DOI] [PubMed] [Google Scholar]
- 14. Ruhr E., Sahl H. G. (1985) Mode of action of the peptide antibiotic nisin and influence on the membrane potential of whole cells and on cytoplasmic and artificial membrane vesicles. Antimicrob. Agents Chemother. 27, 841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kohlrausch U., Höltje J. V. (1991) Analysis of murein and murein precursors during antibiotic-induced lysis of Escherichia coli. J. Bacteriol. 173, 3425–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dai D., Ishiguro E. E. (1988) murH, a new genetic locus in Escherichia coli involved in cell wall peptidoglycan biosynthesis. J. Bacteriol. 170, 2197–2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brötz H., Bierbaum G., Reynolds P. E., Sahl H. G. (1997) The lantibiotic mersacidin inhibits peptidoglycan biosynthesis at the level of transglycosylation. Eur. J. Biochem. 246, 193–199 [DOI] [PubMed] [Google Scholar]
- 18. Brötz H., Bierbaum G., Leopold K., Reynolds P. E., Sahl H. G. (1998) The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother. 42, 154–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Umbreit J. N., Strominger J. L. (1972) Isolation of the lipid intermediate in peptidoglycan biosynthesis from Escherichia coli. J. Bacteriol. 112, 1306–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rick P. D., Hubbard G. L., Kitaoka M., Nagaki H., Kinoshita T., Dowd S., Simplaceanu V., Ho C. (1998) Characterization of the lipid-carrier involved in the synthesis of enterobacterial common antigen (ECA) and identification of a novel phosphoglyceride in a mutant of Salmonella typhimurium defective in ECA synthesis. Glycobiology 8, 557–567 [DOI] [PubMed] [Google Scholar]
- 21. Schneider T., Senn M. M., Berger-Bächi B., Tossi A., Sahl H. G., Wiedemann I. (2004) In vitro assembly of a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II-Gly5) of Staphylococcus aureus. Mol. Microbiol. 53, 675–685 [DOI] [PubMed] [Google Scholar]
- 22. Breukink E., Wiedemann I., van Kraaij C., Kuipers O. P., Sahl H., de Kruijff B. (1999) Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science 286, 2361–2364 [DOI] [PubMed] [Google Scholar]
- 23. Wong K. K., Kuo D. W., Chabin R. M., Waddell S. T., Marsilio F., Leiting B., Pompliano D. L. (1998) Engineering a cell-free murein biosynthetic pathway: combinatorial enzymology in drug discovery. J. Am. Chem. Soc. 120, 13527–13528 [Google Scholar]
- 24. Rubinchik E., Schneider T., Elliott M., Scott W. R., Pan J., Anklin C., Yang H., Dugourd D., Müller A., Gries K., Straus S. K., Sahl H. G., Hancock R. E. (2011) Mechanism of action and limited cross-resistance of new lipopeptide MX-2401. Antimicrob. Agents Chemother. 55, 2743–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schneider T., Kruse T., Wimmer R., Wiedemann I., Sass V., Pag U., Jansen A., Nielsen A. K., Mygind P. H., Raventós D. S., Neve S., Ravn B., Bonvin A. M., De Maria L., Andersen A. S., Gammelgaard L. K., Sahl H. G., Kristensen H. H. (2010) Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science 328, 1168–1172 [DOI] [PubMed] [Google Scholar]
- 26. Rouser G., Fkeischer S., Yamamoto A. (1970) Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 5, 494–496 [DOI] [PubMed] [Google Scholar]
- 27. Christ K., Rüttinger H. H., Höpfner M., Rothe U., Bendas G. (2005) The detection of UV-induced membrane damages by a combination of two biosensor techniques. Photochem. Photobiol. 81, 1417–1423 [DOI] [PubMed] [Google Scholar]
- 28. Christ K., Wiedemann I., Bakowsky U., Sahl H. G., Bendas G. (2007) The role of lipid II in membrane binding of and pore formation by nisin analyzed by two combined biosensor techniques. Biochim. Biophys. Acta 1768, 694–704 [DOI] [PubMed] [Google Scholar]
- 29. Simonis D., Fritzsche J., Alban S., Bendas G. (2007) Kinetic analysis of heparin and glucan sulfates binding to P-selectin and its impact on the general understanding of selectin inhibition. Biochemistry 46, 6156–6164 [DOI] [PubMed] [Google Scholar]
- 30. Ikeda M., Wachi M., Jung H. K., Ishino F., Matsuhashi M. (1991) The Escherichia coli mraY gene encoding UDP-N-acetylmuramoyl-pentapeptide:undecaprenyl-phosphate phospho-N-acetylmuramoyl-pentapeptide transferase. J. Bacteriol. 173, 1021–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ha S., Gross B., Walker S. (2001) E. coli MurG: a paradigm for a superfamily of glycosyltransferases. Curr. Drug Targets Infect. Disord. 1, 201–213 [DOI] [PubMed] [Google Scholar]
- 32. Rohrer S., Berger-Bächi B. (2003) FemABX peptidyl transferases: a link between branched-chain cell wall peptide formation and β-lactam resistance in gram-positive cocci. Antimicrob. Agents Chemother. 47, 837–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mohammadi T., van Dam V., Sijbrandi R., Vernet T., Zapun A., Bouhss A., Diepeveen-de Bruin M., Nguyen-Distèche M., de Kruijff B., Breukink E. (2011) Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 30, 1425–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Soldo B., Lazarevic V., Karamata D. (2002) tagO is involved in the synthesis of all anionic cell-wall polymers in Bacillus subtilis 168. Microbiology 148, 2079–2087 [DOI] [PubMed] [Google Scholar]
- 35. D'Elia M. A., Millar K. E., Beveridge T. J., Brown E. D. (2006) Wall teichoic acid polymers are dispensable for cell viability in Bacillus subtilis. J. Bacteriol. 188, 8313–8316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. El Ghachi M., Derbise A., Bouhss A., Mengin-Lecreulx D. (2005) Identification of multiple genes encoding membrane proteins with undecaprenyl pyrophosphate phosphatase (UppP) activity in Escherichia coli. J. Biol. Chem. 280, 18689–18695 [DOI] [PubMed] [Google Scholar]
- 37. Christ K., Al-Kaddah S., Wiedemann I., Rattay B., Sahl H. G., Bendas G. (2008) Membrane lipids determine the antibiotic activity of the lantibiotic gallidermin. J. Membr. Biol. 226, 9–16 [DOI] [PubMed] [Google Scholar]
- 38. Schneider T., Gries K., Josten M., Wiedemann I., Pelzer S., Labischinski H., Sahl H. G. (2009) The lipopeptide antibiotic friulimicin B inhibits cell wall biosynthesis through complex formation with bactoprenol phosphate. Antimicrob. Agents Chemother. 53, 1610–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Böttiger T., Schneider T., Martínez B., Sahl H. G., Wiedemann I. (2009) Influence of Ca2+ ions on the activity of lantibiotics containing a mersacidin-like lipid II binding motif. Appl. Environ. Microbiol. 75, 4427–4434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nagarajan R. (1991) Antibacterial activities and modes of action of vancomycin and related glycopeptides. Antimicrob. Agents Chemother. 35, 605–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sass V., Schneider T., Wilmes M., Körner C., Tossi A., Novikova N., Shamova O., Sahl H. G. (2010) Human β-defensin 3 inhibits cell wall biosynthesis in Staphylococci. Infect. Immun. 78, 2793–2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xia G., Peschel A. (2008) Toward the pathway of S. aureus WTA biosynthesis. Chem. Biol. 15, 95–96 [DOI] [PubMed] [Google Scholar]
- 43. Brötz-Oesterhelt H., Brunner N. A. (2008) How many modes of action should an antibiotic have? Curr. Opin. Pharmacol. 8, 564–573 [DOI] [PubMed] [Google Scholar]
- 44. Silver L. L. (2007) Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 6, 41–55 [DOI] [PubMed] [Google Scholar]