

Background: CP621–652 is a HIV-1 fusion inhibitor peptide containing the gp41 621QIWNNMT627 motif.
Results: The crystal structure of CP621–652 in complex with T21 was determined and mutational analyses were performed.
Conclusion: The residues Met626 and Thr627 in the gp41 621QIWNNMT627 motif are critical for HIV-1 entry and inhibition.
Significance: Our data provide important information for designing novel HIV fusion inhibitors.
Keywords: Antiviral Agents, Crystal Structure, Fusion Protein, HIV-1, Peptides, Virus Entry, Fusion Inhibitor, Gp41
Abstract
The core structure of HIV-1 gp41 is a stable six-helix bundle (6-HB) folded by its trimeric N- and C-terminal heptad repeats (NHR and CHR). We previously identified that the 621QIWNNMT627 motif located at the upstream region of gp41 CHR plays critical roles for the stabilization of the 6-HB core and peptide CP621–652 containing this motif is a potent HIV-1 fusion inhibitor, however, the molecular determinants underlying the stability and anti-HIV activity remained elusive. In this study, we determined the high-resolution crystal structure of CP621–652 complexed by T21. We find that the 621QIWNNMT627 motif does not maintain the α-helical conformation. Instead, residues Met626 and Thr627 form a unique hook-like structure (denoted as M-T hook), in which Thr627 redirects the peptide chain to position Met626 above the left side of the hydrophobic pocket on the NHR trimer. The side chain of Met626 caps the hydrophobic pocket, stabilizing the interaction between the pocket and the pocket-binding domain. Our mutagenesis studies demonstrate that mutations of the M-T hook residues could completely abolish HIV-1 Env-mediated cell fusion and virus entry, and significantly destabilize the interaction of NHR and CHR peptides and reduce the anti-HIV activity of CP621–652. Our results identify an unusual structural feature that stabilizes the six-helix bundle, providing novel insights into the mechanisms of HIV-1 fusion and inhibition.
Introduction
Cellular entry of human immunodeficiency virus type 1 virus (HIV-1) requires fusion of viral and cellular membranes that are mediated by viral trimeric envelope (env) glycoprotein gp120-gp41 complex (1, 2). The surface subunit gp120 is primarily involved in binding the CD4 receptor and a co-receptor (typically CCR5 or CXCR4) on the target cells, whereas transmembrane subunit gp41 is responsible for the fusion reaction. In the early 1990s, discovery of anti-HIV peptides from gp41 opened a bright avenue for exploring the molecular events of virus-cell membrane fusion (3–5). Several crystallographic studies revealed that the fusogenic core structure of gp41 is a thermostable trimer of hairpin (six-helix bundle structure, 6-HB),4 in which three N-terminal heptad repeat region (NHR) form a central trimeric coiled-coil, whereas three C-terminal heptad repeat regions (CHR) around the NHR pack as antiparallel helices into hydrophobic grooves (5–8). Prominently, the crystal structures identified a deep pocket (∼16-Å long, ∼7-Å wide, and 5–6 Å deep) in the C-terminal region of N-helices, which is formed by a cluster of hydrophobic residues (Fig. 1). Three hydrophobic residues (Trp628, Trp631, and Ile635) from the pocket-binding domain (PBD) (628WMEWEREI635) of the C-helix penetrate into the NHR pocket causing an extensive interaction. It is believed that the pocket can be an ideal target site for developing novel anti-HIV therapeutics (5, 9).
FIGURE 1.

Schematic illustration of HIV-1 gp41 and peptide fusion inhibitors. A, view of the gp41 functional regions. The residue numbers of each region correspond to their positions in gp160 of HIV-1HXB2. FP, fusion peptide; TM, transmembrane domain. B, sequence of NHR- and CHR-derived anti-HIV peptides. The residues corresponding to the NHR pocket region are marked in blue, whereas the residues for the PBD are marked in red. C, the thermal stability and anti-HIV activity of CHR-derived peptides. The data were derived from our previous publication (20).
Peptide inhibitors can bind to the exposed NHR or CHR and block dominant-negatively 6-HB formation that results in inactive fusion (5, 10–11). It has been established that the CHR-derived peptides (C-peptides) are often more potent than that derived from the NHR. T20 (Enfuvirtide, Fuzeon), a C-peptide, has been approved for clinical use as the first member of a new class of anti-HIV drugs, HIV fusion inhibitors (11–13). Because of drug-resistance problem, a series of novel peptide-based fusion inhibitors were developed with improved anti-HIV potency and pharmacokinetic profiles (14–18). Notably, these peptides have been derived primarily from residues 628 to 673 of the gp41 CHR, based on the peptide C34 sequence as a template in particular (17, 19), but little attention was drawn to the upstream sequence of the CHR. We previously found that the 621QIWNNMT627 motif located at the upstream region of the CHR, immediately adjacent to the PBD, plays critical roles for 6-HB formation and inhibition (20). The peptide CP621–652 containing this motif could dramatically improve its thermal stability and anti-HIV activity (Fig. 1), however, the molecular determinants were poorly understood and the 621QIWNNMT627 motif lacks structural information. In our previous publication (20), we discussed that “a crystallographic study on the 6-HB structure formed by CP621–652 and T21 is under the way, and hopefully, this study will provide more detailed information about their interaction. In addition, mutational analyses may also reveal the residues responsible for high affinity interactions between these NHR and CHR peptides.” Here, we report the results from our proposed studies. The crystal structure of the CP621–652·T21 complex discovered that the residues Met626 and Thr627 of the 621QIWNNMT627 motif form an unusual hook-like structure (denoted as M-T hook), a structural feature that has never been observed in the large collection of 6-HB structures of the HIV gp41 core. The structural data suggests that the M-T hook is crucial for stabilizing the interaction between the pocket binding residues from the CHR and the pocket region on the NHR trimer. Our mutagenesis studies validated the importance of both M-T hook residues for the stability of the NHR-CHR interaction, for HIV-1 Env-mediated cell fusion and virus entry, and for the anti-HIV activity of the CP621–652 peptide.
EXPERIMENTAL PROCEDURES
Peptide Synthesis
A panel of peptides, including CP621–652 and its mutants M626A and T627A, T21, N36, and T20 were synthesized by a standard solid-phase Fmoc (N-(9-fluorenyl)methoxycarbonyl) method in Scilight Biotechnology LLC as described previously (14). All peptides were acetylated at the N terminus and amidated at the C terminus. They were purified by reversed phase high-performance liquid chromatography (HPLC) and verified for purity >95% and correct amino acid composition by mass spectrometry. Concentrations of the peptides were determined by UV absorbance and a theoretically calculated molar extinction coefficients (280 nm) of 5500 and 1490 m−1 cm−1 based on the number of tryptophan and tyrosine residues (all the peptides tested contain Trp and/or Tyr), respectively.
Purification, Crystallization, and Structure Determination
The 6-HB containing the CP621–652 and T21 peptides was prepared by dissolving an equal amount (1:1 molar ratio) of peptides in the denaturing buffer (100 mm NaH2PO4, 10 mm Tris-HCl, pH 8.0, 8 m urea). The peptides were refolded by dialysis to yield the 6-HB structure. The resulting 6-HB was purified by size exclusion chromatography (Superdex 75 10/300 GL, GE Healthcare). The 6-HB containing CP621–652·T21 peptides were crystallized by mixing equal volumes (1 μl) of purified sample (∼10 mg/ml) and reservoir solution containing 0.2 m ammonium sulfate, 0.1 m sodium acetate, pH 4.6, 30% (w/v) PEG4000. The cryocooling was achieved by soaking the crystal 30–60 s in reservoir solution containing 15% glycerol. The crystals were flash-frozen in liquid nitrogen before x-ray diffraction experiments. Complete data were collected at the laboratory x-ray source (HighFlux HomeLabTM, Rigaku, wavelength = 1.54 Å). The crystal belonged to the space group of H32, contained one-third of a complete 6-helical bundle (one CP621–652 peptide and one T21 peptide) per asymmetry unit, and diffracted the x-ray to a resolution limit of 1.74 Å. The structures of the 6-HB were solved by molecule replacement (Phaser, CCP4 suite) using the HIV-1 gp41 core structure (Protein Data Bank code 3F4Y) as the search model. The initial electron density map was improved by manual model building (COOT) and refinement using PHENIX (21). The final atomic model has good refinement statistics and stereochemistry qualities (Table 1).
TABLE 1.
Data collection and refinement statistics
| CP621–652/T21 | |
|---|---|
| Data collection | |
| Space group | H32 |
| Cell dimensions | |
| a, b, c (Å) | 44.97, 44.97, 209.24 |
| α, β, γ (°) | 90.00, 90.00, 120.00 |
| X-ray source | RIGAKU MICROMAX-007 HF |
| Wavelength (Å) | 1.54 |
| Data range (Å) | 23.70–1.74 |
| Reflections unique | 8804 |
| Rsyma (last shell) | 0.037 (0.207) |
| I/σI | 33.0 (5.6) |
| Completeness (%) (last shell) | 99.8 (98.7) |
| Redundancy (last shell) | 6.51 (3.84) |
| Refinement | |
| Resolution range (Å) | 23.70–1.74 |
| Reflections F > 0.06 (cross-validation) | 8596 (411) |
| Rworkb/Rfreec (last shell) | 0.1921/0.1968 (0.2063/0.2537) |
| Non-hydrogen protein atoms | 681 |
| Protein | 580 |
| Water | 87 |
| N-acetyl group | 3 |
| C-NH2 group | 1 |
| Acetate | 4 |
| Glycerol | 6 |
| B-factors average | 23.01 |
| Protein | 22.00 |
| N-Acetyl group | 38.16 |
| C-NH2 group | 14.94 |
| Acetate | 30.93 |
| Glycerol | 47.67 |
| Water | 31.88 |
| Root mean square deviation | |
| Bond lengths (Å) | 0.005 |
| Bond angles (°) | 0.832 |
| MolProbity score | 1.67, 82nd percentile |
| % Favored regions and outliers in Ramachandran plot | 100.0, 0.0 |
a Rsym = ΣhklΣi|Ihkl,j − Ihkl|/ΣhklΣjIhkl,j, where Ihkl is the average of symmetry-related observations of a unique reflection.
b Rwork = Σhkl‖Fobs(hkl)| − |Fcalc(hkl)‖/Σhkl|Fobs(hkl)|.
c Rfree = the cross-validation R factor for 5% of reflections against which the model was not refined.
Site-directed Mutagenesis
Two of the HIV-1NL4-3 mutants carrying M626A or T627A substitutions were generated as described previously (22). The mutations were created using double-stranded DNA templates and selection of mutants with DpnI. For each mutation, the two primers contained the desired mutation and occupied the same starting and ending positions on opposite strands of the plasmid DNA. DNA synthesis was carried out by PCR in a 50-μl reaction volume using 1 ng of denatured plasmid template, 50 pm upper and lower primers, and 5 units of high-fidelity thermostable polymerase PrimeStar (Takara, Dalian, China). PCR amplification was carried out for one cycle of denaturation at 98 °C for 5 min, then 18 cycles of 98 °C for 15 s and 68 °C for 15 min, followed a final extension at 72 °C for 10 min. The amplicons were treated with the restriction enzyme DpnI for 1 h at 37 °C. DpnI-resistant molecules, which were rich in the desired mutants, were recovered by transforming Escherichia coli strain DH5α to antibiotic resistance. The successful mutations were confirmed by sequencing.
Cell-Cell Fusion Assays
To evaluate the effect of mutations on HIV-1 Env-driven cell-cell fusion, a sensitive assay was adapted from our previous studies (23, 24). Briefly, 293T effector cells seeded in 6-well plates at 4 × 105 cells per well were transfected with the plasmid encoding HIV-1NL4-3 Env or its mutants (M626A or T627A) in combination with plasmid pGAL4-VP16, which encodes the herpes simplex virus VP16 transactivator fused to the DNA-binding domain of the Saccharomyces cerevisiae transcription factor GAL4. In parallel, U87-CD4-CXCR4 target cells seeded in 48-well plates at 2.5 × 104 cells per well were transfected with pGal5-luc plasmid, which encodes the luciferase reporter gene under control of a promoter containing five GAL4-binding sites. The day after transfection, the effector cells were added to the target cells. After co-culturing for an additional 30 h, the cells were lysed by cell culture lysis buffer, and the luciferase activity was measured by luciferase assay system (Promega, Madison, WI).
To detect the inhibitory activity of CP621–652 and its mutants, cell fusion was monitored using a reporter gene assay based on activation of the HIV LTR-driven luciferase cassette in TZM-bl (Target) cells by HIV-1 tat from HL2/3 (Effector) cells (25). Briefly, the TZM-bl cells were plated in 96-well clusters (1 × 104 per well) and incubated at 37 °C overnight. The target cells were co-cultured with HL2/3 cells (3 × 104/well) for 6 h at 37 °C in the presence or absence of a tested peptide at graded concentrations. Luciferase activity was measured using luciferase assay regents and a Luminescence Counter (Promega) according to the manufacturer's instructions. Background luminescence in TZM-bl cells was determined without addition of HL2/3 cells. The percent inhibition of fusion by the peptides and 50% inhibitory of fusion concentration (IC50) values were calculated as previously described (14).
HIV-1 Pseudovirus and Single-cycle Infection
HIV-1 pseudoviruses were generated as described previously (26, 27). Briefly, 293T cells (5 × 106 cells in 15 ml of growth medium in a T-75 culture flask) were cotransfected with 10 μg of an Env-expressing plasmid and 20 μg of a backbone plasmid pSG3δEnv that encodes a Env-defective, luciferase-expressing HIV-1 genome using Lipofectamine 2000 (Invitrogen). Pseudovirus-containing culture supernatants were harvested 48 h after transfection and filtered at 0.45-μm pore size, and stored at −80 °C in 1-ml aliquots until use. The 50% tissue culture infectious dose (TCID50) of a single thawed aliquot of each pseudovirus batch was determined in TZM-bl cells. The antiviral activity of the peptide CP621–652 or its mutants (M626A and T627A) was determined using TZM-b1 cells. Briefly, the peptides were prepared with 10 series of dilutions in a 3-fold stepwise manner and mixed with 100 TCID50 viruses and incubated 1 h at room temperature. The mixture was added to TZM-bl cells (104/well) and incubation at 37 °C for 48 h, and luciferase activity was measured as described above.
Inhibition of 6-HB Formation by Peptides
A mouse monoclonal antibody (mAb) specific for the gp41 6-HB (NC-1) was obtained from Dr. Shibo Jiang through the ARRRP, Division of AIDS, NIAID, National Institutes of Health. The inhibitory activity of CP621–652 or its mutants (M626A and T627A) on 6-HB formation was measured by a modified ELISA-based method as previously described (20, 27). Briefly, a 96-well polystyrene plate (Costar, Corning Inc., Corning, NY) was coated with NC-1 (2 μg/ml in 0.1 m Tris, pH 8.8). A tested peptide at graded concentrations was mixed with C34-biotin (0.1 μm) and incubated with N36 (0.1 μm) at room temperature for 30 min. The mixture was then added to the NC-1-coated plate, followed by incubation at room temperature for 30 min and washing with a washing buffer (PBS containing 0.1% Tween 20) three times. Then streptavidin-labeled horseradish peroxidase (Invitrogen) and the substrate 3,3,5,5-tetramethylbenzidine (Sigma) were added sequentially. Absorbance at 450 nm (A450) was measured using an ELISA reader (Bio-Rad).
CD Spectroscopy
Circular dichroism (CD) spectroscopy was performed as previously described (20). Briefly, CP621–652 or its mutant peptides (M626A, T627A) were incubated with an equal molar concentration of the NHR-derived peptide N36 at 37 °C for 30 min. The final concentration of each peptide was 10 μm in PBS buffer, pH 7.2. The CD spectra were acquired on Jasco spectropolarimeter (model J-815) using a 1-nm bandwidth with a 1-nm step resolution from 195 to 260 nm at room temperature. The spectra were corrected by subtraction of a blank corresponding to the solvent. Data were averaged over three accumulations. The α-helical content was calculated from the CD signal by dividing the mean residue ellipticity (θ) at 222 nm by the value expected for 100% helix formation (−33,000 degrees·cm2 dmol−1). The thermal denaturation experiment was performed by monitoring the change in ellipticity (θ) at 222 nm at the increasing temperature (20–98 °C) using a temperature controller. The temperature was increased at a rate of 1.2 °C per min; data were acquired at a 1-nm bandwidth at 222 nm at a frequency of 0.25 Hz. The melting curve was smoothened, and the midpoint of the thermal unfolding transition (Tm) values were taken as the maximum of the derivative d[θ]222/dT. The Tm value was detected at a peptide concentration of 10 μm in PBS buffer.
RESULTS
Assembly, Crystallization, and Structure Determination of CP621–652·T21 Complex
To crystallize the CP621–652·T21 complex, we assembled the complex by mixing equal amounts of synthetic peptides pre-dissolved in denaturing buffer. The mixture was dialyzed to allow refolding. The 6-HB formed by CP621–652 and T21 was purified by size-exclusion chromatography. The crystal of CP621–652·T21 complex belonged to the space group of H32 and contained a pair of CP621–652·T21 peptides per asymmetric unit, and diffracted x-ray to a resolution limit of 1.74 Å. The crystal structure was solved by molecular replacement. The finally refined atomic model has excellent refinement statistics and stereochemistry qualities (Table 1). The structure was validated by MolProbity analysis. The MolProbity score is 1.67, with a in rating 82nd percentile among structures of comparable resolution. The Ramachandran plot finds all residues in the favored area.
General View of CP621–652·T21 Structure
CP621–652 and T21 peptides assemble into a typical 6-HB structure as anticipated. Three T21 helices form the central trimeric coiled-coil, whereas three CP621–652 helices wrap around the outside of T21 trimer in an antiparallel orientation (Fig. 2). In the major part of CP621–652, the peptide adopts an α-helical conformation. Interestingly, the helical conformation does not extend all the way to the N-terminal portion of the peptide. The 621QIWNNMT627 motif located upstream of the pocket-binding domain is directed away from the central NHR coiled-coil. There was insufficient electron density to model residues Gln621, Ile622, or Trp623 located at the N terminus of the peptide, indicating that this portion is relatively mobile (Fig. 2).
FIGURE 2.

The 6-helical bundle structure formed by CP621–652·T21. A ribbon model of 6-HB structure formed by CP621–652·T21. The peptides as well as their N and C termini are labeled. Left side, side view of 6-HB; right, top view of 6-HB. The T21 trimer is colored light gray; CP621–652 peptides are colored marine blue. The 621QIWNNMT627 motif at the N-terminal of CP621–652 is highlighted in orange with the labels. Key residues Met626 and Thr627, forming a hook-like structure, are shown in stick model with the labels. The motif does not adopt further α-helical conformations. The N-terminal of CP621–652 points away from the central NHR coiled-coil.
We previously demonstrated that a conserved salt bridge between Asp632 in the CHR and Lys574 in the NHR is critical for HIV-1 entry and inhibition (26, 27). In the crystal structure, we found two conformers for Asp632. The distance between the side chain of conformer B of Asp632 (59% occupancy) and the side chain of Lys574 is 3.95 Å, indicating a typical salt bridge. The carboxylate of conformer A of Asp632 (41% occupancy), which is oriented closer to Lys547, accepts a hydrogen bond from Nζ atom of Lys574 side chain (distance 2.01 Å, angle 137°) (Fig. 3). In addition, we found another hydrogen bond between the side chain of conformer A of Gln567 (41% occupancy) and the side chain of His643 (distance 2.06 Å, angle 149°). Collectively, these inter-helical hydrogen bonds may contribute to the binding affinity of CP621–652 to the T21 coiled-coil trimer.
FIGURE 3.

Hydrogen bond interactions between CP621–652 and T21 peptides confer the thermostability of 6-HB. The 6-HB structure formed by T21 and CP621–652 is placed horizontally with the labels. The N and C termini of the peptides are labeled. The T21 trimer is colored light gray, CP621–652 peptides are colored marine blue, and the 621QIWNNMT627 motifs of CP621–652 are highlighted in orange with the label. Hydrogen bonds are observed between Asp632 and Lys574 and between His643 and Gln567, and are indicated by dash lines.
Met626 and Thr627 Form a Hook-like Structure (M-T Hook) That Stabilizes 6-HB
Regardless of the flexibility of the N-terminal portion of CP621–652 containing the 621QIWNNMT627 motif, the conformation of residues Met626 and Thr627 are well defined. As show in Fig. 4, Thr627 plays a role of terminating the α-helical conformation by rotating the dihedral angle ψ of the residue by ∼180°, so that the N terminus of CP621–652 turns away from the helical portion of the peptide. The unusual conformation of Thr627 is stabilized by a hydrogen bond between the side chain hydroxyl group of Thr627 and the backbone NH group of Glu630 (distance 2.34 Å, angle 168°). This hydrogen bond also caps the downstream α-helix, stabilizing its helical conformation. Consequently, the upstream residue Met626 is positioned above the left side of the hydrophobic pocket on the NHR coiled-coil trimer, allowing its nonpolar side chain to accommodate a hydrophobic groove between Leu568 and Trp571 on T21 and Glu630 and Glu634 on CP621–652 (Fig. 4). Collectively, residues Met626 and Thr627 fold into a unique hook-like structure (we denoted as M-T hook) that may contribute to stabilize the 6-HB structure as the following. Thr627 redirects peptide chain and positions Met626 on top of the hydrophobic pocket; the side chain of Met626 functions as an ideal cover for the hydrophobic pocket below, stabilizing the interaction between the hydrophobic pocket and the pocket-binding domain.
FIGURE 4.

Methionine-threonine (M-T) hook of CP621–652 stabilize the hydrophobic pocket on NHR coiled coil. A, upper part, a ribbon model of the 6-HB structure formed by CP621–652·T21 (positioned horizontally). The T21 trimer is colored light gray; CP621–652 peptides are colored in marine blue. The N and C termini of the peptides are labeled. Lower part, a portion of the above 6-HB structure is magnified in the blue box and is illustrated with a wall-eye stereo image. Residues Thr627 and Met626 form a hook-like structure (highlighted in orange) that stabilizes the interaction between T21 and CP621–652. Thr627 terminates the α-helical conformation of the N-terminal of CP621–652 peptide. The hydroxyl group of the Thr627 side chain accepts a hydrogen bond from the NH group of Glu630, directing the N terminus of the CP621–652 peptide away from the T21 trimer. The side chain of Met626 covers the hydrophobic pocket on the T21 trimer. The M-T hook and residues involving interaction with the MT hook are shown in stick model. Dash lines indicate the hydrogen bonds between the residues. B, a portion of the 6-HB structure formed by CP621–652/T21 is displayed in a refined stick model with the superimposed refined 2Fo − Fc electron density map (1.5 s contour, blue mesh). The atoms are colored by type: carbon, yellow; nitrogen, blue; oxygen, red. The residues on the M-T hook and residues involving the interaction with the M-T hook are labeled.
Met626 and Thr627 Are Essential for Virus Fusion and Entry
To define the function of residues Met626 and Thr627 for HIV-1 infectivity, we substituted them with alanine, respectively, by using a plasmid encoding the HIV-1NL4-3 Env as template. The point mutations were verified by DNA sequencing, and expression and processing of the HIV-1 Env glycoprotein were confirmed by Western blotting (data not shown). We then determined the effects of substitutions on Env-mediated cell-cell fusion and viral single-cycle infection. As shown in Fig. 5A, both M626A and T627A substitutions could completely abolish the function of HIV-1NL4-3 Env to induce the cell-cell fusion. Consistently, whereas the HIV-1NL4-3 pseudovirus carrying a wild-type Env could efficiently enter the target TZM-bl cells, the M626A or T627A variants lost their cell entry completely (Fig. 5B).
FIGURE 5.

HIV-1NL4-3 Env-mediated cell fusion and entry. A, HIV-1NL4-3 Env-mediated cell-cell fusion activity. B, infectivity of HIV-1NL4-3 pseudoviruses on TZM-b1 cells. C, the effect of substitutions of M-T hook residues on HIV-1NL4-3 Env-mediated cell fusion and infection. Both cell fusion and single-cycle infection assays were performed at least three times and the data are presented as mean ± S.D. The control is cell-derived background in the absence of HIV-1 Env (fusion) or pseudoviruses (entry).
Both Met626 and Thr627 are highly conserved amino acids in the gp41 sequence, but several naturally occurring mutations can be identified in HIV-1 isolates, such as M626L, M626T, and T627S. We further replaced them with a panel of conservative and nonconservative residues. Interestingly, the Envs with M626L and M626T maintained a large extent of activity for cell fusion and entry (Fig. 5C), suggesting that these naturally occurring changes might be compatible for the interactions with the hydrophobic pocket. However, substitutions of Met626 with the positively or negatively charged residues (M626R and M626E) or noncharged polar residue (M626G) could effectively disrupt the Env. Of note, a conservative substitution in Thr627 (T627S) resulted in the Env with a marginal activity, albeit this mutation naturally occurred. Similarly, substitutions of Thr627 with the charged residues were also disruptive. These results suggest that Met626 and Thr627 are highly important for the function of HIV-1 Env to mediate cell membrane fusion and infection.
Met626 and Thr627 Are Critical for Anti-HIV Activity of CP621–652
We sought to characterize the roles of Met626 and Thr627 for the anti-HIV activity of the peptide CP621–652. Two peptides that carry M626A or T627A single mutations were therefore synthesized and their anti-HIV activity was evaluated in a direct comparison to the parental peptide. First, the cell-cell fusion inhibition assay showed that CP621–652 could inhibit the HIV-1HXB2 Env-mediated cell fusion with an IC50 of 8.6 nm, but peptides M626A and T627A inhibited the cell fusion at IC50 of 37.8 and 51.2 nm, respectively (Table 2). Furthermore, the virus entry inhibition assay indicated that CP621–652 could inhibit the HIV-1NL4-3 pseudovirus with an IC50 of 5.8 nm, whereas two mutant peptides inhibited the cell entry of pseudovirus at IC50 of 50.3 and 126.2 nm, respectively. Therefore, residues Met626 and Thr627 are critical for the peptide CP621–652 and their single substitutions could significantly impair the anti-HIV activity. The results support our observations from the above crystal structure.
TABLE 2.
Inhibitory activity of wild-type and mutant CP621–652 on HIV-1-mediated cell fusion and entry
Both cell fusion and single-cycle infection assays were performed in triplicate and repeated at least three times. The data are expressed as mean ± S.D.
| Peptide | Cell fusion |
Virus entry |
||
|---|---|---|---|---|
| IC50 | Fold-changea | IC50 | Fold-change | |
| nm | nm | |||
| CP621–652 | 8.6 ± 2.5 | 1 | 5.8 ± 0.6 | 1 |
| M626A | 37.8 ± 8.4 | 4.4 | 50.3 ± 2.6 | 8.7 |
| T627A | 51.2 ± 7.6 | 6 | 126.2 ± 1.9 | 21.8 |
| T20 | 11.5 ± 0.9 | NAb | 22.1 ± 0.8 | NA |
a Compared to the IC50 of wild-type CP621–652.
b NA, not applicable.
Met626 and Thr627 Are Critical for Inhibition of 6-HB Formation
Structures of the HIV-1 gp41 core have also revealed the mechanism of action of peptide fusion inhibitors (5, 10, 17). It is thought that the exogenous C-peptides act by competitive binding to the NHR of gp41 during its conformational change to the fusogenic state (i.e. pre-hairpin conformation) and thus block the 6-HB formation in a dominant-negative fashion. We previously developed an ELISA-based method to detect whether the peptide or small molecule-based fusion inhibitors can physically block 6-HB formation, in which the mAb NC-1 specific for 6-HB was used as a capture and the biotinylated-C34 was used for signal detection (20, 27). Here, we applied this method to further investigate the function of Met626 and Thr627 for 6-HB inhibition. As shown in Fig. 6, CP621–652 could efficiently block the formation of 6-HB as modeled by peptides N36 and C34 at a dose-dependent manner, consistent with our previous data (20). However, M626A and T627A peptides had no inhibitory activity at a concentration as high as 200 μm. The results suggested that these two mutant peptides could not compete off the biotinylated-C34 for binding to N36 and demonstrated that residues Met626 and Thr627 play important roles in inhibition of 6-HB by the peptide inhibitors.
FIGURE 6.
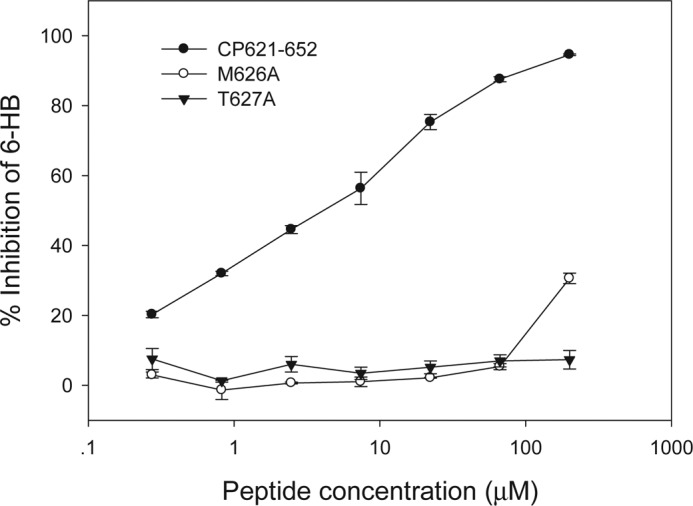
Inhibition of 6-HB formation by wild-type or mutant CP621–652 peptides. CP621–652 could inhibit 6-HB at a dose-dependent manner, but its mutants peptides (M626A and T627A) failed.
Met626 and Thr627 Are Critical for Interaction of NHR and CHR
We further studied the structure and function of the M-T hook residues by CD spectroscopy. The interaction of wild-type and mutant CP621–652 with the NHR-derived peptide N36 was analyzed. The CD spectrum of an equimolar mixture of N36 and CP621–652 peptides showed a typical double minima at 208 and 222 nm (Fig. 7A), which indicates the presence of stable α-helical conformations. In comparison, both M626A and T627A peptides induced significantly less α-helicity. The thermal stability of the N36·CP621–652 complex, defined as the midpoint of the thermal unfolding transition (Tm) values, was found to be 61.1 °C, whereas that of N36·M626A and N36·T627A complexes were 52.1 and 54.0 °C, respectively (Fig. 7B). Therefore, the binding affinity of two mutant peptides decreased significantly, which might explain their inability to block the 6-HB formed by N36 and C34 above. The CD results demonstrated that both M-T hook residues play important roles in the interaction of NHR and CHR.
FIGURE 7.

Interaction of wild-type and mutant CP621–652 with NHR-derived peptide N36. The α-helicity (A) and thermostability (B) of peptide complexes were measured by CD spectroscopy.
DISCUSSION
The primary structure of HIV-1 gp41 contains multiple functional domains (from N- to C terminus): a fusion peptide, the NHR and CHR linked by a loop with a disulfide bond at its basis, a transmembrane region, and a long cytoplasmic tail (Fig. 1). Previous studies have demonstrated that molecular interactions between NHR and CHR are essential for HIV-1-mediated cell fusion and infection (28–33). The crystallographic structures of the HIV-1 gp41 core have revealed the detailed molecular contacts within the trimer of the hairpin structure (6-HB) and a deep hydrophobic pocket in the NHR trimer (6–8), which provides the basis for exploring viral fusion mechanisms and for developing viral entry inhibitors. Unsatisfactorily, all three pioneering structures of the HIV-1 gp41 core were determined by using biosynthetic or synthesized peptide fragments (6–8), in which the loop region sequences between the putative NHR and CHR were completely omitted and thus the structure of the upstream sequence of the PBD or the boundary of a C-helix could not be observed. Recently, several new gp41 6-HB structures were solved for deciphering the mechanism of action of peptide inhibitors, but none of them included the PBD upstream motif (16, 34–35). Therefore, the structure of this region was often deduced to be helical based on the structural analogy of SIV the gp41 core (36–38), in which the C-terminal helix of SIV gp41 extends 5 residues to the loop region with respect to the most complete structure of HIV-1 gp41. However, our present structure of the 6-HB formed by CP621–652 and T21 has demonstrated that the conformation of the heptad 621QIWNNMT627 is flexible, resulting that the structures of most of its residues cannot be well defined by the crystallographic method, except residues Met626 and Thr627. This observation suggests that the upstream motif of PBD does not likely adopt the helical conformation during the fusion process.
More importantly, our studies reveal a unique hook-like structure (M-T hook) comprising Thr626 and Met627, which can stabilize the interaction between the NHR pocket and the CHR PBD. The M-T hook presents a structural feature that has never been observed in the available HIV gp41 core structures. This structural feature has provided a molecular basis for explaining why the 621QIWNNMT627 motif can improve thermal stability of 6-HB significantly (20). Due to the limitation of the crystallographic methods, our current structure does not offer explanations for the roles of other residues within this motif. In other words, we could not rule out the contributions of those residues in thermal stability of 6-HB. The solution structural study or the molecular dynamic study could be useful to elucidate their functions at the molecular level. Indeed, our mutagenesis studies confirmed that the M-T hook residues could determine the function of HIV-1 Env to mediate cell fusion and virus entry. Significantly, single substitutions of M-T hook residues were able to inactivate the virus (Fig. 5). Before and after determination of the gp41 core structure, the functionality of amino acid residues in both NHR and CHR was extensively examined by mutagenesis and biophysical analyses (28–33, 39–42), but little attention was paid to the upstream motif of the pocket-binding domain, such as residues Met626 and Thr627. Given that these two residues are located immediately adjacent to the PBD on the CHR, it is plausible that they could participate in interaction between the NHR pocket and CHR PBD. Our structural and functional data have finely validated their essential roles in HIV-1 infectivity.
In our previous study (20), we found that the CHR peptide lacking the 621QIWNNMT627 motif (CP628–654 in Fig. 1) had no anti-HIV activity at high concentrations, but addition of these heptad residues to the peptide (CP621–652) conferred a potent inhibitory activity on both HIV-1 cell fusion and infection (Fig. 1). During that time, we could not understand the molecular determinants underlying the anti-HIV activity of the CP621–652 peptide, but speculated that interaction between the 621QIWNNMT627 motif and the pocket or its downstream site on the NHR trimer should be the most important determinant. The M-T hook characterized here has confirmed the case. Disruption of the M-T hook by single amino acid substitutions dramatically impaired the peptide for its inhibitory activity in both cell fusion and virus entry, supporting the crystallographic data and the rational inhibitor design. Although most of the reported anti-HIV peptides were derived primarily from the CHR sequence starting from Trp628, there are several potent peptide inhibitors designs, including Met626 and Thr627 at their N termini (43). It would be interesting to obtain structural information of these inhibitors. In our designing of the CP32M inhibitor using the CP621–652 peptide as the template (14), residues Met626 and Thr627 were unchanged, because their substitutions could dramatically reduce the anti-HIV activity of the peptide (data not shown).
Surprisingly, the CP621–652 peptide with single substitutions in the M-T hook residues could not block formation of 6-HB formed by N36 and C34 in our experimental conditions. This result was conflicted with its anti-HIV activity in both cell fusion and single-cycle infection assays. Although the inhibitory activity of mutant peptides reduced significantly, they were still active on the virus. Therefore, we performed a CD spectroscopic study to analyze interaction of CP621–652 and its mutants with the NHR peptide N36. Single substitutions could result in that CP521–652 interacts with N36 with much less α-helicity and thermal stability. Therefore, the M-T hook residues did participate in NHR binding. In this case, Met626 and Thr627 might hook the pocket region tightly and thus stabilize the interaction between the pocket residues on the NHR and the PBD residues projected from the CHR.
In conclusion, we determined the high-resolution crystal structure of the CP621–652·T21 complex, revealing that residues Met626 and Thr627 in the 621QIWNNMT627 motif of CP621–652 form a hook-like structure that stabilizes the interaction of the hydrophobic pocket on the NHR timer. We demonstrated that both M-T hook residues are essential for HIV-1-mediated cell fusion and entry, and play critical roles for the anti-HIV activity of CP621–652. Our findings identify a novel structural feature in the HIV gp41 core, providing a glimpse of the peptide structure upstream of the pocket-binding domain, offering multiple implications for understanding the mechanism for HIV-1 cell fusion and designing novel fusion inhibitors.
This work was supported by Natural Science Foundation of China Grant 30870123 and National Outstanding Youth Award of NSFC Grant 81025009, National 973 program of China Grant 2010CB530100, and National Science and Technology Major Project Grant 2009ZX10004-303.
The atomic coordinates and structure factors (code 3VGX) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- 6-HB
- six-helix bundle
- NHR
- N-terminal heptad repeat
- CHR
- C-terminal heptad repeat
- PBD
- pocket-binding domain.
REFERENCES
- 1. Colman P. M., Lawrence M. C. (2003) The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 4, 309–319 [DOI] [PubMed] [Google Scholar]
- 2. Zhu P., Liu J., Bess J., Jr., Chertova E., Lifson J. D., Grisé H., Ofek G. A., Taylor K. A., Roux K. H. (2006) Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 441, 847–852 [DOI] [PubMed] [Google Scholar]
- 3. Jiang S., Lin K., Strick N., Neurath A. R. (1993) HIV-1 inhibition by a peptide. Nature 365, 113. [DOI] [PubMed] [Google Scholar]
- 4. Wild C. T., Shugars D. C., Greenwell T. K., McDanal C. B., Matthews T. J. (1994) Peptides corresponding to a predictive α-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. U.S.A. 91, 9770–9774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chan D. C., Kim P. S. (1998) HIV entry and its inhibition. Cell 93, 681–684 [DOI] [PubMed] [Google Scholar]
- 6. Chan D. C., Fass D., Berger J. M., Kim P. S. (1997) Core structure of gp41 from the HIV envelope glycoprotein. Cell 89, 263–273 [DOI] [PubMed] [Google Scholar]
- 7. Tan K., Liu J., Wang J., Shen S., Lu M. (1997) Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. U.S.A. 94, 12303–12308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weissenhorn W., Dessen A., Harrison S. C., Skehel J. J., Wiley D. C. (1997) Atomic structure of the ectodomain from HIV-1 gp41. Nature 387, 426–430 [DOI] [PubMed] [Google Scholar]
- 9. Chan D. C., Chutkowski C. T., Kim P. S. (1998) Evidence that a prominent cavity in the coiled-coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. U.S.A. 95, 15613–15617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Eggink D., Berkhout B., Sanders R. W. (2010) Inhibition of HIV-1 by fusion inhibitors. Curr. Pharm. Des. 16, 3716–3728 [DOI] [PubMed] [Google Scholar]
- 11. Ashkenazi A., Wexler-Cohen Y., Shai Y. (2011) Multifaceted action of Fuzeon as virus-cell membrane fusion inhibitor. Biochim. Biophys. Acta 1808, 2352–2358 [DOI] [PubMed] [Google Scholar]
- 12. Kilby J. M., Hopkins S., Venetta T. M., DiMassimo B., Cloud G. A., Lee J. Y., Alldredge L., Hunter E., Lambert D., Bolognesi D., Matthews T., Johnson M. R., Nowak M. A., Shaw G. M., Saag M. S. (1998) Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 4, 1302–1307 [DOI] [PubMed] [Google Scholar]
- 13. Lalezari J. P., Henry K., O'Hearn M., Montaner J. S., Piliero P. J., Trottier B., Walmsley S., Cohen C., Kuritzkes D. R., Eron J. J., Jr., Chung J., DeMasi R., Donatacci L., Drobnes C., Delehanty J., Salgo M. (2003) Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 348, 2175–2185 [DOI] [PubMed] [Google Scholar]
- 14. He Y., Cheng J., Lu H., Li J., Hu J., Qi Z., Liu Z., Jiang S., Dai Q. (2008) Potent HIV fusion inhibitors against Enfuvirtide-resistant HIV-1 strains. Proc. Natl. Acad. Sci. U.S.A. 105, 16332–16337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He Y., Xiao Y., Song H., Liang Q., Ju D., Chen X., Lu H., Jing W., Jiang S., Zhang L. (2008) Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 283, 11126–11134 [DOI] [PubMed] [Google Scholar]
- 16. Nishikawa H., Nakamura S., Kodama E., Ito S., Kajiwara K., Izumi K., Sakagami Y., Oishi S., Ohkubo T., Kobayashi Y., Otaka A., Fujii N., Matsuoka M. (2009) Electrostatically constrained α-helical peptide inhibits replication of HIV-1 resistant to enfuvirtide. Int. J. Biochem. Cell Biol. 41, 891–899 [DOI] [PubMed] [Google Scholar]
- 17. Steffen I., Pöhlmann S. (2010) Peptide-based inhibitors of the HIV envelope protein and other class I viral fusion proteins. Curr. Pharm. Des. 16, 1143–1158 [DOI] [PubMed] [Google Scholar]
- 18. Berkhout B., Sanders R. W. (2011) Molecular strategies to design an escape-proof antiviral therapy. Antiviral Res. 92, 7–14 [DOI] [PubMed] [Google Scholar]
- 19. Naider F., Anglister J. (2009) Peptides in the treatment of AIDS. Curr. Opin. Struct. Biol. 19, 473–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. He Y., Cheng J., Li J., Qi Z., Lu H., Dong M., Jiang S., Dai Q. (2008) Identification of a critical motif for the human immunodeficiency virus type 1 (HIV-1) gp41 core structure. Implications for designing novel anti-HIV fusion inhibitors. J. Virol. 82, 6349–6358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chong H., Xu S., Zhang C., Nie J., Wang Y. (2009) Mutation L33M in the HR1 region of HIV-1 gp41 may play a role in T20 resistance. J. Clin. Virol. 45, 255–258 [DOI] [PubMed] [Google Scholar]
- 23. Bertram S., Glowacka I., Müller M. A., Lavender H., Gnirss K., Nehlmeier I., Niemeyer D., He Y., Simmons G., Drosten C., Soilleux E. J., Jahn O., Steffen I., Pöhlmann S. (2011) Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J. Virol. 85, 13363–13372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Glowacka I., Bertram S., Müller M. A., Allen P., Soilleux E., Pfefferle S., Steffen I., Tsegaye T. S., He Y., Gnirss K., Niemeyer D., Schneider H., Drosten C., Pöhlmann S. (2011) Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 85, 4122–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wexler-Cohen Y., Johnson B. T., Puri A., Blumenthal R., Shai Y. (2006) Structurally altered peptides reveal an important role for N-terminal heptad repeat binding and stability in the inhibitory action of HIV-1 peptide DP178. J. Biol. Chem. 281, 9005–9010 [DOI] [PubMed] [Google Scholar]
- 26. He Y., Liu S., Jing W., Lu H., Cai D., Chin D. J., Debnath A. K., Kirchhoff F., Jiang S. (2007) Conserved residue Lys574 in the cavity of HIV-1 Gp41 coiled-coil domain is critical for six-helix bundle stability and virus entry. J. Biol. Chem. 282, 25631–25639 [DOI] [PubMed] [Google Scholar]
- 27. He Y., Liu S., Li J., Lu H., Qi Z., Liu Z., Debnath A. K., Jiang S. (2008) Conserved salt bridge between the N- and C-terminal heptad repeat regions of the human immunodeficiency virus type 1 gp41 core structure is critical for virus entry and inhibition. J. Virol. 82, 11129–11139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu M., Stoller M. O., Wang S., Liu J., Fagan M. B., Nunberg J. H. (2001) Structural and functional analysis of interhelical interactions in the human immunodeficiency virus type 1 gp41 envelope glycoprotein by alanine-scanning mutagenesis. J. Virol. 75, 11146–11156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mo H., Konstantinidis A. K., Stewart K. D., Dekhtyar T., Ng T., Swift K., Matayoshi E. D., Kati W., Kohlbrenner W., Molla A. (2004) Conserved residues in the coiled-coil pocket of human immunodeficiency virus type 1 gp41 are essential for viral replication and interhelical interaction. Virology 329, 319–327 [DOI] [PubMed] [Google Scholar]
- 30. Shu W., Liu J., Ji H., Radigen L., Jiang S., Lu M. (2000) Helical interactions in the HIV-1 gp41 core reveal structural basis for the inhibitory activity of gp41 peptides. Biochemistry 39, 1634–1642 [DOI] [PubMed] [Google Scholar]
- 31. Wang S., York J., Shu W., Stoller M. O., Nunberg J. H., Lu M. (2002) Interhelical interactions in the gp41 core. Implications for activation of HIV-1 membrane fusion. Biochemistry 41, 7283–7292 [DOI] [PubMed] [Google Scholar]
- 32. Weng Y., Weiss C. D. (1998) Mutational analysis of residues in the coiled-coil domain of human immunodeficiency virus type 1 transmembrane protein gp41. J. Virol. 72, 9676–9682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weng Y., Yang Z., Weiss C. D. (2000) Structure-function studies of the self-assembly domain of the human immunodeficiency virus type 1 transmembrane protein gp41. J. Virol. 74, 5368–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buzon V., Natrajan G., Schibli D., Campelo F., Kozlov M. M., Weissenhorn W. (2010) Crystal structure of HIV-1 gp41 including both fusion peptide and membrane proximal external regions. PLoS Pathog. 6, e1000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yao X., Chong H., Zhang C., Waltersperger S., Wang M., Cui S., He Y. (2012) Broad antiviral activity and crystal structure of HIV-1 fusion inhibitor Sifuvirtide. J. Biol. Chem. 287, 6788–6796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Caffrey M., Cai M., Kaufman J., Stahl S. J., Wingfield P. T., Gronenborn A. M., Clore G. M. (1997) Determination of the secondary structure and global topology of the 44-kDa ectodomain of gp41 of the simian immunodeficiency virus by multidimensional nuclear magnetic resonance spectroscopy. J. Mol. Biol. 271, 819–826 [DOI] [PubMed] [Google Scholar]
- 37. Caffrey M., Cai M., Kaufman J., Stahl S. J., Wingfield P. T., Covell D. G., Gronenborn A. M., Clore G. M. (1998) Three-dimensional solution structure of the 44-kDa ectodomain of SIV gp41. EMBO J. 17, 4572–4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang Z. N., Mueser T. C., Kaufman J., Stahl S. J., Wingfield P. T., Hyde C. C. (1999) The crystal structure of the SIV gp41 ectodomain at 1.47-Å resolution. J. Struct. Biol. 126, 131–144 [DOI] [PubMed] [Google Scholar]
- 39. Cao J., Bergeron L., Helseth E., Thali M., Repke H., Sodroski J. (1993) Effects of amino acid changes in the extracellular domain of the human immunodeficiency virus type 1 gp41 envelope glycoprotein. J. Virol. 67, 2747–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen S. S., Lee S. F., Hao H. J., Chuang C. K. (1998) Mutations in the leucine zipper-like heptad repeat sequence of human immunodeficiency virus type 1 gp41 dominantly interfere with wild-type virus infectivity. J. Virol. 72, 4765–4774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen S. S., Lee C. N., Lee W. R., McIntosh K., Lee T. H. (1993) Mutational analysis of the leucine zipper-like motif of the human immunodeficiency virus type 1 envelope transmembrane glycoprotein. J. Virol. 67, 3615–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ji H., Bracken C., Lu M. (2000) Buried polar interactions and conformational stability in the simian immunodeficiency virus (SIV) gp41 core. Biochemistry 39, 676–685 [DOI] [PubMed] [Google Scholar]
- 43. Dwyer J. J., Wilson K. L., Davison D. K., Freel S. A., Seedorff J. E., Wring S. A., Tvermoes N. A., Matthews T. J., Greenberg M. L., Delmedico M. K. (2007) Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. U.S.A. 104, 12772–12777 [DOI] [PMC free article] [PubMed] [Google Scholar]