

Background: Activated mesangial cells exhibit SMA and contribute to the progression of diabetic nephropathy.
Results: Scleraxis negatively regulated the AGE-induced expression and secretion of BMP4.
Conclusion: Scleraxis and Id1 are involved in the BMP4-SMA pathway and modulate phenotypic changes.
Significance: Deeper insight into the impact of regulatory mechanism of scleraxis-BMP4-Smad1 signal activation might help to prevent diabetic glomerular damage.
Keywords: Bone Morphogenetic Protein (BMP), Differentiation, Signal Transduction, SMAD Transcription Factor, Transforming Growth Factor β (TGFbeta), Diabetic Nephropathy, Phenotype Change, Smooth Muscle α-Actin
Abstract
Activation of mesangial cells (MCs), which is characterized by induction of smooth muscle α-actin (SMA) expression, contributes to a key event in various renal diseases; however, the mechanisms controlling MC differentiation are still largely undefined. Activated Smad1 induced SMA in a dose-dependent manner in MCs. As a direct regulating molecule for SMA, we identified and characterized scleraxis (Scx) as a new phenotype modulator in advanced glycation end product (AGE)-exposed MCs. Scx physically associated with E12 and bound the E-box in the promoter of SMA and negatively regulated the AGE-induced SMA expression. Scx induced expression and secretion of bone morphogenetic protein 4 (BMP4), thereby controlling the Smad1 activation in AGE-treated MCs. In diabetic mice, Scx was concomitantly expressed with SMA in the glomeruli. Inhibitor of differentiation 1 (Id1) was further induced by extended treatment with AGE, thereby dislodging Scx from the SMA promoter. These data suggest that Scx and Id1 are involved in the BMP4-Smad1-SMA signal transduction pathway besides the TGFβ1-Smad1-SMA signaling pathway and modulate phenotypic changes in MCs in diabetic nephropathy.
Introduction
Diabetic nephropathy is a morbid complication associated with diabetes mellitus and is the most common cause of end-stage renal disease. Glomerulosclerosis in diabetic nephropathy is caused by accumulation of extracellular matrix (ECM)2 proteins in the mesangial interstitial space, resulting in the narrowing and obliteration of glomerular capillaries (1) and leading to irreversible progression to end-stage renal disease.
Emerging evidence suggests that the cause of glomerulosclerosis in diabetic nephropathy is phenotypic switching of MCs to an activated state. In response to injury, MCs can transdifferentiate into myofibroblasts, a specialized population of mesenchymal cells that secrete interstitial collagens (i.e. type I and type III collagens) that are not normally present in the mesangial matrix and markedly up-regulate the expression of smooth muscle-like proteins (i.e. SMA) (2–4). The fundamental significance of myofibroblasts and SMA expression is unclear, although these actions may lead to prevention of cell migration and concentrate these cells at the site of injury (5). Furthermore, this phenotypic alteration, which is characterized by SMA gene induction, is a common phenomenon in the process of sclerotic or fibrotic changes in many organs, such as the liver (6), lungs (7), and skin (8). However, the underlying molecular mechanisms for this regulation remain unknown.
Recently, we demonstrated that Smad1 plays a key role in the progression of various renal diseases such as diabetic nephropathy and proliferative glomerulonephritis (4, 9). Smad1 transcriptionally up-regulates ECM proteins (type IV and type I collagens and osteopontin) in the common process of progressing glomerulosclerosis (4). Moreover, induction of Smad1 and SMA expression coincides with the development of glomerulosclerosis in diabetic rats (10). Furthermore, we reported that urinary Smad1 is a novel marker to detect the initial stage of glomerulosclerosis in both type 1 and type 2 diabetic rodents (10, 11). Thus, Smad1 was thought to be closely involved in the phenotypic change of MCs in diabetes. However, there is little information about which molecule stands between Smad1 and SMA, modulating the phenotype of MCs.
The basic helix-loop-helix (bHLH) transcription factors have been implicated in cell fate specification in a variety of cell lineages. Examples of these proteins include the myogenic bHLH factors MyoD and myogenin in muscle development (12–15), neuronal factors such as neurogenin and Mash1 (16), and the TAL gene involved in hematopoiesis (17). Cell type-specific bHLH proteins typically form heterodimers with ubiquitous bHLH proteins, such as E12/E47 (products of the E2A gene), through the HLH domains (18) and enable the basic region to form a bipartite DNA-binding motif that recognizes the so-called E-box sequence CANNTG (19). In addition, four Id proteins, Id1 through Id4, lack the DNA binding activity due to the absence of a basic domain and have been characterized as negative regulators of bHLH transcription factors. The majority of information concerning the role of bHLH proteins in the process of differentiation has been investigated during embryogenesis. Few studies have examined the role of these proteins in adults or in diseases. Although E-box sites are present in the promoter of the SMA gene, no MC-specific bHLH transcription factors have yet been identified. In this study, we identified scleraxis (Scx) as a binding factor to the promoter of SMA and established the mechanism and function of Scx-mediated SMA expression under diabetic conditions.
As described above, in glomerulosclerosis activated MCs produce bone ECM proteins (type I collagen (Col1) and osteopontin) as well as SMA. Bone morphogenetic protein 4 (BMP4) has been implicated in several aspects of embryonic development, from establishment of the basic embryonic body plan to morphogenesis of some organs, including the kidney, by regulating cell proliferation, differentiation, apoptosis, and chemotaxis (20, 21). BMP4 is also involved in the commitment of mesenchymal cells to chondrogenic and osteogenic lineages (22, 23). However, the functional roles of BMP4 in adults or in diseases remain elusive. In this study, we reveal an important role of BMP4 for activation of Smad1-SMA signal transduction in the process of phenotypic alteration in MCs. Moreover, we elucidated the molecular mechanism for negative regulation of SMA by Scx in MCs. However, Scx positively regulated BMP4-induced SMA expression.
EXPERIMENTAL PROCEDURES
Animals
The animals were housed under specific pathogen-free conditions at the animal facility of Tokushima University. All animal experiments were performed in accordance with institutional guidelines, and the Review Board of Tokushima University granted ethical permission for this study. Male C57BL/6 mice, 8 weeks old, weighing 22–24 g were rendered diabetic by the intraperitoneal injection of 50 mg per kg body weight streptozotocin (STZ) in citrate buffer, pH 4.5, for 5 consecutive days. The diabetic state was confirmed 5 days after final injection by measurement of the blood glucose level. All mice that were given STZ had a blood glucose concentration exceeding 400 mg/dl and were considered diabetic (24).
AGE Preparation
AGE-BSA was prepared by the method described previously (4, 25). BSA was incubated with glucose 6-phosphate for >60 days at 37 °C. Control BSA was prepared by incubating BSA without glucose 6-phosphate under the same conditions as those for AGE-BSA. Preparations were tested for endotoxin using an Endospecy ES-24S system (Seikagaku Co., Tokyo, Japan), and no endotoxin was detected. Protein concentrations were determined by the Bradford method using BSA as the standard. Nϵ-(Carboxymethyl) lysine (CML) was purchased from Nippi Protein Engineering (Tokyo, Japan).
Cell Culture
A glomerular MC line was established from glomeruli isolated from normal 4-week-old mice (C57BL/6J×SJL/J) and was identified according to the method described previously (26, 27). The MCs were plated on 100-mm plastic dishes (Nunc Roskilde, Denmark) and maintained in B medium (a 3:1 mixture of minimal essential medium/F-12 modified with trace elements) supplemented with 1 mm glutamine, penicillin at 100 units/ml, streptomycin at 100 mg/ml, and 10% fetal calf serum (FCS) (Irvine Scientific, Santa Ana, CA). The cultured cells fulfilled the generally accepted criteria for glomerular MCs described previously (26). AGE or BSA exposure was carried out as described previously (4). CML was added to the MC culture medium at a final concentration of 100 μg/ml after overnight starvation.
Plasmids
The full-length cDNAs of Smad1 and E12 were obtained using gene-specific primers for reverse transcription-PCR (RT-PCR) and inserted into pcDNA3 or pCMV-Myc (resulting in pcDNA3-Smad1 and Myc-E12). The full-length of Scx cDNA was inserted into p3×FLAG-CMV (Sigma) to construct the NH2-terminal FLAG epitope-tagged Scx (FLAG-Scx). A monoclonal antibody (M2; Sigma) directed toward this epitope was used in the Western blot experiments. For expression studies, the Scx gene was cloned into the pcDNA3 expression plasmid. Mouse constitutively active ALK1 (caALK1) was generated by mutation of Glu-194 into aspartic acid. Site-directed mutagenesis was carried out by a PCR-based approach and was inserted into pCMV-Myc. Constitutively active Smad1 (caSmad1) was obtained by substitution of Ser-463 and Ser-465 of mouse Smad1 vector (kindly provided from Prof. Miyazono, University of Tokyo) with aspartic acid. The constructed plasmids were verified by sequencing. Constitutively active Smad3 (caSmad3) expression vector was kindly provided by Dr. J. Oh (Korea University).
Degenerate PCR and cDNA Cloning
Degenerate PCR primers were designed according to two highly conserved amino acid sequence motifs in the basic region and second helix region of the bHLH proteins. Scx was obtained by the RT-PCR method using the following two degenerate primers: 5′-CCAA(C/T)GC(A/C/G/T)CGIGA(A/G)CG(A/C/G/T)(A/G)-3′ and 5′-CCAG(A/G)TGIG(A/C)(A/G/T)AT(A/G)TA(A/G)CT(A/C/G/T)GA(A/C/G/T)GC-3′, corresponding to the target amino acid sequences of NARER(D/N) and ASSYIAHL, respectively (29, 30). One microgram of total RNA of mouse MCs treated with AGE or BSA was reverse-transcribed with oligo(dT) primer using Moloney murine leukemia virus reverse transcriptase (Superscript II; Invitrogen) in a total volume of 30 μl. RT-negative controls were performed to exclude the possibility of genomic or other DNA contamination. Full-length cDNA clones for Scx were isolated by 5′- and 3′-rapid amplification of cDNA end (RACE) using poly(A)+ RNA from mouse MCs. RACE was performed using a SMART RACE cDNA amplification kit according to the manufacturer's instructions (Clontech).
RT-PCR
Total RNA was extracted from mouse MCs using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. The RNA was reverse-transcribed with oligo(dT) primer using a SuperScript first-strand synthesis kit (Invitrogen) to generate the first-strand cDNA, followed by PCR to detect the expression of Scx and GAPDH. The sequences of the PCR primers were as follows: 5′-GACCGCACCAACAGCGTGAA-3′ and 5′-GTGGACCCTCCTCCTTCTAACTTC-3′ for Scx; 5′-CTCTGGACGTACAACTGGTATTG-3′ and 5′-TGGATGCCCGCTGACTCCAT-3′ for SMA; and 5′-TATGACTCCACTCACGGCAAAT-3′ and 5′-TGCTTCACCACCTTCTTGATGT-3′ for GAPDH. The reaction products were separated on 1.5% agarose gel and stained with ethidium bromide.
Antibodies
We generated polyclonal antibodies (Scx1 and Scx2) raised against a synthetic peptide corresponding to amino acids 169–185 of mouse Scx and confirmed their specificity by dot blot analysis with the antigenic peptide. For Western blot analyses, results using Scx1 were shown in each figure. The anti-E12, anti-E2A, and anti-ALK1 (Santa Cruz Biotechnology, Santa Cruz, CA); anti-Smad1 (Epitomics, Burlingame, CA); anti-pSmad1/5/8, anti-Smad2, anti-Smad3, and anti-pSmad3 (Cell Signaling, Beverly, MA); anti-TGFβ3 (R&D Systems, Minneapolis, MN); anti-TGFβ1, anti-TGFβ2, and anti-SMA (Abcam, Cambridge, UK); anti-FLAG, anti-β-actin, and anti-α-tubulin (Sigma); anti-Myc (Clontech); and anti-Col1 (Rockland Immunochemicals, Gilbertsville, PA) antibodies were used for Western blot analysis and immunoprecipitation assay. The anti-pSmad1 (Cell Signaling), anti-SMA, and anti-Scx1 antibodies were used for immunofluorescent staining. Polyclonal anti-TGFβ1 neutralizing antibody (AB10-NA) and monoclonal anti-BMP4 neutralizing antibody (MAB757) (R&D Systems) were used for inhibition assays.
Western Blotting
Proteins from MCs or mouse total kidneys were resolved by SDS-PAGE, transferred to a nitrocellulose membrane (GE Healthcare), subjected to Western blot analysis using primary antibodies, and detected using an enhanced chemiluminescence detection system (Invitrogen).
EMSA
EMSA was performed using oligonucleotides corresponding to specific E-boxes (E1 5′-GGCAGCTCAGCTGCTTATGGG-3′ and E2 5′-GCCCTCAGAACAACTGCTCAAATG-3′). Briefly, the nuclear pellet was collected by centrifugation (600 × g, 10 min, 4 ºC). The nuclear lysates (100 μg) were prepared and incubated in 25 μl of reaction mixture containing poly(dI/dC), 10× binding buffer (50 μm ZnCl2, 0.25 mm DTT, 20 mm Tris, 60 mm KCl, 1 mm MgCl2, 0.1 mm EDTA, and 10% glycerol), and the labeled oligonucleotide for 30 min at room temperature. The reactions were terminated using 25 μl of the loading dye and loaded onto a 7% gel retardation assay gel. The run was performed at 250 V for 3 h at 4 ºC. At the end of the run, the gel was removed, dried, and exposed in a phosphorimager at room temperature.
Transfection and Co-immunoprecipitation
CHOK1 cells were transfected using FuGENE 6 (Roche Applied Science) according to the manufacturer's protocol. After 48 h of transfection, the cells were washed with PBS, and 1 ml of ice-cold lysis buffer (25 mm Tris-HCl, pH 7.4, 100 mm NaCl, 2 mm EDTA, 0.5% Nonidet P-40, Complete protease inhibitors mixture; Roche Applied Science) was added. The cell lysates were precleared with normal rabbit IgG. Equal amounts of protein were incubated with 10 μl of rabbit anti-FLAG or anti-Myc antibody for 3 h at 4 ºC with constant rotation. The immune complex was isolated with protein G-Sepharose (2 h at 4 ºC), and the immunoprecipitates were washed three times with sample buffer, resuspended in 2× SDS sample buffer containing 2-mercaptoethanol, and boiled for 5 min. After centrifugation, a portion of the samples were separated by SDS-PAGE before Western blot analyses.
Co-immunoprecipitation of Endogenous Scx and E12
Kidneys isolated from STZ-induced diabetic mice and nondiabetic control mice were minced and suspended in lysis buffer containing 150 mm NaCl, 10 mm Tris-HCl, pH 7.5, and 0.1% deoxycholate. The tissue suspension was homogenized, and the lysates were immunoprecipitated with anti-E12 antibody and control immunoglobulin G. The presence of Scx in the immunoprecipitates was determined by Western blotting with anti-Scx antibody.
Reporter Assay
MCs (0.15 × 105) were plated in 24-well plates and 6 h later co-transfected with increasing amounts of Myc-E12 expression plasmid. Trasfections were perfomed using FuGENE 6. Luciferase activities were normalized to Renilla luciferase activities derived from co-transfected pRL-SV40-Luc (Promega).
siRNAs and Transfection
MCs (1.0 × 105) were seeded into 6-well plates (Nunc) and grown until they were 40–60% confluent. The Smad1, Scx, E12, Id1, and ALK1 siRNA (Invitrogen) and the control siRNA (Dharmacon, Lafayette, CO) were combined with INTERFERin reagent (Polyplus Transfection, New York), and the cells were transfected with siRNA (100 nm) according to the recommended protocol.
Histology, Immunohistochemistry, and Immunocytochemistry
Tissues were fixed in methyl Carnoy's solution and were paraffin-embedded. Multiple sections were prepared and stained with periodic acid silver methenamine and periodic acid-Schiff's reagent. Cryopreserved kidney tissues were cut into 5-μm-thick sections and fixed in methanol at 4 ºC for 15 min. To eliminate nonspecific staining, sections were incubated with the appropriate preimmune serum for 30 min at room temperature, followed by incubating with the primary antibodies. For detection, appropriate biotinylated or fluorescein isothiocyanate-conjugated secondary antibodies were used for visualization.
ChIP Assay
ChIP assays were performed essentially as described previously (4). We used anti-E2A antibody (Santa Cruz Biotechnology), anti-Scx antibody, or normal control IgG at 4 ºC overnight. PCR was performed with primers to amplify the region containing the E-box on the SMA promoter. The 5′-primer was 5′-GGAGCTCCCCAATTTGTTG-3′, and the 3′ primer was 5′-CAGCCTCCGCCTCTTACC-3′. Input DNA (2.5%) was used as a template in the PCR.
RESULTS
TGFβ1-induced SMA Expression Besides Activation of Smad3 in Diabetic Conditions
Although we previously reported that AGE induces and phosphorylates Smad1 (4) and that Smad1 and SMA proteins co-exist in the glomeruli of diabetic rats (10), interaction between Smad1 and SMA in MCs in diabetic conditions remained unknown. Induction of SMA as well as Smad1 and pSmad1 was observed in AGE-treated MCs (Fig. 1A). TGFβ has been known to play an important role in the AGE response of the glomeruli (30) and has previously been reported to induce SMA expression in various cells, including MCs (3, 31). Because there are three mammalian isoforms, TGFβ1–3, we examined which isoform is expressed and involved in diabetic kidneys. TGFβ1 expression was markedly up-regulated in diabetic mice. In contrast, alteration of TGFβ2 and TGFβ3 expressions was not observed (Fig. 1B). Similar changes were found in MCs treated with AGE (Fig. 1C). To elucidate the effect of TGFβ1 on the induction of SMA by AGE in MCs, an inhibition assay using the TGFβ1 neutralizing antibody was performed (Fig. 1D). MCs treated with the TGFβ1-neutralizing antibody showed significant reduction of pSmad3 up-regulated by AGE. However, small reduction of Smad1, pSmad1, and SMA expression induced by AGE was observed (Fig. 1D). To confirm the role of Smad3 in AGE-induced SMA expression, siRNA knockdown for Smad3 was carried out. Smad3 knockdown did not appear to affect the expression of SMA in AGE-treated MCs (Fig. 2E). To further examine and confirm the effect of Smad3 on the SMA expression, we performed transient overexpression of constitutively active Smad3 (caSmad3) in MCs, resulting in no increase in the protein levels of SMA (Fig. 2F).
FIGURE 1.
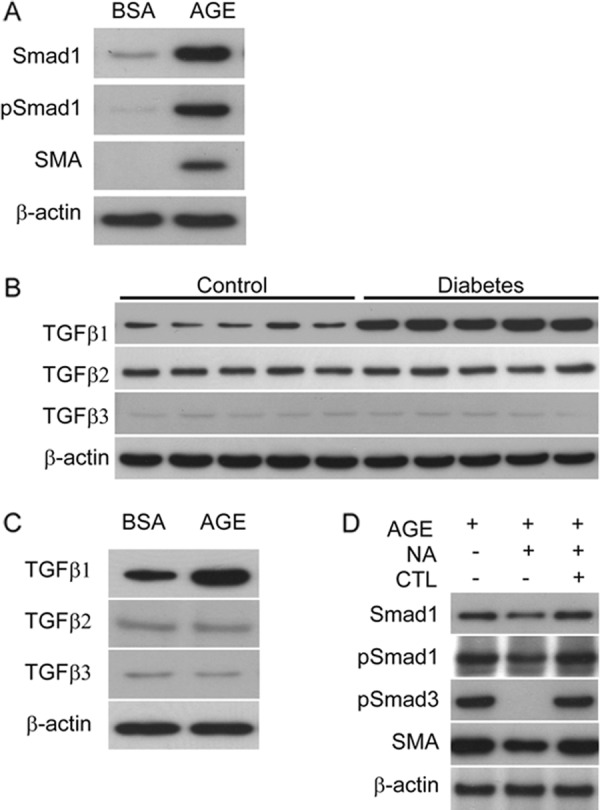
Effects of TGFβ signaling on the induction of Smad1 and SMA by AGE in MCs. A, Western blot of protein extracts prepared from MCs treated with AGE or BSA (10 μg/cm2) for 24 h. One of three independent experiments is shown. B, Western blot analysis for TGFβ isoforms in whole kidney extracts in STZ-induced diabetic and control mice at 32 weeks after treatment (n = 5 in each group). C, TGFβ isoforms proteins were monitored by Western blot analysis in response to a 24-h treatment with BSA or AGE (10 μg/cm2). One of three independent experiments is shown. D, MCs were treated with neutralizing antibody for TGFβ1 (10 μg/ml) or control normal IgY for 24 h after a 6-h exposure to AGE (10 μg/cm2). Equal amounts of cell lysates were subjected to Western blot. One of three independent experiments is shown. β-Actin was used as a loading control.
FIGURE 2.

Activated Smad1 induces SMA expression in MCs. A and B, after 24 h of transfection with increasing amounts of the Smad1 expression plasmid (pcDNA3-Smad1) in the absence (A) or presence (B) of TGFβ1 stimulation for 1 h, Western blot analyses were performed. C, Western blot analyses of MCs 24 h after transfection with increasing amounts of the caSmad1 expression plasmid. D, effect of co-transfection of caALK1 and pcDNA3-Smad1 for 24 h on Smad1-SMA signaling pathway in MCs. Whole cell lysates were subjected to Western blot. E, MCs were treated with Smad3 siRNA or scrambled control siRNA (CTL) for 48 h under the exposure to AGE or BSA (10 μg/cm2). Equal amounts of cell lysates were subjected to Western blot. F, Western blot analysis of MCs 24 h after transfection with increasing amounts of the caSmad3 expression plasmid. α-Tubulin or β-actin was used as a loading control. One of at least three independent experiments is shown in each panel.
Smad1 Is Involved in TGFβ1-induced SMA Expression in MCs
Next, we investigated whether Smad1 protein up-regulates SMA protein in MCs. MCs were transiently transfected with a plasmid encoding Smad1 in the absence or presence of TGFβ1 (Fig. 2, A and B). Induction of SMA was observed in parallel with the induction of Smad1 protein only when stimulated with TGFβ1. These results indicate that Smad1 activation is important for the induction of SMA. Moreover, constitutively active Smad1 induced SMA protein in a dose-dependent manner (Fig. 2C). We previously demonstrated that activin receptor-like kinase 1 (ALK1) transduces TGFβ1 signaling to Smad1 in AGE-treated MCs (4). Transiently, co-transfection assay revealed that Smad1 overexpression in MCs notably induced SMA protein along with phsophorylation of Smad1 by constitutively active ALK1 (Fig. 2D). These data suggest that induction and phosphorylation of Smad1 are determinants of SMA expression in MCs.
Scx Is Isolated as a bHLH Factor and Is Induced under AGE Exposure in MCs
Next, to elucidate the underlying mechanisms in the direct regulation of SMA expression in response to Smad1 activation, we focused our attention on the E-box sites in the promoter of SMA gene. The bHLH transcription factors belong to a larger superfamily of HLH transcription factors that contains more than 240 proteins expressed in eukaryotic organisms ranging from yeast to humans (32). To identify MC-specific bHLH transcription factors that bind to the E-box motif, we performed degenerate PCR screening using primer sets targeting the conserved sequences within various bHLH subfamilies. We carried out the screening for mouse MCs treated with AGE for induction of SMA expression to isolate a clone that encodes a specific transcription factor (Fig. 3A). We identified this clone as the cDNA that encodes Scx using the RACE method. Scx was originally identified as a bHLH transcription factor that is expressed in the sclerotome, in mesenchymal precursors of bone and cartilage, and in connective tissues (33, 34). From these reports, Scx has been implicated in the regulation of the development of collagen-rich tissues such as tendons and ligaments. However, the function and significance of Scx expression in MCs have remained unknown.
FIGURE 3.

Identification and regulation of Scx expression by AGE in MCs. A, PCR products obtained using degenerate primers for the HLH transcription factors family from cDNA prepared from MCs treated with BSA or AGE (10 μg/cm2) for 24 h were analyzed on an acrylamide gel. M, marker; N, negative control. B, after treatment with BSA or AGE (1 or 10 μg/cm2) for the indicated time, the specific mRNA expression levels were determined by using RT-PCR analysis. GAPDH amplification was used as a control. One of three independent experiments is shown. C, Scx protein was monitored by Western blot analysis in response to a 24- or 72-h treatment with BSA or AGE (1 or 10 μg/cm2). One of three independent experiments is shown. D, immunofluorescence analysis of mouse MCs treated with BSA or AGE (10 μg/cm2) for the indicated time. The cells were counterstained with DAPI, and the nuclei were identified. Data from one of three representative experiments is shown. After exposure with CML or vehicle alone (Ctl) for 72 h, the expression levels of mRNA (E) and protein (F) were examined. GAPDH and β-actin were used as a loading control for RT-PCR and Western blot, respectively. One of at least three independent experiments is shown in each panel.
Significant induction of both the Scx mRNA and protein levels was observed in AGE-treated MCs using RT-PCR and Western blot analysis, respectively (Fig. 3, B and C). In contrast, BSA treatment did not induce expression of Scx. Similar results were seen in CML-treated MCs (Fig. 3, E and F). Because nuclear localization is important for transcription factor function, we next examined the issue of whether the translocation of Scx is affected by AGE treatment in MCs (Fig. 3D). Scx protein was induced and distributed throughout MCs with a preferential cytoplasmic localization after a 24-h incubation in the presence of AGE. Furthermore, the nuclear accumulation of Scx in response to AGE was observed in the cells 72 h after AGE stimulation, whereas BSA treatment led to little expression of Scx. These findings indicate that the induction and subsequent translocation of Scx to the nucleus are regulated by AGE exposure in MCs.
Scx Associates with E12
Because Scx was reported to bind the E-box consensus sequence from muscle creatine kinase enhancer as a heterodimer with E12 based on the results of a gel mobility shift assay (35), we first examined whether Scx physically interacts with E12 in mammalian cells by performing co-immunoprecipitation experiments. FLAG-Scx was co-immunoprecipitated with Myc-E12 only when both were co-expressed. We also confirmed the expression of E12 protein by immunoblot analysis using anti-E12 antibody (Fig. 4A). Furthermore, both Scx and E12 were localized in the nucleus as detected by DAPI counterstaining in cultured MCs. Scx protein was partially co-localized with E12 in the nucleus (Fig. 4B). The co-localization was confirmed via co-immunoprecipitation assays (Fig. 4C). In subsequent experiments, we validated the endogenous association of Scx and E12 in the diabetic mouse kidney by co-immunoprecipitation (Fig. 4D). From these results, we confirmed that Scx and E12 could physiologically engage in a specific interaction under diabetic conditions.
FIGURE 4.

Scx physically associated with E12. A, co-immunoprecipitation of FLAG-Scx and Myc-E12 from co-transfected CHOK1 cells lysate was detected by Western blot. IP, immunoprecipitation. IB, immunoblot. B, immunofluorescence of AGE-treated MCs with antibodies against the Scx (green) and E12 (red) protein showing the localization of the two proteins in the nucleus. White arrows indicated partial co-localization of Scx and E12 proteins. The cells were counterstained with DAPI to visualize the nuclei. C, Western blot of immunoprecipitates from AGE-treated MCs lysates using a polyclonal anti-E12 antibody or control IgG. The reaction mixture was loaded as input. D, kidney lysates from diabetic or nondiabetic (Control) mice were immunoprecipitated (IP) with anti-E12 antibody and control IgG. The presence of Scx in the immunoprecipitates was determined by Western blot with anti-Scx antibody. The control antibody does not immunoprecipitate Scx. Aliquots of the lysates from diabetic kidney were loaded as positive controls (PC). One of five independent experiments is shown in each panel.
Scx Negatively Regulates SMA Expression through Direct Binding to the Promoter
Considering the primary structure, the Scx protein was expected to bind the E-box in the promoter of the SMA gene, which is a common target of the bHLH transcription factors (18). Kumar et al. (36) clearly demonstrated that there are two E-boxes in the SMA promoter, at −214 bp (E1) and −252 bp (E2), which act as positive regulatory elements in mice. A labeled oligonucleotide containing the putative E-box, E1 or E2, was examined to determine whether Scx would bind to E-boxes in a gel mobility shift assay. Interestingly, E2 successfully bound to the nuclear extracts, but E1 did not. Unlabeled oligonucleotides competed effectively with the labeled E-box for binding to nuclear extracts (Fig. 5A). Anti-Scx antibodies were found to supershift the labeled E2 oligonucleotide-nuclear protein complex. Addition of an anti-E12 antibody also resulted in the supershift of the complex (Fig. 5B). Taken together, these data demonstrated the existence of an Scx-binding E-box in the promoter region of the SMA gene. Next, we tested the effects of Scx on the expression of SMA. Transient overexpression of Scx in cultured MCs without any stimulation failed to induce SMA expression (data not shown). Conversely, we found that forced expression of Scx in AGE-treated MCs was able to strongly repress the expression of the SMA protein and mRNA (Fig. 5, C and D), suggesting that Scx is a unique negative regulator of SMA in MCs exposed to AGE. Next, to explore the mechanisms for induction of SMA protein expression, we examined transiently overexpression of E12 protein in MCs, because E12 is a ubiquitous bHLH protein. Expression of SMA protein was increased in an E12-dependent manner (Fig. 5E). Moreover, we tested the transcriptional activity of the SMA promoter by transfecting to MCs with E12 expression plasmids. Luciferase activity of the SMA promoter was increased in a dose-dependent manner (Fig. 5F). These data indicate that E12 itself contributes to the induction of SMA protein.
FIGURE 5.

Negative regulation of SMA through the Scx-E-box binding. A, EMSA to examine the binding of Scx to the E-boxes (E1 and E2) of the SMA promoter. Labeled E-box oligonucleotides were mixed with nuclear extract from mouse MCs treated with AGE and yielded shifted bands (arrow). Cold competition with excess unlabeled E2 probe abrogated the shifted bands. One of four independent experiments is shown. B, supershift demonstrates the presence of Scx and E12 proteins and E-box complexes. Incubation of extracted nuclear proteins with Scx antibodies (Scx1 (S1) and Scx2 (S2)) and an E12 antibody induced strong supershifted bands, although the mixture between these antibodies and E2 probe without nuclear extract showed no bands. One of four independent experiments is shown. C, Western blot; D, RT-PCR analyses of SMA expression in whole cell lysates and total RNAs, respectively, from MCs transfected with increasing amounts of the FLAG-tagged Scx expression plasmid. α-Tubulin and GAPDH served as loading controls, respectively. One of three independent experiments is shown. E, Western blot analyses of MCs 24 h after transfection with increasing amounts of the Myc-E12 expression plasmid. α-Tubulin was used as a loading control. One of three independent experiments is shown. F, luciferase activities in lysates prepared 36 h post-transfection were measured. The activity of the reporter plasmid alone was arbitrary given a value of 1, and the activities of the other transfections were adjusted relative to this assay. All reporter assays were performed in triplicate, and standard deviations (S.D.) are denoted by the bars. CTL, control.
Co-existence of Scx and SMA in Diabetic Nephropathy Mice
As Scx was induced by AGE stimulation in cultured MCs, we subsequently examined the glomerular expression of Scx in the model of diabetic nephropathy mice. Kidneys from the diabetic or nondiabetic mice at 38 weeks after STZ or vehicle injection were tested for Scx staining. At this stage, a substantial mesangial expansion was detected in the diabetic mice. Notably, the glomeruli of the diabetic kidneys showed significantly greater numbers of Scx-positive cells with nuclear pattern than did the renal glomeruli of the control mice (Fig. 6). At the same time, positive immunostaining for pSmad1 and SMA, corresponding to the diabetic change in STZ-induced diabetic mice, was observed as in our previous reports (Fig. 6) (4, 10, 11). In addition, induction of Col1, which is known to be regulated by Scx, was also observed in STZ-induced diabetic mice. These histological observations raised a question about the mechanism of the concurrent expression of SMA, Col and Scx in diabetic glomeruli.
FIGURE 6.

Detection of Scx and SMA in diabetic nephropathy mice. STZ-induced diabetic (right panels) and control mice (left panels) were dissected at 32 weeks after treatment (n = 6 in each group). Light microscopy showed mesangial matrix expansion in STZ mice by periodic acid-Schiff's (PAS) staining. An increase in the expression of pSmad1, SMA, and Scx was noted by immunohistochemical staining in diabetic glomeruli in STZ mice. Nuclear staining of Scx in glomeruli in STZ mice was observed. Nuclei of the cells were stained by DAPI (blue). Representative microscopic appearance of the glomerulus is shown. Original magnification for all panels was ×400.
Smad1-SMA Signaling Pathway Is Strongly Activated by BMP4 in MCs
To address the above question, we first focused our attention on the regulatory mechanism for the AGE-induced Smad1 expression and phosphorylation. The neutralization of TGFβ1 was not enough to suppress the SMA expression in AGE-treated MCs (Fig. 1D), implying that another molecule is involved in the induction and activation of Smad1 under AGE exposure. Accordingly, we examined the effects of BMP4 in the induction of SMA protein expression. Induction and secretion of BMP4 were observed in AGE-treated MCs. BSA did not show these effects (Fig. 7A). BMP4 notably phosphorylated Smad1 and induced SMA in a dose-dependent manner (Fig. 7B). In addition, BMP4 strongly induced Smad1 protein expression in parallel with SMA induction (Fig. 7B). The induction of SMA by TGFβ1 and BMP4 was clearly blocked by knockdown using siRNA for Smad1 (Fig. 7C). Although TGFβ1 is known to play an important role in the AGE response of the glomeruli (37), these results suggest that not only TGFβ1-Smad1 but also BMP4-Smad1 signal plays a very important role in the phenotype alteration of MCs.
FIGURE 7.

BMP4 induced SMA expression through Smad1 activation in MCs. A, Western blot of equal amounts of total cell lysate protein or conditioned medium prepared from MCs treated with AGE or BSA (10 μg/cm2) for 24 h. B, Western blot analysis of Smad1, pSmad1, and SMA protein expression in total cell lysates from MCs treated with BMP4 at the indicated concentrations for 12 h. C, MCs were treated with scrambled or Smad1 siRNA. Scrambled siRNA (CTL) was used as a control in the experiments. After the siRNA transfection and successive 48 h of incubation, MCs were treated with 10 ng/ml TGFβ1 or BMP4 for an additional 12 h. Equal amounts of cell lysates were subjected to Western blotting using antibodies against Smad1, pSmad1, and SMA. β-Actin was used as a loading control. One of three independent experiments is shown.
Relationship between BMP4 and Scx under AGE Stimulation
To address the question of the co-induction of BMP4 and Scx by AGE, we examined the relationship between BMP4 and Scx in MCs. BMP4 alone did not induce Scx in MCs (Fig. 8A). In contrast, Scx markedly increased BMP4 expression in a dose-dependent manner in MCs (Fig. 8B). Next, we examined the effects of AGE stimulation on the link between BMP4 and Scx in MCs. To elucidate the effect of BMP4 on the induction of SMA by AGE in MCs, an inhibition assay using the BMP4 neutralizing antibody was performed. MCs treated with the BMP4-neutralizing antibody showed significant reduction of pSmad1 and SMA up-regulated by AGE. However, no reduction of Scx expression induced by AGE was observed (Fig. 8C). To confirm the role of Scx in AGE-induced BMP4 expression, siRNA knockdown for Scx was carried out. Scx knockdown inhibited the up-regulation of BMP4 expression by AGE in MCs (Fig. 8D). These results indicate that Scx regulated SMA expression through dual pathways (direct negative regulation and BMP4-mediated positive regulation) in AGE-treated MCs. Moreover, Col1 expression was positively controlled by Scx in AGE-treated MCs (Fig. 8, B and D). Thus co-expression of SMA, Col1, and Scx can be achieved in diabetic conditions.
FIGURE 8.
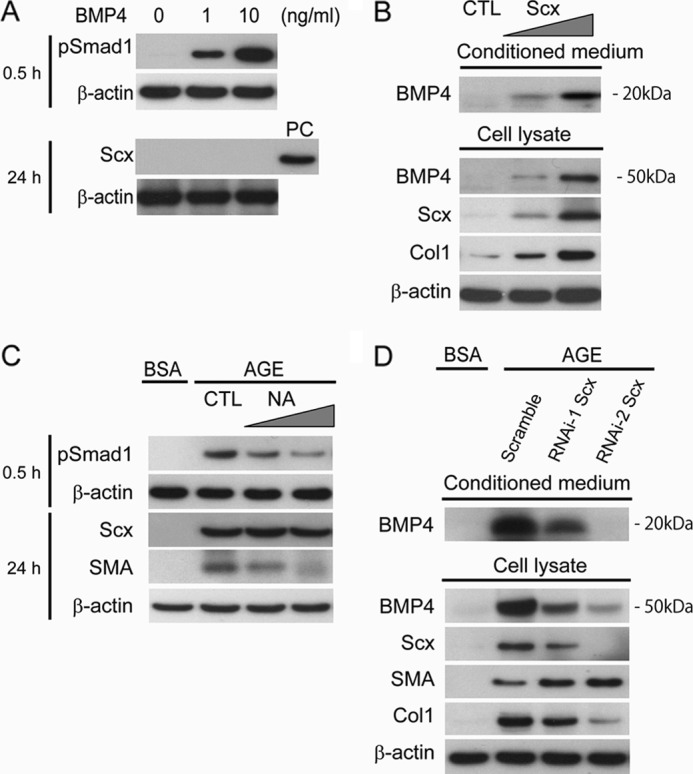
Scx up-regulated the expression and secretion of BMP4 in AGE-treated MCs. A, Western blot analysis of pSmad1 and Scx proteins in the BMP4 stimulation at the indicated concentrations. PC, positive control. B, Western blot analysis of BMP4 expression in conditioned medium and whole cell lysates from MCs at 24 h after transfection with increasing amounts of the Scx expression plasmid (pcDNA3-Scx). Empty vector was used as a control (CTL). C, MCs were treated with neutralizing antibody (NA) for BMP4 (10 μg/ml) or control normal IgG (CTL) for 24 h after a 6-h exposure to AGE or BSA (10 μg/cm2). Equal amounts of cell lysates were subjected to Western blot. One of three independent experiments is shown. D, MCs were treated with siRNAs for Scx for 48 h after a 6-h exposure to AGE or BSA (10 μg/cm2). Scrambled siRNA was used as a control in the experiments. Equal amounts of cell lysates or conditioned medium were subjected to Western blot. One of three independent experiments is shown. β-Actin was used as a loading control in each experiment.
Id1 Modulates the Balance between Scx and SMA Expression under AGE Stimulation
To further understand the regulatory mechanism for co-expression of SMA, Col1, and Scx, we investigate the negative feedback mechanism in a diabetic condition. Although AGE up-regulated both Scx and SMA expression, a time-dependent induction was observed only for SMA expression. Up-regulation of E12 was found in the same fashion as that of SMA. In contrast, Col1 expression was not affected in the prolongation of AGE stimulation (Fig. 9A). Knockdown of E12 showed the reduction of SMA and no effects on Scx and Col1 expressions under AGE stimulation (Fig. 9B). Next, we examined the transcriptional regulation of SMA via the E-box (E2) in its promoter by means of a ChIP assay. Interestingly, binding of Scx to the E-box (E2) was observed at 24 h of AGE stimulation, but at 72 h, the binding effect was almost diminished (Fig. 9C). Inhibitors of differentiation (Ids) and bHLH proteins dictate in an opposite manner cellular programs of differentiation in various cell types, and BMPs can induce the expression of Ids (38, 39). Several reports have demonstrated that BMP, but not TGFβ, strongly activates the Id1 promoter in a Smad1-dependent manner (40–43). Therefore, to understand the molecular mechanism underlying the co-existence of Scx, SMA, and Col1 in diabetic conditions, we investigated the role of Id1 in the AGE-stimulated induction of Scx and SMA in MCs. Id1 was induced by AGE in a time-dependent manner (Fig. 9D) similar to the induction of SMA in MCs (Fig. 9A). These data suggest the possibility that Id1 modulates SMA expression through negative regulation for Scx under AGE stimulation. Knockdown of Id1 revealed that Scx significantly suppressed SMA expression in the absence of Id1 protein under AGE stimulation (Fig. 9E). Furthermore, a ChIP assay after Id1 knockdown showed the persistence of the binding effect of Scx until 72 h post-AGE stimulation (Fig. 9F). Collectively, these results suggest that Id1 may serve an important effector function for the control of SMA expression via Scx under diabetic conditions.
FIGURE 9.
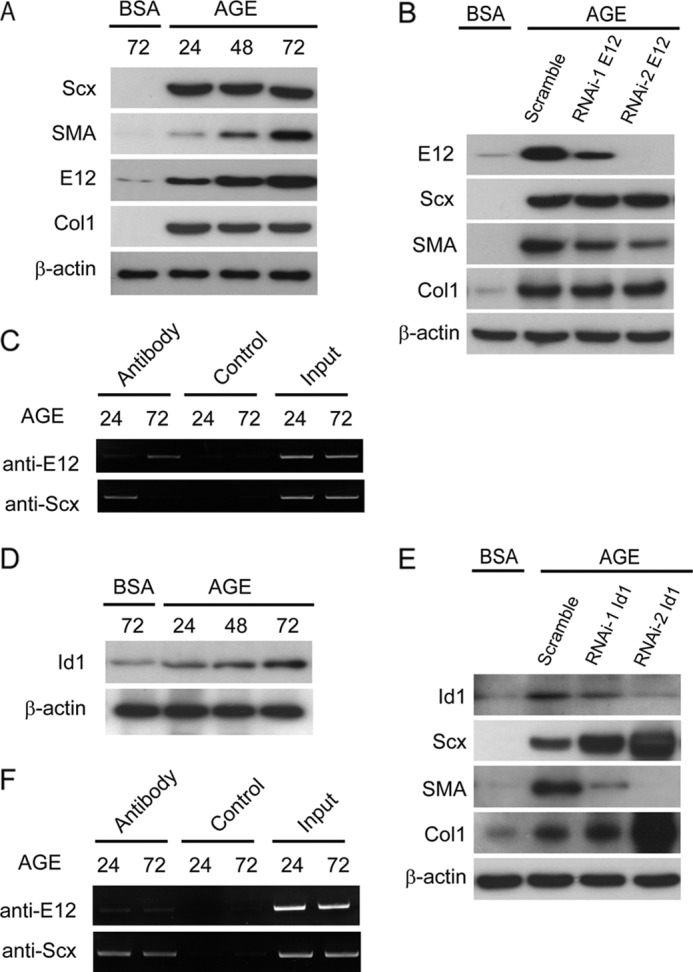
Id1 promoted SMA expression through the inhibition of Scx expression at the downstream signaling pathway of AGE in MCs. A, Scx and SMA proteins were monitored by Western blot analysis in response to BSA or AGE (10 μg/cm2) for the indicated times. B, MCs were treated with siRNAs for E12 for 48 h after a 6-h exposure to AGE or BSA (10 μg/cm2). Scrambled siRNA was used as a control in the experiments. Equal amounts of cell lysates were subjected to Western blot. C, ChIP assay was carried out on MCs treated with BSA or AGE (10 μg/cm2) for the indicated times. ChIP PCR products were amplified from input-positive controls (Input), a negative control normal rabbit IgG (Control), and antibodies specific for E12 or Scx. PCR was performed by using primers for an E-box-containing motif, E2. D, Western blot analysis of MCs treated with BSA or AGE (10 μg/cm2) for the indicated times. E, MCs were treated with Id1 siRNAs (RNAi-1 or -2) or scrambled control siRNA for 48 h after 6-h the exposure to AGE or BSA (10 μg/cm2). Equal amounts of cell lysates were subjected to Western blot. F, ChIP assay for MCs exposed to AGE (10 μg/cm2) for 24 or 72 h and treated with siRNAs for Id1 for the last 18 or 48 h, respectively. The PCR was performed as described as above. β-Actin was used as a loading control in each experiment. One of three independent experiments is shown.
Relationship between BMP4 Pathway and TGFβ1 Pathway in the Regulation of SMA Induction
We further investigated whether BMP4 and TGFβ1 exhibited a synergistic role in the induction of SMA in MCs. The high dose but not the low dose of TGFβ1 and BMP4 showed an additional effect on the induction of SMA (Fig. 10, A and B). As ALK1 transduces TGFβ1 signaling to Smad1 in MCs (4), we performed knockdown assay using siRNA for ALK1 in TGFβ1-treated MCs. Induction of ALK1 expression by TGFβ1 was much higher in the higher dose stimulation. Knockdown of ALK1 blocked the TGFβ1-induced expression of SMA (Fig. 10C). Moreover, we carried out the neutralization assay using TGFβ1- and BMP4-neutralizing antibodies. Although mutual inhibition in these two pathways was not shown (Fig. 10, D and E), co-treatment with these neutralizing antibodies synergistically suppresses SMA induction in MCs under AGE stimulation (Fig. 10F), suggesting that both TGFβ1 and BMP4 are induced by AGE stimulation. Collectively, activation of both BMP4 pathway and TGFβ1 pathway is involved in the synergistic induction of SMA in AGE-treated MCs.
FIGURE 10.

Effects of BMP4 and TGFβ1 in the induction of SMA in MCs. A and B, Western blot analysis of pSmad1, pSmad3, and SMA protein expression in total cell lysates from MCs treated with TGFβ1 or BMP4 at the indicated concentrations for 0.5 or 24 h. C, MCs were pretreated with siRNA for ALK1 for 48 h before a 24-h exposure to TGFβ1. Scrambled siRNA (Scr) was used as a control in the experiments. Equal amounts of cell lysates were subjected to Western blot. D and E, MCs were treated with TGFβ1-neutralizing antibody (NA) or BMP4-neutralizing antibody (10 μg/ml) for 1 or 24 h after a 1-h exposure to TGFβ1 or BMP4 (10 ng/ml). Normal IgY or normal IgG was used as a control, respectively. Induction of SMA and pSmad1 protein expression was monitored in Western blot. F, MCs were treated with TGFβ1-neutralizing antibody or BMP4-neutralizing antibody (10 μg/ml) for 1 or 24 h after a 6-h exposure to AGE or BSA (10 μg/cm2). α-Tubulin was used as a loading control in each experiment. One of three independent experiments is shown.
DISCUSSION
Diverse cells and tissues react to injury with similar responses, including impaired cell differentiation and development of sclerosis and fibrosis from increased ECM accumulation. Regardless of whether the responsible cells are derived from fibroblasts in the skin, lungs (44), or heart or from hepatic stellate cells (45), MCs (2, 46), or even transdifferentiated epithelial cells (47, 48), activation of SMA expression is a common phenomenon. In diabetic nephropathy, activated dedifferentiated MCs, expressing SMA, secrete collagens and lead to ECM accumulation and subsequent glomerular obliteration. In this study, we described novel pathways regulating the phenotypic alteration in MCs under diabetic conditions. Thus far, TGFβ1 has been considered to be an important and critical mediator in the process of induction of SMA (31); however, the molecular mechanism of the intracellular signal transduction has remained elusive. Whereas there are three isoforms of TGFβ (TGFβ1, -β2, and -β3), TGFβ1 alone was up-regulated in diabetic conditions. We previously found a co-existence of Smad1 and SMA in the sclerotic region in diabetic nephropathy rats and mice (10, 11). In this study, we demonstrated that the SMA protein was induced by both BMP4 and TGFβ1 in a Smad1-dependent way.
As Smad3 is a well known intermediate in the TGFβ signaling pathway (50), we asked whether Smad3 mediated the induction of SMA expression in an overexpression assay using constitutively active Smad3 and in a knockdown assay using siRNA for Smad3. Neither of them showed the alteration of SMA expression in MCs. TGFβ1 is able to activate two distinct TGF-β type I receptors and signal transduction pathways as follows: the ALK5/Smad2/3 pathway and the ALK1/Smad1/5-regulated pathway (51). Accordingly, we examined the expression of ALK1 in MCs under exposure to AGEs. We previously demonstrated that ALK1 is induced in AGE-treated MCs (4). In this study, transient overexpression of constitutively active ALK1 could induce SMA in parallel with activation of Smad1 in MCs without AGE stimulation. Furthermore, it is generally known that Smad1 is also directly phosphorylated by BMPs through type I and II BMP receptors (52). Notably, AGE induced the expression and secretion of BMP4. Subsequently BMP4 phosphorylated and activated Smad1 and induced SMA expression in MCs. BMP4 induced much higher levels of Smad1 protein expression than did TGFβ1, revealing a novel aspect of the regulatory mechanism of the phenotypic modulation of MCs. AGE also activates TGFβ1 signaling pathway leading to phosphorylation of Smad1 via ALK1. Thus, the BMP4-Smad1 signal, besides TGFβ1-Smad1 signal, has an additional effect on SMA induction and is closely involved in the phenotypic change.
Previously, Kumar et al. (36) demonstrated an essential role of two E-boxes for SMA expression by in vivo analysis using promoter-LacZ transgenic mice. However, the direct regulatory mechanisms for SMA expression via its E-boxes in MCs have remained elusive. In addition, there has been no information about MC-specific bHLH factors. We therefore performed a degenerate PCR screening for AGE-stimulated MCs and identified Scx as a key candidate molecule for modulation of the cell phenotype under diabetic conditions, because it is well known that AGE is linked to the phenotypic alteration of MCs in diabetic nephropathy. Induction of Scx was also observed in CML-treated MCs. Because CML is recognized by receptor for advanced glycation end products (53), this induction of Scx may be mediated by the receptor for advanced glycation end product pathway. Scx was originally named after its expression in the sclerotome, which plays a critical role in the development of chondrogenic cell lineages during embryogenesis (42). Similar to other bHLH transcription factors, Scx and E12 physically interacted as a heterodimer and co-localized in the nucleus of MCs. Thus, judging from the observed expression of Scx, AGE-induced Scx was thought to act as a transcriptional factor for some target genes containing E-boxes in the promoter. Gel mobility shift assays showed that Scx binds to an E-box motif (E2) in the promoter of SMA. Scx acted as a negative regulator for SMA induction in MCs. In contrast, AGE also up-regulated the expression of E12 protein that may be involved in the induction of SMA. These results raised the possibility that Scx may have a suppressive effect on phenotypic changes in response to the emergence of SMA induced by overexpressed E12 under AGE stimulation. In addition, Scx is known to be expressed in tendons, ligaments, and other dense connective tissues and to promote the secretion of Col1 (33, 54, 55). Glomerulosclerosis in diabetic nephropathy is characterized by a prolonged and gradual decline in renal function, caused by a progressive accumulation of ECM, including Col1, and subsequent glomerular obliteration (4, 56). Accordingly, Scx may be involved in the irreversible sclerotic change in diabetic glomerulopathy.
Terminally differentiated cell lineages basically manifest a tendency to stabilize the original phenotype for tissue formation or organ function, although they have the tendency to change in response to current environmental conditions as an adaptation. In fact, it is well recognized that MCs derived from mesenchymal cells dedifferentiated into other mesenchymally derived cell lineages, such as osteoblasts and chondrocytes and tenocytes, which represent the expressions of Col1, osteopontin, Sox9, and Cbfa1 (57–59). This mixture of different phenotypes is also observed in glomeruli of human diabetic glomerulosclerosis (60, 61). Similar mixture was reported in hepatic fibrosis (62). However, little is known about how the phenotypic alteration in terminally differentiated cells was regulated. In this study, different mesenchymally derived cell lineage-like phenotypes appeared in damaged glomeruli of diabetic mice at the same time. We previously demonstrated that Smad1 transcriptionally up-regulates these osteoblastic matrix proteins, such as Col1 and osteopontin, in MCs (4). The intermediate mesoderm is the source of all kidney tissue in the normal development, and the mesoderm patterning program is controlled by the gradient of BMP4 as a morphogen (63, 64). Therefore, here we examined the relationship between Scx and BMP4 in AGE-exposed MCs. Transient overexpression of Scx induced BMP4 in a dose-dependent manner. Thus, it is speculated that BMP4 controls phenotypic alteration in MCs to adapt to the critical changes of microenvironments in the diabetic kidney. BMPs, including BMP4, are indispensable factors for a normal kidney development and basically induce irreversible differentiation of perivascular mesenchymal type cells into osteoprogenitor cells (65). Under normal circumstances, BMP4 is supposed to control the cell phenotype of various cells during their development (66). However, under diabetic conditions, it is considered that the regulation of BMP4 signaling is not controlled properly. Hence, deregulated BMP4 signal transduction may turn out to fail to organize the structure and to restore its original function, further underscoring the complexity of gene expression in the diabetic glomeruli.
In ChIP analyses, along with prolonged treatment with AGE, binding of Scx to the E-box was diminished. These results point to another regulatory mechanism between the BMP4-SMA signaling pathway and Scx expression under exposure to AGEs. The actions of the bHLH proteins are inhibited by the dominant negative HLH proteins, Id1–4. Among the Ids, Id1 was previously reported to be expressed in MCs (49), but the target molecule of Id1 was not identified. Several recent studies have revealed that Id proteins are one of the most crucial targets of BMPs and that they may be responsible for the exhibition of the biological activities of BMPs (42). These findings support the possibility that Id1 may positively regulate BMP signaling by sequestering the E-protein from Scx to halt its negative regulation for SMA expression in MCs. We report here that Id1 was strongly induced by AGE in a time-dependent manner. Induction of Id1 by AGE is considered to block the activation of Scx and subsequently enhance the expression levels of SMA in MCs. Taken together, the results discussed herein suggest that the balance of expression levels between Id1 and Scx could be an important factor for modulating exacerbation or improvement of diabetic nephropathy. The balance may also contribute to renal remodeling or reconstitution of injured renal glomeruli, especially MCs (Fig. 11).
FIGURE 11.
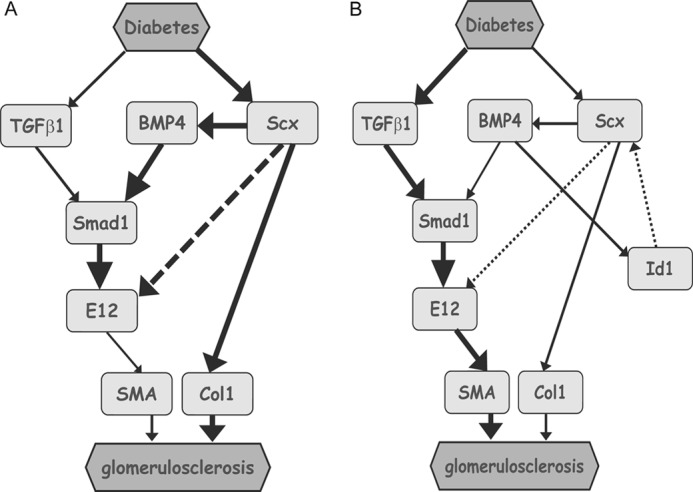
Proposed model for phenotype alteration by Scx, BMP4/Smad1, and Id1 in diabetic nephropathy. Following kidney injury in diabetes, MCs undergo activation, which connotes a transition from quiescent cells, and secrete interstitial collagens, including Col1, and exhibit smooth muscle-like proteins such as SMA. Key mediator proteins underlying these effects are shown. The state of activated MCs in diabetic conditions is regulated by Scx activation. The activation of the Scx-BMP4-Smad1 signal may lead to diabetic glomerulosclerosis, regardless of whether negative feedback of Id1 is present (B) or not (A). Continuous lines and dashed lines represent positive and negative regulations, respectively.
As SMA is also a marker of phenotypic alterations in other tissues that lead to fibrosis and/or sclerosis, such as liver, pancreas, lungs, and skin, the activation of the BMP4-Smad1 signaling pathway and the additional interplay between Scx and Id1 may contribute to the progressive exacerbation in many fibrotic or sclerotic diseases. Further elucidation of the mechanism of this complexity in phenotype alteration may reveal novel findings that could lead to more effective therapeutic approaches.
Acknowledgments
We are grateful to Dr. K. Miyazono (University of Tokyo) for Smad1 expression vector and Dr. J. Oh (Korea University) for the constitutively active Smad3 expression vector. We also thank A. Sakurai, T. Sasaki, and S. Hayashi for their technical support.
This work was supported by The Kidney Foundation of Japan Grant JKFB09-41, funds from The Mitsui Sumitomo Welfare Foundation, Grants-in-aid for Scientific Research 19590973 and 21591033, and Japan Science and Technology Agency.
- ECM
- extracellular matrix
- AGE
- advanced glycation end product
- bHLH
- basic helix-loop-helix
- CML
- carboxymethyl lysine
- Id
- inhibitor of differentiation
- MC
- mesangial cell
- Scx
- scleraxis
- SMA
- smooth muscle α-actin
- STZ
- streptozotocin
- Col1
- type I collagen
- RACE
- 5′- and 3′-rapid amplification of cDNA end.
REFERENCES
- 1. Klahr S., Schreiner G., Ichikawa I. (1988) The progression of renal disease. N. Engl. J. Med. 318, 1657–1666 [DOI] [PubMed] [Google Scholar]
- 2. Johnson R. J., Iida H., Alpers C. E., Majesky M. W., Schwartz S. M., Pritzi P., Gordon K., Gown A. M. (1991) Expression of smooth muscle cell phenotype by rat mesangial cells in immune complex nephritis. α-Smooth muscle actin is a marker of mesangial cell proliferation. J. Clin. Invest. 87, 847–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Desmoulière A., Geinoz A., Gabbiani F., Gabbiani G. (1993) Transforming growth factor-β1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Abe H., Matsubara T., Iehara N., Nagai K., Takahashi T., Arai H., Kita T., Doi T. (2004) Type IV collagen is transcriptionally regulated by Smad1 under advanced glycation end product (AGE) stimulation. J. Biol. Chem. 279, 14201–14206 [DOI] [PubMed] [Google Scholar]
- 5. Rønnov-Jessen L., Petersen O. W. (1996) A function for filamentous α-smooth muscle actin. Retardation of motility in fibroblasts. J. Cell Biol. 134, 67–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramadori G., Veit T., Schwögler S., Dienes H. P., Knittel T., Rieder H., Meyer zum Büschenfelde K. H. (1990) Expression of the gene of the α-smooth muscle-actin isoform in rat liver and in rat fat-storing (ITO) cells. Virchows Arch. B. Cell Pathol. Incl. Mol. Pathol. 59, 349–357 [DOI] [PubMed] [Google Scholar]
- 7. Zhang K., Rekhter M. D., Gordon D., Phan S. H. (1994) Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am. J. Pathol. 145, 114–125 [PMC free article] [PubMed] [Google Scholar]
- 8. Sundberg C., Ivarsson M., Gerdin B., Rubin K. (1996) Pericytes as collagen-producing cells in excessive dermal scarring. Lab. Invest. 74, 452–466 [PubMed] [Google Scholar]
- 9. Takahashi T., Abe H., Arai H., Matsubara T., Nagai K., Matsuura M., Iehara N., Yokode M., Nishikawa S., Kita T., Doi T. (2005) Activation of STAT3/Smad1 is a key signaling pathway for progression to glomerulosclerosis in experimental glomerulonephritis. J. Biol. Chem. 280, 7100–7106 [DOI] [PubMed] [Google Scholar]
- 10. Matsubara T., Abe H., Arai H., Nagai K., Mima A., Kanamori H., Sumi E., Takahashi T., Matsuura M., Iehara N., Fukatsu A., Kita T., Doi T. (2006) Expression of Smad1 is directly associated with mesangial matrix expansion in rat diabetic nephropathy. Lab. Invest. 86, 357–368 [DOI] [PubMed] [Google Scholar]
- 11. Mima A., Arai H., Matsubara T., Abe H., Nagai K., Tamura Y., Torikoshi K., Araki M., Kanamori H., Takahashi T., Tominaga T., Matsuura M., Iehara N., Fukatsu A., Kita T., Doi T. (2008) Urinary Smad1 is a novel marker to predict later onset of mesangial matrix expansion in diabetic nephropathy. Diabetes 57, 1712–1722 [DOI] [PubMed] [Google Scholar]
- 12. Weintraub H., Davis R., Tapscott S., Thayer M., Krause M., Benezra R., Blackwell T. K., Turner D., Rupp R., Hollenberg S. (1991) The myoD gene family. Nodal point during specification of the muscle cell lineage. Science 251, 761–766 [DOI] [PubMed] [Google Scholar]
- 13. Weintraub H. (1993) The MyoD family and myogenesis. Redundancy, networks, and thresholds. Cell 75, 1241–1244 [DOI] [PubMed] [Google Scholar]
- 14. Hasty P., Bradley A., Morris J. H., Edmondson D. G., Venuti J. M., Olson E. N., Klein W. H. (1993) Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364, 501–506 [DOI] [PubMed] [Google Scholar]
- 15. Nabeshima Y., Hanaoka K., Hayasaka M., Esumi E., Li S., Nonaka I., Nabeshima Y. (1993) Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 364, 532–535 [DOI] [PubMed] [Google Scholar]
- 16. Guillemot F., Lo L. C., Johnson J. E., Auerbach A., Anderson D. J., Joyner A. L. (1993) Mammalian achaete-scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell 75, 463–476 [DOI] [PubMed] [Google Scholar]
- 17. Begley C. G., Aplan P. D., Denning S. M., Haynes B. F., Waldmann T. A., Kirsch I. R. (1989) The gene SCL is expressed during early hematopoiesis and encodes a differentiation-related DNA-binding motif. Proc. Natl. Acad. Sci. U.S.A. 86, 10128–10132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Murre C., McCaw P. S., Baltimore D. (1989) A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and Myc proteins. Cell 56, 777–783 [DOI] [PubMed] [Google Scholar]
- 19. Massari M. E., Murre C. (2000) Helix-loop-helix proteins. Regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 20, 429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hogan B. L. (1996) Bone morphogenetic proteins. Multifunctional regulators of vertebrate development. Genes Dev. 10, 1580–1594 [DOI] [PubMed] [Google Scholar]
- 21. Miyazaki Y., Oshima K., Fogo A., Hogan B. L., Ichikawa I. (2000) Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. J. Clin. Invest. 105, 863–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Erlebacher A., Filvaroff E. H., Gitelman S. E., Derynck R. (1995) Toward a molecular understanding of skeletal development. Cell. 80, 371–378 [DOI] [PubMed] [Google Scholar]
- 23. Reddi A. H. (1998) Role of morphogenetic proteins in skeletal tissue engineering and regeneration. Nat. Biotechnol. 16, 247–252 [DOI] [PubMed] [Google Scholar]
- 24. Makibayashi K., Tatematsu M., Hirata M., Fukushima N., Kusano K., Ohashi S., Abe H., Kuze K., Fukatsu A., Kita T., Doi T. (2001) A vitamin D analog ameliorates glomerular injury on rat glomerulonephritis. Am. J. Pathol. 158, 1733–1741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Doi T., Vlassara H., Kirstein M., Yamada Y., Striker G. E., Striker L. J. (1992) Receptor-specific increase in extracellular matrix production in mouse mesangial cells by advanced glycosylation end products is mediated via platelet-derived growth factor. Proc. Natl. Acad. Sci. U.S.A. 89, 2873–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davies M. (1994) The mesangial cell. A tissue culture view. Kidney Int. 45, 320–327 [DOI] [PubMed] [Google Scholar]
- 27. Abe H., Iehara N., Utsunomiya K., Kita T., Doi T. (1999) A vitamin D analog regulates mesangial cell smooth muscle phenotypes in a transforming growth factor-β type II receptor-mediated manner. J. Biol. Chem. 274, 20874–20878 [DOI] [PubMed] [Google Scholar]
- 28.Deleted in proof
- 29. Narumi O., Mori S., Boku S., Tsuji Y., Hashimoto N., Nishikawa S., Yokota Y. (2000) OUT, a novel basic helix-loop-helix transcription factor with an Id-like inhibitory activity. J. Biol. Chem. 275, 3510–3521 [DOI] [PubMed] [Google Scholar]
- 30. Yang C. W., Vlassara H., Peten E. P., He C. J., Striker G. E., Striker L. J. (1994) Advanced glycation end products up-regulate gene expression found in diabetic glomerular disease. Proc. Natl. Acad. Sci. U.S.A. 91, 9436–9440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Orlandi A., Ropraz P., Gabbiani G. (1994) Proliferative activity and α-smooth muscle actin expression in cultured rat aortic smooth muscle cells are differently modulated by transforming growth factor-β1 and heparin. Exp. Cell Res. 214, 528–536 [DOI] [PubMed] [Google Scholar]
- 32. Atchley W. R., Fitch W. M. (1997) A natural classification of the basic helix-loop-helix class of transcription factors. Proc. Natl. Acad. Sci. U.S.A. 94, 5172–5176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brent A. E., Schweitzer R., Tabin C. J. (2003) A somitic compartment of tendon progenitors. Cell 113, 235–248 [DOI] [PubMed] [Google Scholar]
- 34. Brent A. E., Tabin C. J. (2004) FGF acts directly on the somitic tendon progenitors through the Ets transcription factors Pea3 and Erm to regulate scleraxis expression. Development 131, 3885–3896 [DOI] [PubMed] [Google Scholar]
- 35. Cserjesi P., Brown D., Ligon K. L., Lyons G. E., Copeland N. G., Gilbert D. J., Jenkins N. A., Olson E. N. (1995) Scleraxis. A basic helix-loop-helix protein that prefigures skeletal formation during mouse embryogenesis. Development 121, 1099–1110 [DOI] [PubMed] [Google Scholar]
- 36. Kumar M. S., Hendrix J. A., Johnson A. D., Owens G. K. (2003) Smooth muscle α-actin gene requires two E-boxes for proper expression in vivo and is a target of class I basic helix-loop-helix proteins. Circ. Res. 92, 840–847 [DOI] [PubMed] [Google Scholar]
- 37. Li J. H., Huang X. R., Zhu H. J., Oldfield M., Cooper M., Truong L. D., Johnson R. J., Lan H. Y. (2004) Advanced glycation end products activate Smad signaling via TGF-β-dependent and independent mechanisms. Implications for diabetic renal and vascular disease. FASEB J. 18, 176–178 [DOI] [PubMed] [Google Scholar]
- 38. Ogata T., Wozney J. M., Benezra R., Noda M. (1993) Bone morphogenetic protein 2 transiently enhances expression of a gene, Id (inhibitor of differentiation), encoding a helix-loop-helix molecule in osteoblast-like cells. Proc. Natl. Acad. Sci. U.S.A. 90, 9219–9222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hollnagel A., Oehlmann V., Heymer J., Rüther U., Nordheim A. (1999) Id genes are direct targets of bone morphogenetic protein induction in embryonic stem cells. J. Biol. Chem. 274, 19838–19845 [DOI] [PubMed] [Google Scholar]
- 40. Korchynskyi O., ten Dijke P. (2002) Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem. 277, 4883–4891 [DOI] [PubMed] [Google Scholar]
- 41. Valdimarsdottir G., Goumans M. J., Rosendahl A., Brugman M., Itoh S., Lebrin F., Sideras P., ten Dijke P. (2002) Stimulation of Id1 expression by bone morphogenetic protein is sufficient and necessary for bone morphogenetic protein-induced activation of endothelial cells. Circulation 106, 2263–2270 [DOI] [PubMed] [Google Scholar]
- 42. Viñals F., Reiriz J., Ambrosio S., Bartrons R., Rosa J. L., Ventura F. (2004) BMP-2 decreases Mash1 stability by increasing Id1 expression. EMBO J. 23, 3527–3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Itoh F., Itoh S., Goumans M. J., Valdimarsdottir G., Iso T., Dotto G. P., Hamamori Y., Kedes L., Kato M., ten Dijke P. (2004) Synergy and antagonism between Notch and BMP receptor signaling pathways in endothelial cells. EMBO J. 23, 541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kuhn C., McDonald J. A. (1991) The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am. J. Pathol. 138, 1257–1265 [PMC free article] [PubMed] [Google Scholar]
- 45. Lee K. S., Buck M., Houglum K., Chojkier M. (1995) Activation of hepatic stellate cells by TGFα and collagen type I is mediated by oxidative stress through c-myb expression. J. Clin. Invest. 96, 2461–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takeoka H., Iehara N., Uematsu-Yanagita M., Abe H., Sunamoto M., Yamada Y., Kita T., Doi T. (1998) A multifunctional transcription factor (A1p145) regulates the smooth muscle phenotype in mesangial cells. Biochem. Biophys. Res. Commun. 252, 290–295 [DOI] [PubMed] [Google Scholar]
- 47. Sappino A. P., Schürch W., Gabbiani G. (1990) Differentiation repertoire of fibroblastic cells. Expression of cytoskeletal proteins as marker of phenotypic modulations. Lab. Invest. 63, 144–161 [PubMed] [Google Scholar]
- 48. Strutz F., Müller G. A., Neilson E. G. (1996) Transdifferentiation. A new angle on renal fibrosis. Exp. Nephrol. 4, 267–270 [PubMed] [Google Scholar]
- 49. Simonson M. S., Rooney A., Herman W. H. (1993) Expression and differential regulation of Id1, a dominant negative regulator of basic helix-loop-helix transcription factors, in glomerular mesangial cells. Nucleic Acids Res. 21, 5767–5774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moustakas A., Souchelnytskyi S., Heldin C. H. (2001) Smad regulation in TGF-β signal transduction. J. Cell Sci. 114, 4359–4369 [DOI] [PubMed] [Google Scholar]
- 51. Oh S. P., Seki T., Goss K. A., Imamura T., Yi Y., Donahoe P. K., Li L., Miyazono K., ten Dijke P., Kim S., Li E. (2000) Activin receptor-like kinase 1 modulates transforming growth factor-β1 signaling in the regulation of angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 97, 2626–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Massagué J., Wotton D. (2000) Transcriptional control by the TGF-β/Smad signaling system. EMBO J. 19, 1745–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kislinger T., Fu C., Huber B., Qu W., Taguchi A., Du Yan S., Hofmann M., Yan S. F., Pischetsrieder M., Stern D., Schmidt A. M. (1999) Nϵ-(Carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 274, 31740–31749 [DOI] [PubMed] [Google Scholar]
- 54. Schweitzer R., Chyung J. H., Murtaugh L. C., Brent A. E., Rosen V., Olson E. N., Lassar A., Tabin C. J. (2001) Analysis of the tendon cell fate using Scleraxis, a specific marker for tendons and ligaments. Development 128, 3855–3866 [DOI] [PubMed] [Google Scholar]
- 55. Petersen W., Hohmann G., Stein V., Tillmann B. (2002) The blood supply of the posterior tibial tendon. J. Bone Joint Surg. Br. 84, 141–144 [DOI] [PubMed] [Google Scholar]
- 56. Mauer S. M., Steffes M. W., Ellis E. N., Sutherland D. E., Brown D. M., Goetz F. C. (1984) Structural-functional relationships in diabetic nephropathy. J. Clin. Invest. 74, 1143–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Adler S., Striker L. J., Striker G. E., Perkinson D. T., Hibbert J., Couser W. G. (1986) Studies of progressive glomerular sclerosis in the rat. Am. J. Pathol. 123, 553–562 [PMC free article] [PubMed] [Google Scholar]
- 58. Nagasaki T., Ishimura E., Shioi A., Jono S., Inaba M., Nishizawa Y., Morii H., Otani S. (1997) Osteopontin gene expression and protein synthesis in cultured rat mesangial cells. Biochem. Biophys. Res. Commun. 233, 81–85 [DOI] [PubMed] [Google Scholar]
- 59. Sumi E., Iehara N., Akiyama H., Matsubara T., Mima A., Kanamori H., Fukatsu A., Salant D. J., Kita T., Arai H., Doi T. (2007) SRY-related HMG box 9 regulates the expression of Col4a2 through transactivating its enhancer element in mesangial cells. Am. J. Pathol. 170, 1854–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Oldfield M. D., Bach L. A., Forbes J. M., Nikolic-Paterson D., McRobert A., Thallas V., Atkins R. C., Osicka T., Jerums G., Cooper M. E. (2001) Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Invest. 108, 1853–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yamamoto T., Nakamura T., Noble N. A., Ruoslahti E., Border W. A. (1993) Expression of transforming growth factor β is elevated in human and experimental diabetic nephropathy. Proc. Natl. Acad. Sci. U.S.A. 90, 1814–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Friedman S. L. (2000) Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 275, 2247–2250 [DOI] [PubMed] [Google Scholar]
- 63. Pourquié O., Fan C. M., Coltey M., Hirsinger E., Watanabe Y., Bréant C., Francis-West P., Brickell P., Tessier-Lavigne M., Le Douarin N. M. (1996) Lateral and axial signals involved in avian somite patterning. A role for BMP4. Cell 84, 461–471 [DOI] [PubMed] [Google Scholar]
- 64. Tonegawa A., Funayama N., Ueno N., Takahashi Y. (1997) Mesodermal subdivision along the mediolateral axis in chicken controlled by different concentrations of BMP-4. Development 124, 1975–1984 [DOI] [PubMed] [Google Scholar]
- 65. Urist M. R., DeLange R. J., Finerman G. A. (1983) Bone cell differentiation and growth factors. Science 220, 680–686 [DOI] [PubMed] [Google Scholar]
- 66. Tang M. K., Leung A. K., Kwong W. H., Chow P. H., Chan J. Y., Ngo-Muller V., Li M., Lee K. K. (2000) Bmp-4 requires the presence of the digits to initiate programmed cell death in limb interdigital tissues. Dev. Biol. 218, 89–98 [DOI] [PubMed] [Google Scholar]