

Background: Cholesterol regulation of large conductance, Ca2+- and voltage-gated K+ (BK) channels has widespread pathophysiological consequences.
Results: Cholesterol-channel recognition involves hydrophobic and hydrophilic interactions and several cholesterol recognition/interaction amino acid consensus motifs in the BK channel long C-end.
Conclusion: Cholesterol regulation of BK channels involves specific channel protein-sterol recognition.
Significance: we provide for the first time the structural basis of BK channel cholesterol sensitivity.
Keywords: Cholesterol, Ion Channels, Membrane Bilayer, Membrane Lipids, Potassium Channels, CRAC Domain, Lipid-Protein Interaction, MaxiK Channel, Lipid Bilayer
Abstract
Large conductance, Ca2+- and voltage-gated K+ (BK) channel proteins are ubiquitously expressed in cell membranes and control a wide variety of biological processes. Membrane cholesterol regulates the activity of membrane-associated proteins, including BK channels. Cholesterol modulation of BK channels alters action potential firing, colonic ion transport, smooth muscle contractility, endothelial function, and the channel alcohol response. The structural bases underlying cholesterol-BK channel interaction are unknown. Such interaction is determined by strict chemical requirements for the sterol molecule, suggesting cholesterol recognition by a protein surface. Here, we demonstrate that cholesterol action on BK channel-forming Cbv1 proteins is mediated by their cytosolic C tail domain, where we identified seven cholesterol recognition/interaction amino acid consensus motifs (CRAC4 to 10), a distinct feature of BK proteins. Cholesterol sensitivity is provided by the membrane-adjacent CRAC4, where Val-444, Tyr-450, and Lys-453 are required for cholesterol sensing, with hydrogen bonding and hydrophobic interactions participating in cholesterol location and recognition. However, cumulative truncations or Tyr-to-Phe substitutions in CRAC5 to 10 progressively blunt cholesterol sensitivity, documenting involvement of multiple CRACs in cholesterol-BK channel interaction. In conclusion, our study provides for the first time the structural bases of BK channel cholesterol sensitivity; the presence of membrane-adjacent CRAC4 and the long cytosolic C tail domain with several other CRAC motifs, which are not found in other members of the TM6 superfamily of ion channels, very likely explains the unique cholesterol sensitivity of BK channels.
Introduction
Cholesterol (CLR)2 is a major constituent of plasma membranes in eukaryotes and crucial in membrane organization, sorting, dynamics, and function (1). In particular, CLR plays a critical role in regulating the activity of membrane-associated proteins, including ion channels (2–7).
Large conductance, Ca2+- and voltage-gated K+ (BK) channels belong to the TM6 superfamily of ion channel proteins. BK channels are ubiquitously expressed in cell membranes and regulate a wide variety of processes, including neuronal excitability, neurotransmitter release, neurosecretion, tuning of cochlear hair cells, smooth muscle tone, immune responses, apoptosis, and brain tumor metastasis (7–10). Moreover, CLR modulation of BK currents has been linked to changes in neuroendocrine GH3 cell action potential firing rate (11), ion transport in colonic epithelial cells (12), membrane smooth muscle excitability and uterine contractility (13), endothelial function (14), vascular myocyte signaling (15), endothelium-dependent and -independent vasodilation (16), and BK channel responses to ethanol and eventual alcohol-induced cerebrovascular constriction (17). Despite the wide range of pathophysiological implications of CLR action on BK currents, which usually results in reduced ionic current (7), the mechanisms and structural bases underlying the CLR-BK channel interaction remain unknown.
Functional BK channels result from the tetrameric association of channel-forming α subunits (Fig. 1a), encoded by the Slo1 or KCNMA1 gene (9, 18). Using native BK or recombinant Slo1 channel proteins, it has been repeatedly demonstrated that CLR at concentrations found in natural membranes leads to a concentration-dependent reduction in BK channel steady-state activity (channel open probability; Po) (19–21). This CLR effect has been classically attributed to altered channel function secondary to changes in the physical properties of the bulk lipid bilayer upon CLR insertion and direct interaction with bilayer lipids (19, 20, 22–24). In a recent structure-activity relationship study of CLR and analogs on recombinant Slo1 channels cloned from rat cerebral artery myocytes (Cbv1) and reconstituted into a bare, two-species phospholipid bilayer, we demonstrated that CLR inhibition of BK Po was defined by strict structural requirements, including the β configuration of a CLR single polar group at C3 and, more importantly, enantiospecificity of the CLR molecule, suggesting CLR recognition by a protein surface. Thus, we hypothesized that the BK channel-forming subunit contained a region(s) that specifically sensed membrane CLR presence, leading to BK Po reduction (25).
FIGURE 1.

The Slo1 CTD confers cholesterol sensitivity to the BK channel. a, schematic representation of a BK channel α (Slo1) subunit. Four Slo1 subunits (Cbv1 isoform) form functional BK channels in native and model membranes. Truncation between Ile-322 and Ile-323 to render the construct Trcbv1S6 is indicated by a zigzag line. Shown are single channel records from WT Cbv1 (b) and Trcbv1S6 (c) after channel reconstitution into CLR-free (control) or CLR-containing POPE/POPS (3:1) (w/w) bilayers. For this and all other figures, ionic currents from constructs were recorded in the same bilayer type with transbilayer voltage set to 0 mV in symmetric 1 mm Ca2+. The arrows indicate base line (channel closed states). d, average channel Po of WT Cbv1 and Trcbv1S6 in CLR-free versus CLR-containing bilayers. For this and all other figures, n (shown in parentheses) represents the number of bilayers tested. *, different from WT Cbv1 in the absence of CLR (p = 0.0002). e, averaged inhibition of WT Cbv1 and Trcbv1S6 activity by CLR as percentage of Po in CLR-free bilayers. *, different from WT Cbv1 (p = 0.0001). Error bars, S.E.
In this study, we combined truncations and point mutagenesis, single channel electrophysiology on channel proteins reconstituted into model membranes, and computational dynamics on the Cbv1 cytosolic C tail to identify and characterize the existence of several domains in the Cbv1 cytosolic tail that are responsible for providing CLR sensitivity to BK channels. Reconstitution of BK Slo1 subunits cloned from cerebral artery myocytes into a two-species bilayer system minimized possible proteic and lipidic membrane contaminants in CLR-BK channel interaction. In addition, this system faithfully reproduces the regulation of native BK channel function by CLR reported with native channels in natural cell membranes (17, 22). We identify seven CLR recognition/interaction amino acid consensus (CRAC) motifs in the BK channel cytosolic tail domain (CTD) as determinants of the overall CLR sensitivity of the channel. However, the CRAC motif spanning residues 444–453 and proximal to the bilayer (CRAC4) is key for CLR recognition, with its signature central tyrosine (Tyr-450) being required. Such recognition involves hydrophobic interactions between sterol and the CRAC4-containing surface of the BK protein rather than extensive hydrogen bonding with the CLR hydroxyl group.
EXPERIMENTAL PROCEDURES
Computational Dynamics
Atomistic molecular dynamics (MD) simulations were run via the AMBER 10 software package (26) on intracellular portions of the BK channel α (Cbv1) subunit and interacting CLR using the ff99SB and gaff force fields, which are optimized for proteins and organic molecules, respectively. To help elucidate the nature of the interactions between CLR and the channel protein, simulations of the full CTD (residues 331–1059) and the truncated intracellular domain (residues 331–456) polypeptides were compared with that of Trcbv1-CRAC4 Y450F and Trcbv1-CRAC4 K453A. The MD starting structures of the channel protein were based on the crystallized portion of the BK channel CTD (Protein Data Bank entry 3NAF) (27). The crystallized sequence was aligned with its full sequence from the UniProt database (accession number Q12791), and Molecular Operating Environment (MOE 2010.10) software was used to create a three-dimensional homology model for the missing loops from residues 617 to 658, 667 to 683, and 834 to 871 in the PDB crystal structure. The model was geometry-optimized using the OPLS-AA force field (26) to a root mean square gradient of 0.5 kcal·mol−1·Å−1. The truncation was made after Ile-456, which is consistent with the truncation made for patch clamp experiments on recombinant channel protein. The preparatory files for the CLR molecule were generated in an antechamber using semiempirical AM1-BCC charges (26), which have been parameterized to reproduce HF/6–31* RESP geometry and atom charges. Counterions were added via Tleap to neutralize the systems. An additional salt concentration was created using 50 K+ and 50 Cl− ions, and the systems were solvated with TIP3P water. CLR was manually placed above the CRAC4 motif with the sterol methyl groups facing the motif and the CLR hydroxyl placed in the vicinity of Lys-453 with the CLR hydrophobic tail near Tyr-450, as described in previous models (4, 28).
Two rounds of minimizations were run: 1,000 steps with the protein and CLR fixed and 5,000 steps with the molecules unrestrained. A restrained warm-up MD was run at constant volume for 100 ps, with a restraint constant of 10.0 kcal·mol−1·Å−2 on the protein and CLR molecules. During this simulation, the temperature was raised to 300 K using the Langevin thermostat with a collision frequency of 1.0 ps−1. Water and ion densities were equilibrated via restrained NPT ensemble MD simulations (p = 1.0 bar) for 2 ns, using the same restraint, thermostat, and collision settings as during the warm-up. After 2 ns, the restraint was lifted, and production runs were recorded, using a time step of 2 fs. All MD simulations used the SHAKE algorithm to constrain covalently bonded hydrogen atoms and the particle mesh Ewald method (26) to calculate long range electrostatic interactions, using a cut-off distance of 12.0 Å. Histidines were represented as HIE (neutral charge: hydrogenated Nϵ, aromatic Nδ).
Molecular Biology and Cell Culture
BK channel-forming α subunit cDNA (Cbv1; AY330293) was cloned from rat cerebral artery myocytes as described previously (29). Using appropriate primers, several PCR-based Cbv1 constructs were engineered. PCR initialization started at 95 °C for 2 min, followed with five cycles (94 °C for 30 s, 48–50 °C for 30 s, 68 °C for 1.5–2 min) and 30 cycles (94 °C for 30 s, 55–59 °C for 30 s, 68 °C for 1.5–2 min), and final elongation at 72 °C for 5 min. PCRs were stopped by chilling at 4 °C for 10 min. The PCR-amplified products and pcDNA3.1 plasmid vector digested with BamHI and XhoI were purified in an agarose gel using a gel extraction kit (Qiagen, Valencia, CA). Ligation was performed using T4 DNA ligase (New England Biolabs) in a 3:1 insert-vector reaction at 16 °C overnight. The ligation mixture was then transformed in MAX Efficiency DH5α competent cells (Invitrogen). Putative clones were screened by PCR on the colonies under PCR conditions described above. Positive clones were checked by restriction analysis using BamHI for linearization. Then BamHI and XhoI were used to cleave the insert from the vector. Correct ligation and insertion were verified by automated sequencing at the University of Tennessee Health Science Center Molecular Research Center.
HEK293 cells transiently transfected with Cbv1 using Lipofectamine 2000 (Invitrogen) were grown to confluence, pelleted, and resuspended on ice in 10 ml of buffer solution: 30 mm KCl, 2 mm MgCl2, 10 mm HEPES, 5 mm EGTA, pH 7.2. A membrane preparation was obtained using a sucrose gradient as described previously (20), and aliquots were stored at −80 °C.
Ionic Current Recording following Channel Reconstitution into Lipid Bilayer
CLR was dissolved in chloroform and then introduced into a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (POPS) 3:1 (w/w) mixture. Final CLR concentrations were 10, 15, and 20% (w/w), which approximately correspond to 16, 25, and 33 mol %, respectively. Lipid mixtures, whether containing CLR or not (control), were dried under N2 gas and resuspended in 25 mg/ml decane (20). Vertical bilayers (80–120 picofarads) were formed by painting the lipid mix across a 200-μm diameter hole in a deldrin cup (Warner Instruments, Hamden, CT).
Fusion between membrane preparation vesicles and the bilayer was promoted by osmosis, with the cis chamber (to which the membrane preparation was added) being hyperosmotic to the trans chamber solution. Electrophysiological recording solutions consisted of the following: cis, 300 mm KCl, 10 mm HEPES, 1 mm HEDTA, 2 mm CaCl2 (free Ca2+ ≅ 1 mm), pH 7.2; trans, 30 mm KCl, 10 mm HEPES, 1 mm HEDTA, 2 mm CaCl2 (free Ca2+ ≅ 1 mm), pH 7.2. Nominal free Ca2+ in solution was calculated using the MaxChelator Sliders program (C. Patton, Stanford University) and validated experimentally as described elsewhere (30). The trans chamber was held at ground while the cis chamber was held at potentials relative to ground. Only channels with their intracellular Ca2+-sensors oriented toward the cis chamber were considered for experimentation.
Ion currents were obtained during 2–3 min of continuous recording at 0 mV using a Warner BC-525D amplifier, low pass-filtered at 1 kHz using the 4-pole Bessel filter built into the amplifier, and sampled at 5 kHz with Digidata 1322A/pCLAMP 8 (Molecular Devices, Sunnyvale, CA). For proper comparisons with previous data obtained by us (17, 20, 21, 25) and others (19, 22, 31), studies were conducted at room temperature (20–25 °C).
We used Po as an index of channel steady-state activity. Po was calculated using a built-in routine in Clampfit 9.2 (Molecular Devices). Data plotting and further analysis were conducted using Origin 7.0 (Originlab, Northampton, MA) and InStat 3.0 (GraphPad, La Jolla, CA).
Chemicals
CLR, POPS (sodium salt), and POPE were purchased from Avanti Polar Lipids (Alabaster, AL). All other chemicals and reagents were purchased from Sigma (St. Louis, MO).
Statistical Analysis
Groups of data are shown as mean ± S.E. Normal distribution of data were determined by the Kolmogorov-Smirnov's test. Multicomparisons across different protein constructs in the absence or presence of CLR were conducted with analysis of variance, followed by Bonferroni's test to determine statistical difference between individual means.
RESULTS
Cholesterol Sensitivity of BK Channels Requires Their Cytosolic Tail Domain
CLR-sensing regions in ion channel proteins have been attributed to transmembrane segments (e.g. acetylcholine nicotinic (AchNic) receptors (2, 3) and TRPV1 channels (32)) as well as cytosolic C tail regions (e.g. mitochondrial translocator (TSPO) proteins (33) and 2TM inwardly rectifying K+ (Kir) channels (5, 34)). Then, to begin to identify the Cbv1 region(s) that confers CLR sensitivity to BK channels, we first compared bilayer CLR action on the activity of BK channel-forming subunits cloned from rat cerebral artery myocytes (WT Cbv1) versus the construct Trcbv1S6, which was engineered by truncation immediately after S6 (i.e. between Ile-322 and Ile-323). Trcbv1S6 retains the basic S1–S6 common to all KVs channels of the TM6 superfamily and the NH2-S0 end characteristic of Slo1 (including Cbv1) while lacking the long CTD (Fig. 1a), another characteristic feature of Slo1 channels (18, 35). Single channel protein function was evaluated under identical recording conditions after channel reconstitution into POPE and POPS 3:1 (w/w) bilayers. In 6 of 6 experiments, WT Cbv1 Po in the presence of 33 mol % CLR was consistently decreased by ∼30% when compared with that in control, CLR-free bilayers (Fig. 1, b, d, and e) (p = 0.0002). This CLR action was neither dependent on activating intracellular Ca2+ (supplemental Fig. S1) nor accompanied by any noticeable change in channel unitary current amplitude (Fig. 1b). In this bilayer type, WT Cbv1 Po was also significantly decreased by ∼20 and 25% (p = 0.0002) in response to bilayer introduction of 16 and 25 mol % CLR, respectively (Fig. 2c). Decreased WT Cbv1 Po in the presence of membrane CLR at molar fractions found in cell membranes is consistent with previous findings from recombinant human SLO1 (20) and WT Cbv1 (25) channels reconstituted into the same binary bilayer, native rat brain BK channels reconstituted into POPE/POPS (55:45) (w/w) bilayers (19), and native BK channels in myocyte membranes exposed to CLR-depleting treatment (22).
FIGURE 2.

The CRAC4-containing CTD region is necessary for BK channel activity to respond to membrane cholesterol. a, scheme showing the cholesterol recognition/interaction amino acid consensus (CRAC) motifs identified along the WT Cbv1 sequence, which have been numbered 1–10 from the Cbv1 N end. Zigzag lines indicate points of truncation. b, single channel records from Trcbv1-CRAC4 constructs. c, averaged inhibition of WT Cbv1 and Trcbv1-CRAC4 channel activity by 16 and 25 mol % CLR. *, different from CLR-induced inhibition of WT Cbv1 (p = 0.0016 and p = 0.0001 for 16 and 25 mol % CLR, respectively). Averaged CLR-induced reduction of activity for different constructs in the presence of 16 (d) and 25 mol % CLR (e) when compared with CLR inhibition of WT Cbv1 channel activity *, significantly different from CLR-induced inhibition in WT Cbv1: Trcbv1S6 (p = 0.0001, p = 0.0001), Trcbv1-C8 (p = 0.026, p = 0.002), Trcbv1-C6 (p = 0.029, p = 0.033), and Trcbv1-C4 (p = 0.002, p = 0.0001); p values are for 16 and 25 mol % CLR. n values are shown in parentheses. Error bars, S.E.
Reconstitution of Trcbv1S6 channels into POPE/POPS (3:1) (w/w) bilayers resulted in a functional receptor that retained basic features of BK channels, including increased Po with positive transmembrane voltage and blockade by paxilline, a selective BK channel blocker (supplemental Fig. S2). However, as expected from a Slo1 construct that lacks the long C tail (36, 37), Trcbv1S6 channels expressed very poorly in cell membranes and incorporated into the bilayer with much more difficulty than WT Cbv1. In addition, probably because the Ca2+-sensing regions associated with “regulatory of conductance for potassium” (RCK) domains were missing (Fig. 1a), the Ca2+ sensitivity of Trcbv1S6 channels was very low, typically requiring 1 mm internal free Ca2+ (Fig. 1c) to raise Po to values comparable with those of WT Cbv1 under physiological micromolar Ca2+, where the effect of a channel inhibitor (i.e. CLR) could be evaluated. Finally, when evaluated at 0 mV in 300/30 mm intracellular K+/extracellular K+, Trcbv1S6 channels displayed a unitary current amplitude smaller than that of WT Cbv1 (Fig. 1, compare c with b). This reduced current amplitude is probably related to the lack of the two RCK domains in Trcbv1S6 (see Fig. 1a and also Refs. 18 and 38).
In contrast to WT Cbv1 channels, Trcbv1S6 Po in CLR-containing bilayers was indistinguishable from that in CLR-free bilayers (Fig. 1, c and d). Moreover, Trcbv1S6 channels failed to respond to a physiologically relevant range of CLR concentrations in the bilayer: 16, 25 mol % (Fig. 2, d and e) and 33 mol % (Fig. 1, c–e). These data demonstrate that the BK region expanding from the N terminus to the channel TM “core” (S0–S6) is not sufficient to empower the channel with its characteristic CLR sensitivity. Rather, the cytosolic tail of WT Cbv1 is necessary for CLR sensing by BK channels. The fact that Trcbv1S6 channels were still able to activate with positive voltage and/or internal Ca2+ (albeit at nonphysiological levels) suggests that their CLR insensitivity is not the consequence of a nonselective disruption of their gating machinery caused by CTD elimination.
A Cholesterol Recognition/Interaction Amino Acid Consensus in Proximal Region of CTD Emboldens BK Channels with CLR Sensitivity
We next decided to pinpoint possible CLR-interacting domains in the Cbv1 sequence that could contribute to the differential CLR sensitivity of WT Cbv1 and Trcbv1S6. A wide variety of structurally unrelated domains that participate in CLR recognition have been identified in both cytosolic and transmembrane proteins (2, 3, 5, 39, 40). However, we initially focused on the CRAC motif because 1) it is defined by a relatively lax sequence (-(L/V)X1–5YX1–5(R/K)) (4), and 2) a CRAC domain in a cytosolic region participates in CLR sensing by another ionotropic receptor, the mitochondrial translocator protein TSPO (formerly known as peripheral benzodiazepine receptor) (41). Primary sequence analysis of WT Cbv1 from its N end to C tail revealed 10 CRAC motifs (termed CRAC1 to 10 from the N to C end location; see Fig. 2a). CRAC1 to 3 are located in the channel transmembrane core and, thus, are common to WT Cbv1 and Trcbv1S6, whereas CRAC4 to 10 are located in the CTD and, thus, absent in Trcbv1S6.
The presence of a CRAC motif in a protein or peptide, however, is not sufficient for either CLR recognition or CLR modulation of receptor activity, requiring experimental validation (4, 39). Because CLR presence in cell membrane but not in cytosolic solution effectively reduces BK channel activity (7), and CRAC4 is very likely the CTD CRAC motif closest to the membrane, we first evaluated the CLR sensitivity of Trcbv1-CRAC4, a construct that was truncated downstream to CRAC4 (i.e. the CRAC located at the proximal end of Cbv1 CTD). Similar to the Trcbv1S6 construct (Fig. 1a), Trcbv1-CRAC4 lacks the CTD RCKs (Fig. 2a) that embolden BK channels with sensitivity to physiological levels (low micromolar) of internal Ca2+ (18, 35, 42–44). Thus, Trcbv1-CRAC4 responses to CLR were evaluated in the presence of [Ca2+]i = 1 mm, as done with Trcbv1S6. In sharp contrast to Trcbv1S6, the Trcbv1-CRAC4 channel was highly sensitive to the presence of CLR: Po decreased by 37 and 39% from its value in CLR-free bilayers when bilayer CLR reached 16 and 25 mol %, respectively (Fig. 2, b and c). Moreover, the CLR sensitivity of Trcbv1-CRAC4 was higher than that of WT Cbv1 (Fig. 2b), the reduction of Po reaching ∼150% of steroid action on WT Cbv1 (Fig. 2, d and e). These data indicate that a short CTD including CRAC4 emboldens Trcbv1-CRAC4 channels with remarkable CLR sensitivity.
To further test the role of CTD in CLR sensitivity, we introduced a series of truncations downstream of CRAC4, removing two CRACs at a time: immediately distal to CRAC6 (Trcbv1-CRAC6 construct) and to CRAC8 (Trcbv1-CRAC8 construct) (Fig. 2a). In contrast to Trcbv1S6 and similar to WT Cbv1 and Trcbv1-CRAC4, both Trcbv1-CRAC6 and Trcbv1-CRAC8 channels were CLR-sensitive (Fig. 2, d and e), supporting the notion that CRAC4 and/or the linker between this domain and S6 is necessary in providing CLR sensitivity to BK channels. However, deletion of CTD regions that included CRAC7 to 10 significantly (p = 0.029 and p = 0.033) reduced the CLR response of BK channel-like constructs: Trcbv1-CRAC6 Po inhibition in response to 16 and 25% mol % CLR was reduced to 52 and 59% of the inhibition evoked by these CLR levels on WT Cbv1 (Fig. 2, d and e). On the other hand, deletion of CTD regions that include CRAC9 and 10 in CTD also reduced the CLR response of BK-like constructs: Trcbv1-CRAC8 Po inhibition in response to 16 and 25 mol % CLR was reduced to 63 and 67% of the inhibition evoked by these CLR levels on WT Cbv1 (p = 0.026 and p = 0.002) (Fig. 2, d and e). Collectively, these data indicate that although CRAC4 and/or the CRAC4-S6 linker is a key determinant of BK channels' CLR sensitivity, Cbv1 CTD sequences distal to CRAC4 also contribute to the overall CLR sensitivity of this family of channels (see “Relative contribution of other CRACs in Cbv1 CTD to the CLR sensitivity of BK channels” and “Discussion”).
To determine whether Trcbv1-CRAC4 sensitivity to CLR is indeed determined by the CRAC motif, we took advantage of the structure-activity relationship of CRAC domains in model peptides that interact with membrane CLR (4, 39) and introduced systematic point mutations to Trcbv1-CRAC4, rendering a variety of channel constructs where CLR sensitivity was probed (Fig. 3a). First, the conservative substitutions Leu-Val and Lys-Arg in CRAC do not usually alter CLR interaction with the CRAC-containing protein (4, 39). Indeed, our data show that Trcbv1-CRAC4 K453R channels were as CLR-sensitive as their Trcbv1-CRAC4 counterparts (Fig. 3b). In contrast, nonconservative substitution of basic Lys in CRAC4 with the nonpolar and shorter Ala rendered Trcbv1-CRAC4 K453A channels, which were totally insensitive to CLR (Fig. 3b). These data underscore the importance of a long basic residue at the C-terminal end of the CRAC4 motif in determining the CLR sensitivity of BK-like Trcbv1-CRAC4 channels.
FIGURE 3.

Modification of BK channel CLR sensitivity by amino acid substitutions in Cbv1 CRAC4. a, primary alignment of the various constructs engineered by point mutagenesis that targeted signature residues in CRAC4 and the linker between S6 and CRAC4. Amino acids within CRAC4 and the S6-CRAC4 linker region that have been targeted by point mutagenesis are shown in red, with their replacements given in blue. b, averaged CLR-induced inhibition of activity observed in Trcbv1-CRAC4 constructs, including different amino acid point substitutions and compared with CLR-induced inhibition of Trcbv1-CRAC4. *, different from CLR-induced inhibition of Trcbv1-CRAC4: Trcbv1-CRAC4 Y450F (p = 0.0001); Trcbv1-CRAC4 V444A (p = 0.0001); Trcbv1-CRAC4 K453A (p = 0.0023). c, single channel records showing CLR modulation of channel activity from constructs where Phe substituted for the signature central Tyr in CRAC4 (Trcbv1-CRAC4 Y450F) and Y429 in the S6-CRAC4 linker (Trcbv1-CRAC4 Y429F). n values are shown in parentheses. Error bars, S.E.
It is noteworthy that the substitution of Val-444 with the shorter Ala resulted in Trcbv1-CRAC4 channels that were resistant to CLR-induced inhibition (Fig. 3b). This result also supports the involvement of a CRAC sequence in CLR action on BK channels because Val or Leu (but not Ala) at the N-terminal end of the CRAC motif is usually required for CLR recognition and/or sequestration (4, 39). Finally, we probed whether Tyr-450, the central residue in CRAC4 of Trcbv1 contributed to the overall CLR sensitivity. A central Tyr seems to be a CRAC signature (4), this residue playing a significant role in CLR modulation of ionotropic TSPO proteins (41). Thus, we next probed the CLR sensitivity of Trcbv1-CRAC4 Y450F, where Phe matched the overall volume of Tyr. Underscoring the unique role of Tyr in CLR-sensing, Trcbv1-CRAC4 Y450F channels displayed comparable Po in CLR-containing and CLR-free bilayers (Fig. 3, b and c). In sharp contrast, a Tyr to Phe substitution in the short linker between S6 and CRAC4 (position 429; Fig. 3a) consistently failed to alter the CLR sensitivity of Trcbv1-CRAC4 channels (Fig. 3, b and c). These results indicate that a Phe substitution of the central, signature Tyr inside CRAC4 but not of a nearby Tyr outside CRAC4, abolishes CLR sensitivity of BK-like Trcbv1-CRAC4 channels. Collectively, our mutagenesis studies demonstrate that the CLR sensitivity of BK-like Trcbv1-CRAC4 channels is determined by the key residues that define a CRAC motif.
To explain the differential role of Tyr and Phe in CLR recognition by CRAC4 and identify the intervening bonds involved in CLR-ion channel recognition, we conducted computational MD studies. Full-length WT Cbv1 CTD, Trcbv1 CTD-CRAC4, Trcbv1 CTD-CRAC4 Y450F, and Trcbv1 CTD-CRAC4 K453A constructs were simulated in triplicates for 5 ns each, giving a 15-ns total simulation time for each construct. All simulations started with CLR in the same position relative to the protein (Fig. 4a). For each of the atoms selected on CLR (Fig. 4b), the percentage of simulation time spent near residues of the protein (i.e. residence time) was calculated using the aggregate results from the three trajectories. Table 1 shows contacts between specific atoms of CLR and protein residues with the highest residence percentages.
FIGURE 4.

Cholesterol and its residence sites within Cbv1 CTD-CRAC4 and vicinity. a, representative snapshot of CLR starting position for MD simulations. Cholesterol is shown in green; Trcbv1 CTD-CRAC4 backbone is shown as a blue ribbon; WT full-length Cbv1 CTD is composed of yellow and blue ribbon structures. b, cholesterol structure with selected atoms labeled as presented under “Results.” c, representative snapshot of MD simulation of CLR interaction with WT Cbv1 CTD shows that the CLR molecule is significantly drifted away from its starting point, yet it remains in the vicinity of the CRAC4 domain. Representative snapshots of MD simulation of CLR interaction with Trcbv1 CTD-CRAC4 (d) and Trcbv1 CTD-CRAC4 Y450F (e) show that CLR hydroxyl hydrogen bonds with Lys-453. f, representative snapshot of MD simulation of CLR interaction with Trcbv1 CTD-CRAC4 K453A showing the CLR molecule drifted away from position 453, with the CLR steroid nucleus in the vicinity of Tyr-450.
TABLE 1.
Residence times (in percentage) for selected CLR atoms on various Cbv1 CTD constructs
| CLR atoms | WT Cbv1 CTD |
Trcbv1 CTD-CRAC4 |
Trcbv1 CTD-CRAC4 Y450F |
Trcbv1 CTD-CRAC4 K453A |
||||
|---|---|---|---|---|---|---|---|---|
| Residue | Percentage | Residue | Percentage | Residue | Percentage | Residue | Percentage | |
| % | % | % | % | |||||
| C19 | Val-339 | 10.97 | His-451 | 39.89 | His-451 | 61.81 | Pro-452 | 20.19 |
| C18 | Glu-417 | 15.23 | His-451 | 43.22 | Phe-450 | 74.03 | Tyr-450 | 21.51 |
| H17 | His-451 | 52.99 | Glu-417 | 43.59 | Glu-417 | 30.29 | Tyr-450 | 65.45 |
| H25 | Tyr-450 | 31.01 | Tyr-450 | 31.65 | Tyr-450 | 43.31 | Pro-408 | 46.84 |
| H3 | Lys-453 | 35.61 | Val-339 | 16.77 | Lys-453 | 16.93 | Glu-417 | 56.99 |
| OH | Lys-453 | 14.46 | Lys-453 | 13.54 | Lys-453 | 27.43 | Glu-417 | 32.90 |
| H9 | His-451 | 48.37 | Glu-417 | 40.97 | Glu-417 | 16.33 | Glu-417 | 57.98 |
The location of the residues in Table 1 and their associated residence times demonstrate similarities between the WT Cbv1 CTD and Trcbv1 CTD-CRAC4 simulations (supplemental Movies S1 and S2) as well as differences between these and the Y450F and K453A mutant simulations (supplemental Movies S3 and S4) (see also Fig. 4, c–f). In the WT Cbv1 CTD and Trcbv1 CTD-CRAC4 simulations, H25 in the lateral chain of CLR showed a lower residence time near Tyr-450 when compared with Trcbv1 CTD-CRAC4 Y450F; CLR H25 in WT Cbv1 CTD and Trcbv1 CTD-CRAC4 simulations spent ∼31% of the time near Tyr-450, whereas this residence time is substantially increased in Trcbv1 CTD-CRAC4 Y450F, reaching 43% (Table 1). Additionally, in both WT Cbv1 CTD and Trcbv1 CTD-CRAC4 simulations, H17 of CLR showed the lowest mobility, which was evidenced by residence times (52.99 and 43.59%, respectively) greater than the other CLR atoms tracked. In contrast, CLR H17 in Trcbv1 CTD-CRAC4 Y450F simulations was not the point of lowest mobility. Highest residence times of this construct were detected for C18 and C19 of CLR at 74.03 and 61.81%, respectively (Table 1). Overall, the Trcbv1 CTD-CRAC4 Y450F simulations showed a lower mobility of the CLR molecule when compared with WT Cbv1 CTD and Trcbv1 CTD-CRAC4 simulations. The entropic penalty (45) associated with this decreased CLR mobility could contribute to the lack of CLR sensitivity observed experimentally with the Trcbv1 CTD-CRAC4 Y450F ion channel construct. Moreover, both WT Cbv1 CTD and Trcbv1 CTD-CRAC4 simulations, which correspond to CLR-sensitive ion channel constructs, showed more CLR mobility when compared with those from mutants; this is evidenced by the lack of any single residence time in excess of 53% for WT Cbv1 CTD and Trcbv1 CTD-CRAC4 constructs. In contrast, longer residence times for several positions are found for both Trcbv1 CTD-CRAC4 Y450F and Trcbv1 CTD-CRAC4 K453A (Table 1), their corresponding Trcbv1 channel constructs showing blunted CLR sensitivity.
However, Trcbv1 CTD-CRAC4 K453A simulations showed CLR interactions different from all other simulations, with the CLR steroid nucleus residing more frequently near Tyr-450 and Glu-417 (Table 1 and supplemental Movie S4). This behavior may be explained by the loss of transient hydrogen bonding between CLR hydroxyl and Lys-453, which is observed in both Trcbv1 CTD-CRAC4 and Trcbv1 CTD-CRAC4 Y450F. This hydrogen bonding would help to keep the CLR molecule closer to Lys-453 (supplemental Movies S2 and S3). Indeed, simulations of Trcbv1 CTD-CRAC4 K453A do not show hydrogen bonding interactions between CLR and residue 453, and in all three triplicates, CLR consistently inverted from its starting position (Fig. 4a) to have its methyl groups facing away from the CRAC domain. The CRAC4 K453A simulations further demonstrate the shifted position of the sterol with H25 (an atom in the hydrophobic tail of CLR) residing near Pro-408 for 46.84% of simulation time (Table 1). Thus, the repositioning of the CLR molecule that occurs early in the MD simulations (supplemental Movie S4) and the entropic penalty associated with a decreased CLR mobility could both contribute to the lack of CLR sensitivity observed in the Trcbv1-CRAC4 K453A channel construct.
In addition to overall mobility and positioning of the CLR molecule, MD data provide atomistic insights on intervening bonds that participate in CLR recognition by BK channels. First, the importance of the CLR single hydroxyl group in monohydroxysterol inhibition of BK channel activity (25) raises the question of whether hydrogen bonds between CLR hydroxyl and Cbv1 CRAC residues participate in CLR inhibition of channel activity. In both WT Cbv1 CTD and Trcbv1 CTD-CRAC4 simulations, CLR hydroxyl spent little time at any given residue under investigation, with a highest residence time of 14% (Table 1). In contrast, this time reached 27 and 33% in Trcbv1 CTD-CRAC4 Y450F and Trcbv1 CTD-CRAC4 K453A. It is also noteworthy that the residence times for C18 and C19 in the WT Cbv1 CTD construct were low: 15.23 and 10.97%, respectively. This indicates that the CLR methyl groups spent little time near CRAC domain residues. This change in position for CLR in WT Cbv1 CTD simulations gives unfavorable angles and distances for hydrogen bonding between CLR hydroxyl and Lys-453. Indeed, the hydrogen bond at this position is hardly observed in WT Cbv1 CTD simulations (supplemental Movie S1). Considering that the corresponding ion channel construct is CLR-sensitive, hydrogen bonding is not necessary to support CLR sensitivity of full-length WT Cbv1. In addition, hydrogen bonding between CLR and Lys-453 is observed at different time points of MD simulation of the CLR-insensitive Trcbv1 CTD-CRAC4 Y450F construct (supplemental Movie S3). Thus, hydrogen bonding is not sufficient to support CLR sensitivity of Trcbv1 channels.
Notably, transient hydrogen bond formation between CLR and Cbv1 is observed in the CLR-sensitive Trcbv1 CTD-CRAC4 (supplemental Movie S2). This bonding, however, is required to position the CLR molecule in the vicinity of CRAC motif-forming residues because loss of this hydrogen bond in the K453A mutant resulted in the CLR molecule drifting away from CRAC4 (see above). In addition to differential hydrogen bonding contributions, the hydrophobic interactions between the Lys side chain and CLR, which were reduced in Trcbv1 CTD-CRAC4 K453A, could contribute to the difference in CLR positioning found in the K453A construct and described above.
Collectively, the MD simulations provide atomic insights relevant to the experimentally observed differences in CLR sensitivity for the Cbv1 constructs. First, simulations of both Trcbv1 CTD-CRAC4 Y450F and Trcbv1 CTD-CRAC4 K453A show reduced CLR mobility when compared with simulations of WT Cbv1 CTD and Trcbv1 CTD-CRAC4, a decreased CLR mobility probably providing an entropic penalty disfavoring CLR binding. Second, the loss of a hydrogen bonding partner at position 453 in Trcbv1 CTD-CRAC4 K453A alters the position of CLR on the CRAC4 domain. Finally, the increased CLR sensitivity of Trcbv1 CTD-CRAC4, when compared with that of full-length WT Cbv1 CTD, could be explained by the loss of the Lys-453 hydrogen bonding partner Glu-479 (Fig. 4a). Consistent with the latter interpretation, Lys-453 is more mobile in Trcbv1 CTD-CRAC4 due to the absence of the hydrogen bond between these two residues. This enhanced mobility increases the number of transient hydrogen bonding events between CLR hydroxyl and Lys-453, which consequently delays the CLR inversion from its starting position in the simulations (supplemental Movie S2) and thus enforces the presence of the CLR molecule in the vicinity of CRAC4 domain, eventually favoring CLR sensitivity of the channel protein. A possible role of hydrogen bonding versus other structural determinants in the different quantitative effect of the T450F substitution on CLR responses of truncated versus full-length Cbv1 channels, however, remains to be experimentally determined.
Y450F Substitution in CRAC4 Blunts Cholesterol Sensitivity of Full-length BK Channels
After establishing the critical role of CRAC4 and its Tyr-450 in providing CLR sensitivity to BK-like Trcbv1-CRAC4 channels, we decided to probe whether this central Tyr, a signature for CRAC motifs (4, 39), could also be a major contributor to the CLR sensitivity of full-length WT Cbv1 channels. Full-length Cbv1 Y450F channels were mildly inhibited by CLR presence in the bilayer, even when CLR concentrations were raised to 25–33 mol % (Fig. 5a). This result was replicated in several independent bilayers (Fig. 5b). Indeed, CRL inhibition of full-length Cbv1 Y450F was significantly smaller (p = 0.0244) than that observed with WT Cbv1 (Fig. 5b). These data indicate that the Tyr to Phe substitution in CRAC4 effectively blunts CLR inhibition of full-length Cbv1 channels. Interestingly, the effect of the Y450F substitution in blunting the CLR sensitivity of full-length Cbv1 (Fig. 5b) was less robust than the effect of the same substitution on Trcbv1-CRAC4 channels (Fig. 3, b and c), which totally blunted the CLR sensitivity of the truncated channel. These findings are consistent with those from Fig. 2, d and e, underscoring that regions distal to CRAC4 contribute to the overall CLR sensitivity of BK channels.
FIGURE 5.
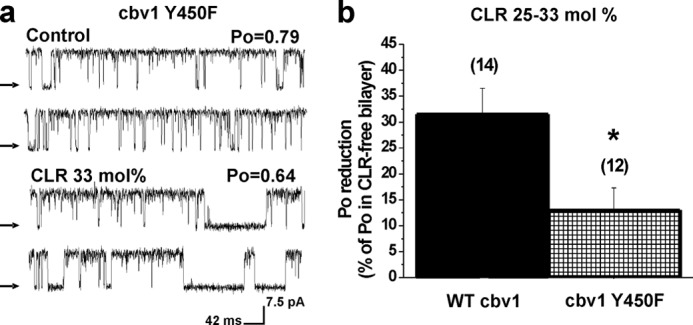
Phenylalanine substitution of Tyr-450 in full-length Cbv1 significantly reduces BK channel sensitivity to membrane cholesterol. a, single channel records from full-length, Cbv1, Y450F channels in CLR-free and 25–33 mol % CLR-containing bilayers. b, averaged inhibition of WT Cbv1 versus Cbv1, Y450F channel activity by 25–33 mol % CLR. *, different from CLR-induced inhibition of WT Cbv1 (p = 0.0244). n values are shown in parentheses. Error bars, S.E.
Contribution of CTD CRACs Distal from CRAC4 to CLR Sensitivity of BK Channels
After documenting that 1) the signature Tyr is critical for the CLR sensitivity of both full-length and Trcbv1-CRAC4 channels (Figs. 3 and 5) and 2) regions distal to CRAC4 contribute to the overall sensitivity of BK channels (Fig. 2, d and e), the next step was to determine whether CRAC motifs in WT Cbv1 CTD that are distal to CRAC4 actually contribute to the overall CLR sensitivity of BK channels. Because 1) a central Tyr is a signature of CRAC sequences (4) and 2) we found that a Tyr to Phe substitution in CRAC4 drastically blunted the CLR sensitivity of BK channels (Figs. 3 (a–c) and 5), we tested the aforementioned hypothesis by introducing Tyr to Phe substitutions in two CRACs distal to CRAC4 at a time, mimicking the truncation strategy that we followed previously (Figs. 2a and 6a).
FIGURE 6.
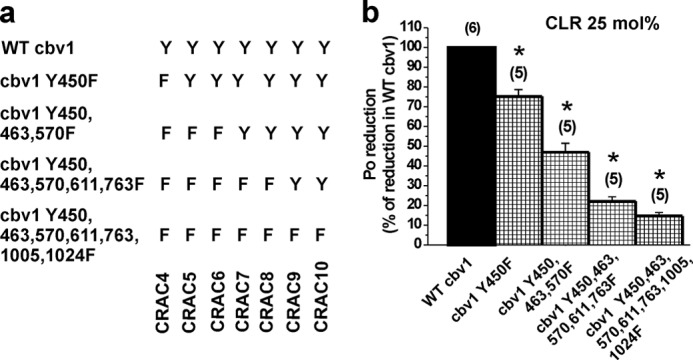
Cumulative substitution of Tyr to Phe in full-length Cbv1 gradually reduces CLR action. a, schematic representation of Tyr to Phe substitutions throughout Cbv1 CTD CRACs (i.e. CRAC4 to 10). b, averaged CLR-induced inhibition of activity in Cbv1 channel constructs containing Tyr to Phe substitutions compared with CLR-induced inhibition of WT Cbv1. *, different from CLR-induced inhibition in WT Cbv1: Cbv1 Y450F (p = 0.026); Cbv1 Y450F,Y463F,Y570F (p = 0.0006); Cbv1 Y450F,Y463F,Y570F,Y611F,Y763F (p = 0.0006); Cbv1 Y450F,Y463F,Y570F,Y611F,Y763F,Y1005F,Y1024F (p = 0.0001). n values are shown in parentheses. Error bars, S.E.
The resulting full-length channel constructs with one or more central Tyr to Phe substitutions within a CRAC domain distal to CRAC4 showed some CLR sensitivity but a much smaller sensitivity than what is characteristic of WT Cbv1 (Fig. 6b). Remarkably, the figure shows that the more Tyr to Phe substitutions we introduced within the CTD CRACs, the more BK channel refractoriness to CLR modulation resulted. Furthermore, Cbv1-Y450F,Y463F,Y570F,Y611F,Y762F,Y1005F,Y1024F was barely sensitive to CLR (Fig. 6b), a refractoriness that was close to the CLR-resistant Trcbv1S6 (Fig. 1e). Collectively, these data and the results shown previously indicate that although Tyr-450 and CRAC4 are the major determinants of CLR sensitivity of BK channels, Tyr residues located in CRACs distal to CRAC4 also contribute to CLR action on BK channel activity.
DISCUSSION
The combination of MD, site-directed mutagenesis, and single channel electrophysiology on recombinant BK channel-forming Cbv1 protein allowed us to determine for the first time the structural bases of CLR inhibition of BK channels, which involve identification of CLR-sensing regions in the Cbv1 protein, advance of a CLR-Cbv1 CRAC4 docking model, and unveiling of the chemical interactions between sterol ligand atoms and ion channel receptor residues that lead to modification of BK channel function.
Confirming previous studies (20, 22, 25), the present data document that BK channel activity reduction by CLR at molar fractions found in natural membranes does not require cell organelles, cytosolic signals, channel accessory proteins, or a complex lipid environment. Rather, BK channel-forming Cbv1 subunits and a two phospholipid species suffice. Thus, the actual location of the CLR-sensing region in the Cbv1 protein was the first question we addressed. Data from truncated Cbv1 constructs and Tyr to Phe substitutions within Cbv1 CRAC4 to 10 demonstrate that 1) the CLR-sensing region of the Cbv1 protein is contained within the CTD (Fig. 1, a–e), 2) all CRACs in the Cbv1 CTD contribute to the overall CLR sensitivity of the channel, and 3) in the absence of more distal CRACs, CRAC4 is sufficient to provide CLR sensitivity to BK channels (Figs. 2 and 6).
CLR-sensing regions have been identified in several ion channel proteins, including TRPV1 (32), AchNic receptors (2, 3), and Kir2.1 channels (34). TRPV1 and Cbv1 are structurally and functionally related; both channel-forming proteins belong to the TM6 superfamily of ion channels, arrange in homotetrameric complexes to conform functional ion channels, and behave as outward rectifiers (9, 43). However, CLR-sensing regions in TRPV1 and Cbv1 clearly differ; in the former, the CLR docking site has been mapped to a groove between transmembrane S5 and the voltage-sensing domain of an adjacent subunit (32), whereas in Cbv1, the CLR-sensing region(s) that leads to modification of channel activity is 1) of cytosolic topology (CRAC4 to 10) and 2) probably self-contained within a single Cbv1 monomer. Regarding AChNic receptors, at least 15 CLR sites have been mapped, yet all of them are located within TM regions. Moreover, in the absence of CLR, the AchNic receptor does not gate appropriately (2). Although we identified CLR recognition sequences in the Cbv1 TM6 core (i.e. CRAC1 to 3; Fig. 2a), the CLR insensitivity of Trcbv1S6 (Fig. 1) indicates that CRAC1 to 3 are not sufficient to provide CLR sensitivity to BK channels. Whether CLR bound to Cbv1 CRACs1–3 is necessary to support basic BK channel gating remains to be formally tested. However, it is interesting to point out that Cbv1 and other Slo1 channels are fully operational in terms of gating and conduction when reconstituted into artificial, CLR-free phospholipid bilayers (20, 25, 31) (Fig. 1b, top two traces), where any possible contaminant CLR is probably diluted into the bulk bilayer phospholipid (20, 46). Finally, as found for the 2TM Kir2.1 channel (34), Cbv1 CLR-sensing regions are mapped to the channel cytosolic C tail. The proposed residues involved in CLR sensing by Cbv1 CTD follow the general sequence that defines a CLR consensus motif: [4.39–4.43(R/K)]-[4.50(Trp/Tyr)]-[4.46 (I/V/L)]-[2.41(Phe, Tyr)] (according to the Ballesteros-Weinstein nomenclature), which is not found in the CLR-sensing cytosolic region of Kir2.1 channels (34). Thus, it seems that the presence of seven CRACs in the long Cbv1 CTD as determinants of ion channel CLR sensitivity represents a distinct structural feature of Slo1/Slo proteins, which could explain the high CLR sensitivity of BK channels reported across cell types and species (7).
Several proteins known to bind CLR, however, do contain the CRAC sequence. These include the HIV1 transmembrane protein gp41 (47), the σ1 receptor (48), TSPO (33, 41), and the peripheral myelin P0 protein (49, 50). CRAC sequences in the σ1 receptor and the myelin P0 protein are located at the cytosolic membrane inner leaflet interface (48, 49). Likewise, the sequence LWYIK, which is critical for HIV gp41 recognition of CLR, is located immediately adjacent to the protein TM region (47). CRAC sequences have been mapped next to the last TM domain in MAG, plasmolipin, and PMP22 proteins (51), although their participation in CLR modification of protein function remains to be determined. Comparison of CLR sensitivity between WT Cbv1, full-length Cbv1 Y450F, Trcbv1-CRAC4 Y450F, and Trcbv1-CRAC4 K453A (Figs. 3 and 5) points to CRAC4 as a critical contributor to the overall CLR sensitivity of Cbv1 channels. Crystallographic data map Slo1 CRAC4 at the N end of the CTD (35, 44), a location that is similar to the TSPO protein CRAC sequence (41). Thus, the protein location and critical importance of CRAC4 in regulating overall CLR sensitivity of BK channels seems to fit a widespread design in transmembrane proteins, where CRAC domains are located in close proximity to the cell membrane inner leaflet and thus may be particularly important in mediating CLR modification of protein function. Moreover, we mapped CRAC motifs immediately distal to the last TM sequence in human Kv1.3, Kv1.5, and Kv11.1 and rat Kv2.1, all of these constructs reported as CLR-sensitive (5, 52). Therefore, the critical role of a CRAC motif immediately distal to the last TM sequence in determining ion channel sensitivity to membrane CLR may not be restricted to BK channels but could be expanded to other members of the TM6 superfamily of ion channels.
Consistent with previous data obtained with short model peptides that interact with membrane CLR (4, 39), our point mutagenesis and MD data from a complex, oligomeric transmembrane ionotropic receptor indicate that the three positions required by the CRAC4 motif to sense the presence of CLR are Leu/Val, Tyr, and Lys/Arg (Figs. 3 and 4). In TSPO, the CLR-interacting residues are Val, Tyr, and Arg (41), whereas Val, Tyr, and Lys appear in the Cbv1 CRAC4 (Fig. 3a) sequence, both sequences satisfying the criteria for the CLR consensus motif (see above). Accordingly, the K453R substitution in CRAC4 does not alter the CLR sensitivity of Cbv1 channels (Fig. 3c).
The fact that the Y450F substitution in CRAC4 alone and/or CRACs distal to CRAC4, but not in nearby regions outside a CRAC domain (Figs. 3, 5, and 6), drastically decreases the CLR sensitivity of truncated and full-length BK channels underscores the critical role of the central Tyr in CRAC sequences for CLR recognition. It has been previously reported with myelin P0 proteins that the substitution of LFYLIR with LFSLIL in the cytosolic end CRAC disrupts protein-CLR interactions (50). Likewise, substitution of CRAC central Tyr to Ser and Pro alters CLR interactions with TSPO protein (41) and Aggregatibacter actinomycetemcomitans cytolethal distending toxin (53), respectively. In contrast to these nonconservative substitutions, we substituted Tyr-450 in Cbv1 CRAC4 with Phe, which has a lateral side volume similar to that of Tyr. The drastic effect of this conservative substitution on BK channel CLR sensitivity is explained by MD data. First, in both full-length WT Cbv1 and Trcbv1 CTD-CRAC4, but not in the Trcbv1 CTD-CRAC4 Y450F construct, H25 in the CLR lateral chain shows a high residence near Tyr-450 (Fig. 4b and Table 1). Second, simulations of Y450F constructs show decreased mobility of CLR (Table 1), which is very likely to provide an entropy penalty (45) for CLR presumed binding.
In agreement with models of CLR interaction with model peptides and TSPO proteins (4, 39, 41), WT Cbv1 CTD and Trcbv1 CTD-CRAC4 simulations demonstrate the importance of a positively charged Arg/Lys residue, which acts as a hydrogen bond acceptor for the 3β-hydroxyl group of CLR. The increased mobility of Lys-453 in truncated constructs could explain the experimentally observed increase in the CLR sensitivity of Trcbv1 CTD-CRAC4 compared with WT by allowing more transient hydrogen bonding events between CLR and Lys-453, as observed in the Trcbv1 CTD-CRAC4 simulations. However, our model does not display the previously proposed CLR 3β-hydroxyl group hydrogen bonding with the hydroxyl of Tyr (4, 39). Additionally, in the CLR-recognizing motif of the β2 adrenergic receptor, an aromatic amino acid has been proposed to interact with the D ring of CLR (28), but our model shows that Tyr-450 interacts with the aliphatic chain more than with the D ring of CLR, except in the Trcbv1 CTD-CRAC4 K453A simulations.
A remarkable finding from our study is that cumulative substitution of central Tyr residues in CRAC motifs distal to CRAC4 progressively decreases the CLR sensitivity of Cbv1 channels, indicating that CRAC5 to 10 in the Cbv1 CTD contribute to the overall CLR sensitivity of BK channels. For both closed (35) and open (44) channel states, crystallographic data place the BK CTD in the cytosolic aqueous compartment. Thus, in the CLR-unligated channel, CRAC5 to 10 should be located considerably far away from the bilayer inner surface. Therefore, the question arises of how membrane CLR, which is primarily embedded in the bilayer hydrophobic core (54, 55) can reach those Cbv1 CTD distal sites. Answers to this question remain speculation. Diffusion of free CLR from the bilayer inner leaflet to distal cytosolic regions of the BK channel CTD via the aqueous phase is energetically unfavored, considering the well known hydrophobicity and poor solubility of CLR in the aqueous medium of the cell. Indeed, CLR application via the aqueous phase fails to inhibit human SLO1 channels (56), which also contain CRACs. In contrast, human SLO1 channels following reconstitution into POPS/POPE 3:1 (w/w) are CLR-sensitive (20). Thus, it appears that CLR access to Slo1 CRACs directly from the aqueous face does not alter channel function. An intriguing possibility is that CLR modulation of BK channel function requires a major conformational change in the CTD with movement of the C tail toward the bilayer inner surface, a process that could cost higher energetic penalties to CLR-containing CRACs. It is also possible that CLR could be “shuttled” from the bilayer to nearby CRAC4, and from this to progressively distal CRACs, with consequent progressive decrement in CLR availability (possibly associated to progressive energetic cost in moving CLR downstream) to the following CRAC. The fact that the Y450F mutation alters CLR sensitivity more in Cbv1 truncated after CRAC4 than in full-length Cbv1 (Fig. 3b versus Fig. 5b), however, suggests that CLR still can interact with the distal CRAC sites in the presence of an altered CRAC4 that does not fully sense CLR. Future studies are clearly needed to distinguish between these and other possibilities for the supplemental role of CTD CRACs 5–10 in the BK channel overall CLR sensitivity.
The model we present here for CLR-BK channel interactions supported by our experimental data constitutes the first identification and description of the structural elements involved in BK channel protein-CLR recognition resulting in regulation of channel function. Given the large number of residues and positions identified in the current study along BK channel CTD that determine the overall CLR response of BK channels, our data may prove useful for future identification of polymorphisms that could contribute to genetic variability in CLR sensitivity and modified tissue function via BK channel regulation. Finally, structural information from the current study could be used to design novel compounds that interfere with CLR-BK channel association in order to alter CLR modulation of BK channels that results in pathology.
Acknowledgment
We thank Maria Asuncion-Chin (Department of Pharmacology, University of Tennessee Health Science Center) for excellent technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants 2R37 AA011560 and R01 HL104632 (to A. M. D.). This work was also supported by a University of Tennessee Health Science Center Neuroscience Institute postdoctoral fellowship (to A. K. S.).

This article contains supplemental Figs. S1 and S2 and Movies S1–S4.
- CLR
- cholesterol
- AchNic
- acetylcholine nicotinic
- BK
- Ca2+/voltage-gated K+
- CRAC
- cholesterol recognition/interaction amino acid consensus motif
- CTD
- cytosolic C tail domain
- Kir
- inwardly rectifying K+
- MD
- molecular dynamics
- POPE
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine
- RCK
- regulatory of conductance for potassium
- TM
- transmembrane
- Po
- channel open probability.
REFERENCES
- 1. Mouritsen O. G., Zuckermann M. J. (2004) What's so special about cholesterol? Lipids 39, 1101–1113 [DOI] [PubMed] [Google Scholar]
- 2. Barrantes F. J. (2004) Structural basis for lipid modulation of nicotinic acetylcholine receptor function. Brain Res. Brain Res. Rev. 47, 71–95 [DOI] [PubMed] [Google Scholar]
- 3. Barrantes F. J. (2010) Cholesterol effects on nicotinic acetylcholine receptor. Cellular aspects. Subcell. Biochem. 51, 467–487 [DOI] [PubMed] [Google Scholar]
- 4. Epand R. M. (2006) Cholesterol and the interaction of proteins with membrane domains. Prog. Lipid Res. 45, 279–294 [DOI] [PubMed] [Google Scholar]
- 5. Levitan I., Fang Y., Rosenhouse-Dantsker A., Romanenko V. (2010) Cholesterol and ion channels. Subcell. Biochem. 51, 509–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jafurulla M., Tiwari S., Chattopadhyay A. (2011) Identification of cholesterol recognition amino acid consensus (CRAC) motif in G-protein-coupled receptors. Biochem. Biophys. Res. Commun. 404, 569–573 [DOI] [PubMed] [Google Scholar]
- 7. Dopico A. M., Bukiya A. N., Singh A. K. (2012) Differential contribution of BK subunits to nongenomic regulation of channel function by steroids. in Cholesterol Regulation of Ion Channels and Receptors (Barrantes F. J., Levitan I., eds) John Wiley & Sons, Inc., in press [Google Scholar]
- 8. Liang F., Schulte B. A., Qu C., Hu W., Shen Z. (2005) Inhibition of the calcium- and voltage-dependent big conductance potassium channel ameliorates cisplatin-induced apoptosis in spiral ligament fibrocytes of the cochlea. Neuroscience 135, 263–271 [DOI] [PubMed] [Google Scholar]
- 9. Salkoff L., Butler A., Ferreira G., Santi C., Wei A. (2006) High-conductance potassium channels of the SLO family. Nat. Rev. Neurosci. 7, 921–931 [DOI] [PubMed] [Google Scholar]
- 10. Sontheimer H. (2008) An unexpected role for ion channels in brain tumor metastasis. Exp. Biol. Med. 233, 779–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin M. W., Wu A. Z., Ting W. H., Li C. L., Cheng K. S., Wu S. N. (2006) Changes in membrane cholesterol of pituitary tumor (GH3) cells regulate the activity of large-conductance Ca2+-activated K+ channels. Chin. J. Physiol. 49, 1–13 [PubMed] [Google Scholar]
- 12. Lam R. S, Shaw A. R., Duszyk M. (2004) Membrane cholesterol content modulates activation of BK channels in colonic epithelia. Biochim. Biophys. Acta 1667, 241–248 [DOI] [PubMed] [Google Scholar]
- 13. Shmygol A., Noble K., Wray S. (2007) Depletion of membrane cholesterol eliminates the Ca2+-activated component of outward potassium current and decreases membrane capacitance in rat uterine myocytes. J. Physiol. 581, 445–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang X. L., Ye D., Peterson T. E., Cao S., Shah V. H., Katusic Z. S., Sieck G. C., Lee H. C. (2005) Caveolae targeting and regulation of large conductance Ca2+-activated K+ channels in vascular endothelial cells. J. Biol. Chem. 280, 11656–11664 [DOI] [PubMed] [Google Scholar]
- 15. Prendergast C., Quayle J., Burdyga T., Wray S. (2010) Cholesterol depletion alters coronary artery myocyte Ca2+ signaling in a stimulus-specific manner. Cell Calcium 47, 84–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jeremy R. W., McCarron H. (2000) Effect of hypercholesterolemia on Ca2+-dependent K+ channel-mediated vasodilatation in vivo. Am. J. Physiol. Heart Circ. Physiol. 279, H1600–H1608 [DOI] [PubMed] [Google Scholar]
- 17. Bukiya A. N., Vaithianathan T., Kuntamallappanavar G., Asuncion-Chin M., Dopico A. M. (2011) Smooth muscle cholesterol enables BK β1 subunit-mediated channel inhibition and subsequent vasoconstriction evoked by alcohol. Arterioscler. Thromb. Vasc. Biol. 31, 2410–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee U. S., Cui J. (2010) BK channel activation. Structural and functional insights. Trends Neurosci. 33, 415–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang H. M., Reitstetter R., Mason R. P., Gruener R. (1995) Attenuation of channel kinetics and conductance by cholesterol. An interpretation using structural stress as a unifying concept. J. Membr. Biol. 143, 51–63 [DOI] [PubMed] [Google Scholar]
- 20. Crowley J. J., Treistman S. N., Dopico A. M. (2003) Cholesterol antagonizes ethanol potentiation of human brain BKCa channels reconstituted into phospholipid bilayers. Mol. Pharmacol. 64, 365–372 [DOI] [PubMed] [Google Scholar]
- 21. Bukiya A. N., Vaithianathan T., Toro L., Dopico A. M. (2008) The second transmembrane domain of the large conductance, voltage- and calcium-gated potassium channel β1 subunit is a lithocholate sensor. FEBS Lett. 582, 673–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bolotina V., Omelyanenko V., Heyes B., Ryan U., Bregestovski P. (1989) Variations of membrane cholesterol alter the kinetics of Ca2+-dependent K+ channels and membrane fluidity in vascular smooth muscle cells. Pflugers Arch. 415, 262–268 [DOI] [PubMed] [Google Scholar]
- 23. Morris C. E., Juranka P. F. (2007) Lipid stress at play. Mechanosensitivity of voltage-gated channels. Curr. Top. Membr. 59, 297–337 [DOI] [PubMed] [Google Scholar]
- 24. Lundbaek J. A. (2008) Lipid bilayer-mediated regulation of ion channel function by amphiphilic drugs. J. Gen. Physiol. 131, 421–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bukiya A. N., Belani J. D., Rychnovsky S., Dopico A. M. (2011) Specificity of cholesterol and analogs to modulate BK channels points to direct sterol-channel protein interactions. J. Gen. Physiol. 137, 93–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Case D. A., Cheatham T. E., 3rd, Darden T., Gohlke H., Luo R., Merz K. M., Jr., Onufriev A., Simmerling C., Wang B., Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wu Y., Yang Y., Ye S., Jiang Y. (2010) Structure of the gating ring from the human large-conductance Ca2+-gated K+ channel. Nature 466, 393–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hanson M. A., Cherezov V., Griffith M. T., Roth C. B., Jaakola V. P., Chien E. Y., Velasquez J., Kuhn P., Stevens R. C. (2008) A specific cholesterol binding site is established by the 2.8 Å structure of the human β2-adrenergic receptor. Structure 16, 897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jaggar J. H., Li A., Parfenova H., Liu J., Umstot E. S., Dopico A. M., Leffler C. W. (2005) Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ. Res. 97, 805–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dopico A. M. (2003) Ethanol sensitivity of BKCa channels from arterial smooth muscle does not require the presence of the β1-subunit. Am. J. Physiol. Cell Physiol. 284, C1468–C1480 [DOI] [PubMed] [Google Scholar]
- 31. Yuan C., O'Connell R. J., Jacob R. F., Mason R. P., Treistman S. N. (2007) Regulation of the gating of BKCa channel by lipid bilayer thickness. J. Biol. Chem. 282, 7276–7286 [DOI] [PubMed] [Google Scholar]
- 32. Picazo-Juárez G., Romero-Suárez S, Nieto-Posadas A., Llorente I., Jara-Oseguera A., Briggs M., McIntosh T. J., Simon S. A., Ladrón-de-Guevara E., Islas L. D., Rosenbaum T. (2011) Identification of a binding motif in the S5 helix that confers cholesterol sensitivity to the TRPV1 ion channel. J. Biol. Chem. 286, 24966–24976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li H., Yao Z., Degenhardt B., Teper G., Papadopoulos V. (2001) Cholesterol binding at the cholesterol recognition/interaction amino acid consensus (CRAC) of the peripheral-type benzodiazepine receptor and inhibition of steroidogenesis by an HIV TAT-CRAC peptide. Proc. Natl. Acad. Sci. U.S.A. 98, 1267–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Epshtein Y., Chopra A. P., Rosenhouse-Dantsker A., Kowalsky G. B., Logothetis D. E., Levitan I. (2009) Identification of a C-terminus domain critical for the sensitivity of Kir2.1 to cholesterol. Proc. Natl. Acad. Sci. U.S.A. 106, 8055–8060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yuan P., Leonetti M. D., Pico A. R., Hsiung Y., MacKinnon R. (2010) Structure of the human BK channel Ca2+-activation apparatus at 3.0 Å resolution. Science 329, 182–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Piskorowski R., Aldrich R. W. (2002) Calcium activation of BKCa potassium channels lacking calcium bowl and RCK domains. Nature 420, 499–502 [DOI] [PubMed] [Google Scholar]
- 37. Wang S. X., Ikeda M., Guggino W. B. (2003) The cytoplasmic tail of large conductance, voltage- and Ca2+-activated K+ (MaxiK) channel is necessary for its cell surface expression. J. Biol. Chem. 278, 2713–2722 [DOI] [PubMed] [Google Scholar]
- 38. Jiang Y., Pico A., Cadene M., Chait B. T., MacKinnon R. (2001) Structure of the RCK domain from the E. coli K+ channel and demonstration of its presence in the human BK channel. Neuron 29, 593–601 [DOI] [PubMed] [Google Scholar]
- 39. Epand R. M., Thomas A., Brasseur R., Epand R. F. (2010) Cholesterol interaction with proteins that partition into membrane domains. An overview. Subcell. Biochem. 51, 253–278 [DOI] [PubMed] [Google Scholar]
- 40. Gimpl G. (2010) Cholesterol-protein interaction. Methods and cholesterol reporter molecules. Subcell. Biochem. 51, 1–45 [DOI] [PubMed] [Google Scholar]
- 41. Jamin N., Neumann J. M., Ostuni M. A., Vu T. K., Yao Z. X., Murail S., Robert J. C., Giatzakis C., Papadopoulos V., Lacapère J. J. (2005) Characterization of the cholesterol recognition amino acid consensus sequence of the peripheral-type benzodiazepine receptor. Mol. Endocrinol. 19, 588–594 [DOI] [PubMed] [Google Scholar]
- 42. Cox D. H. (2005) The BKCa channel's Ca2+-binding sites, multiple sites, multiple ions. J. Gen. Physiol. 125, 253–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Latorre R., Brauchi S. (2006) Large conductance Ca2+-activated K+ (BK) channel. Activation by Ca2+ and voltage. Biol. Res. 39, 385–401 [DOI] [PubMed] [Google Scholar]
- 44. Yuan P., Leonetti M. D., Hsiung Y., MacKinnon R. (2012) Open structure of the Ca2+ gating ring in the high-conductance Ca2+-activated K+ channel. Nature 481, 94–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Edwards A. A., Mason J. M., Clinch K., Tyler P. C., Evans G. B., Schramm V. L. (2009) Altered enthalpy-entropy compensation in picomolar transition state analogues of human purine nucleoside phosphorylase. Biochemistry 48, 5226–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moczydlowski E., Alvarez O., Vergara C., Latorre R. (1985) Effect of phospholipid surface charge on the conductance and gating of a Ca2+-activated K+ channel in planar lipid bilayers. J. Membr. Biol. 83, 273–282 [DOI] [PubMed] [Google Scholar]
- 47. Vincent N., Genin C., Malvoisin E. (2002) Identification of a conserved domain of the HIV-1 transmembrane protein gp41 which interacts with cholesteryl groups. Biochim. Biophys. Acta 1567, 157–164 [DOI] [PubMed] [Google Scholar]
- 48. Palmer C. P., Mahen R., Schnell E., Djamgoz M. B., Aydar E. (2007) Sigma-1 receptors bind cholesterol and remodel lipid rafts in breast cancer cell lines. Cancer Res. 67, 11166–11175 [DOI] [PubMed] [Google Scholar]
- 49. Luo X., Sharma D., Inouye H., Lee D., Avila R. L., Salmona M., Kirschner D. A. (2007) Cytoplasmic domain of human myelin protein zero likely folded as β-structure in compact myelin. Biophys. J. 92, 1585–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Saher G., Quintes S., Möbius W., Wehr M. C., Krämer-Albers E. M., Brügger B., Nave K. A. (2009) Cholesterol regulates the endoplasmic reticulum exit of the major membrane protein P0 required for peripheral myelin compaction. J. Neurosci. 29, 6094–6104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Saher G., Simons M. (2010) Cholesterol and myelin biogenesis. Subcell. Biochem. 51, 489–508 [DOI] [PubMed] [Google Scholar]
- 52. Balijepalli R. C., Delisle B. P., Balijepalli S. Y., Foell J. D., Slind J. K., Kamp T. J., January C. T. (2007) Kv11.1 (ERG1) K+ channels localize in cholesterol- and sphingolipid-enriched membranes and are modulated by membrane cholesterol. Channels 1, 263–272 [DOI] [PubMed] [Google Scholar]
- 53. Boesze-Battaglia K., Brown A., Walker L., Besack D., Zekavat A., Wrenn S., Krummenacher C., Shenker B. J. (2009) Cytolethal distending toxin-induced cell cycle arrest of lymphocytes is dependent upon recognition and binding to cholesterol. J. Biol. Chem. 284, 10650–10658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bittman R. (1997) Has nature designed the cholesterol side chain for optimal interaction with phospholipids? Subcell. Biochem. 28, 145–171 [DOI] [PubMed] [Google Scholar]
- 55. Loura L. M., Prieto M. (1997) Dehydroergosterol structural organization in aqueous medium and in a model system of membranes. Biophys. J. 72, 2226–2236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. King J. T., Lovell P. V., Rishniw M., Kotlikoff M. I., Zeeman M. L., McCobb D. P. (2006) β2 and β4 subunits of BK channels confer differential sensitivity to acute modulation by steroid hormones. J. Neurophysiol. 95, 2878–2888 [DOI] [PubMed] [Google Scholar]