

Background: The mechanism by which interaction between Aβ and Zn2+ induces Aβ aggregation and cell toxicity is elusive.
Results: Zn2+ and Aβ40 form metastable neurotoxic oligomers.
Conclusion: Aβ40 binding to Zn2+ leads to formation of small neurotoxic oligomers that become benign upon further self-assembly.
Significance: We provide a structure-function analysis of Zn2+-stabilized Aβ40, a neurotoxic species that may contribute to the pathology in AD.
Keywords: Aggregation, Alzheimer's Disease, Amyloid, Biophysics, Zinc, Aggregate Structure, Toxicity
Abstract
The roles of metal ions in promoting amyloid β-protein (Aβ) oligomerization associated with Alzheimer disease are increasingly recognized. However, the detailed structures dictating toxicity remain elusive for Aβ oligomers stabilized by metal ions. Here, we show that small Zn2+-bound Aβ1–40 (Zn2+-Aβ40) oligomers formed in cell culture medium exhibit quasi-spherical structures similar to native amylospheroids isolated recently from Alzheimer disease patients. These quasi-spherical Zn2+-Aβ40 oligomers irreversibly inhibit spontaneous neuronal activity and cause massive cell death in primary hippocampal neurons. Spectroscopic and x-ray diffraction structural analyses indicate that despite their non-fibrillar morphology, the metastable Zn2+-Aβ40 oligomers are rich in β-sheet and cross-β structures. Thus, Zn2+ promotes Aβ40 neurotoxicity by structural organization mechanisms mediated by coordination chemistry.
Introduction
The predominant proteinaceous component of amyloid plaques, a pathological hallmark of Alzheimer disease (AD)4, is amyloid β-protein (Aβ). Aβ is generated by sequential enzymatic cleavage of the amyloid β-protein precursor (APP) by β- and γ-secretases (1). The two major forms of Aβ produced from APP contain 40 (Aβ40) or 42 (Aβ42) amino acid residues. The cause of AD is believed to be pathological accumulation and self-association of Aβ into neurotoxic oligomers. These impair synaptic communication and trigger a cascade of events, leading to neurodegeneration in susceptible brain areas (2, 3). Aβ oligomers, rather than Aβ fibrils, are considered the primary neurotoxic agents acting in AD (4, 5).
Various factors and events that modulate Aβ toxicity include disruption of metal homeostasis, disruption of critical metabolic processes, inflammation, and oxidative stress. These factors make working in this field extremely challenging; in particular, elucidating the molecular mechanisms governing the pathology of Aβ misfolding and aggregation. For example, Zn2+ is highly enriched in AD plaques compared with healthy, aged-matched brain tissues (6–9). Certain neurons in the mammalian cerebral cortices store up to millimolar Zn2+ concentrations and release Zn2+ in (sub)millimolar pulses from their presynaptic terminals (10). Synaptic Zn2+ may play a critical role in mediating toxicity induced by Aβ oligomers (11). Specifically, transgenic mouse models of AD demonstrated significantly reduced loads of cerebral senile plaques and precipitated Aβ when the gene for the synaptic vesicle transporter ZnT3 (SLC30A3) was knocked out (12). In vitro, Zn2+ interacts with Aβ40, inducing its aggregation within milliseconds (13, 14). It was shown that Zn2+ interacts with soluble Aβ40 oligomers and alters their stability (15). Importantly, physiological conditions, such as the presence of NaCl, significantly accelerate Zn2+-induced Aβ aggregation (16). The rapid interaction of Zn2+ with Aβ40 results in formation of quasi-spherical aggregates. With aging, these aggregates increase in size significantly and do not convert into fibrils (17). Interaction of Zn2+ with Aβ40 increases the exposure of hydrophobic surfaces in Aβ40 (18).
Zn2+-induced Aβ aggregates were proposed as neurotoxic agents disrupting synaptic communication (11, 13). However, the molecular determinants driving neurotoxicity remain elusive. We thus set out to perform a detailed structure-toxicity analyses of Aβ40 in the presence of Zn2+ in serum-free culture media. Here, we report the self-assembly pathways and structure-toxicity interplay of Zn2+-induced, quasi-spherical metastable Aβ40 oligomers (Zn2+-Aβ40). These oligomers possess conformational characteristics typical of fibrillar structures, yet their morphology is non-fibrillar. They irreversibly affect spontaneous calcium activity and neuron viability.
EXPERIMENTAL PROCEDURES
Reagents
All reagents were purchased from Sigma-Aldrich (Israel) unless mentioned otherwise. All reagents were of analytical grade. Purified deionized water was prepared using a Milli-Q water-purification system (Millipore, Billerica, MA).
Aβ40 Sample Preparation
Aβ40 was purchased from rPeptide and prepared for experiments as described previously (14). Briefly, Aβ40 was dissolved at 665 μm (defined by UV-Vis spectroscopy with ϵ292 = 2300 M−1 cm−1) in 10 mm NaOH, sonicated for 1 min in a Branson 1510 bath sonicator, and centrifuged for 10 min at 12,000 × g at 4 °C to precipitate large aggregates. Concentrations of stock solutions prepared this way were occasionally confirmed by amino acid analysis. The differences in the concentrations determined by absorption and amino acid analysis were < 8%. The stock solutions were diluted to 230 μm in 10 mm MOPS (pH 6.9 ± 0.1). These solutions did not scatter near-UV light at 300 nm, suggesting that they did not contain large aggregates or fibrils. Moreover, Aβ40 preparations were hardly distinguishable from the control (buffer) by transmission electron microscopy (TEM) examination at t = 0 (supplemental Fig. S1a). In such preparations, Aβ40 monomers coexist with low-molecular-weight oligomers (19). Zn2+-Aβ40 oligomers were prepared by adding 0.01 m ZnCl2 (in 10 mm MOPS) to the Aβ40 solution. For fibril preparation, 230 μm Aβ40 was incubated for 7 days at 37 °C without agitation. Fibril formation was examined by TEM (supplemental Fig. S2).
In TEM, atomic force microscopy (AFM), CD, thioflavin T (ThT) fluorescence, and cell-culture experiments, the final Aβ40 and Zn2+ concentrations were 10 and 20 μm, respectively (Aβ concentration calculated for the monomeric protein). We estimated the concentration of free Zn2+ in our preparations by using data published by Bush et al. (13), assuming two binding sites for Zn2+ on Aβ:
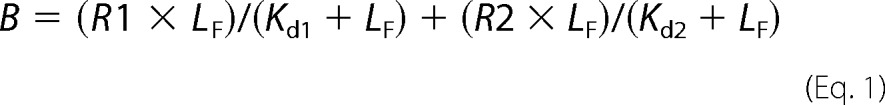 |
where B is the concentration of Zn2+-Aβ40 oligomers, LF is the concentration of free Zn2+, R1 and R2 are the concentrations of each Zn2+-binding site, and Kd1 and Kd2 are the respective dissociation constants.
Accordingly, the concentration of free Zn2+ in our preparations is estimated at 5.15 μm. These calculations suggest that under the conditions we used, each Aβ40 molecule binds ∼ 1.5 Zn2+ ions.
TEM, AFM, CD, and ThT experiments were performed in serum-free cell-culture medium (129 mm NaCl, 4 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 10 mm glucose, and 10 mm HEPES (pH 7.4) with osmolarity adjusted to 320 mOsm) using dilutions identical to those used in cell culture experiments. Fibril suspensions in MOPS were sonicated for 20 s, centrifuged for 20 min at 2000 × g, washed three times in 10 mm MOPS, and finally added to neuronal cultures. All morphologies were prepared in MOPS and comprised 8.7% of the final volume of culture medium.
Electron Microscopy
Images were acquired using a Tecnai 12 transmission electron microscope (FEI, Eindhoven, The Netherlands) operated at 120 kV. Micrographs were taken using a MegaView III charge-coupled device camera (SIS, Münster, Germany). Aliquots of different Aβ40 preparations were adsorbed for 1 min onto carbon-coated copper grids and negatively stained with 1% (w/v) uranyl acetate for 30 s. TEM images were analyzed using ImageJ.
Atomic Force Microscopy
Aliquots (5 μl) of freshly prepared Zn2+-Aβ40 were spotted onto freshly cleaved mica (Ted Pella, Inc., Redding, CA), incubated at room temperature for 3 min, rinsed with Milli-Q water, and air-dried. At least five different regions on the mica surface were examined.
Images were collected using a MultiMode AFM with a Nanoscope V controller (Veeco Metrology LLC, Santa Barbara, CA) equipped with an E-scanner in tapping mode at 22–24 °C. All images were recorded using silicon microcantilevers (OMCL-AC240TS-W2, Olympus) with a spring constant of ∼2 N/m (manufacturer-specified) and at a scan rate of 1–3 Hz. The target amplitude was 300 mV with a set point of ∼230 mV for all measurements. Images were acquired in different scan directions and at different scales to verify the consistency of the evaluated structures.
Profiles of Zn2+-Aβ40 oligomers were acquired from the AFM images, and the corresponding aggregate heights were calculated, binned, normalized, and plotted using Matlab (MathWorks, Inc., Natick, MA).
Circular Dichroism Spectroscopy
Room-temperature CD spectra of 10 μm Zn2+-free Aβ40 or Zn2+-Aβ40 solutions in serum-free media were measured at t = 0 h and t = 2 h in 2-mm path-length quartz cuvettes (Helma, Jena, Germany) in the spectral range of 200–260 nm. Spectra were recorded using a JASCO 815 spectropolarimeter at a 100-nm/min scan rate with 0.2-nm resolution. For each sample, four spectra were acquired and averaged. The background was subtracted, and the spectra were smoothed using OriginPro 8.0.
X-ray Powder Diffraction
MOPS stock solutions containing Zn2+-Aβ40 were centrifuged at 12,000 × g for 10 min at 4 °C. The pellet was washed gently three times with 1 ml of Milli-Q water containing 1 μm ZnCl2, placed as a thick film onto a silicon zero-background sample holder, and air-dried for 30 min at room temperature. X-ray diffraction measurements were carried out in reflection mode using a TTRAX III θ-θ diffractometer (Rigaku, Japan) equipped with a rotating copper anode operating at 50 kV and 200 mA and a scintillation detector.
Parallel-beam optics (angle divergence ∼0.05°) formed by a multilayered mirror (Rigaku, Cross Beam Optics attachment, Japan) were used to obtain high-quality data from a dried-drop sample at low diffraction angles. Specular diffraction (θ/2θ scan) was performed under ambient conditions from 2° to 30°. The average measurement time was ∼5 h. After 10 h, sample degradation was observed. The cross-section of the x-ray beam was 1 × 5 mm2, and the angular divergence of the reflected beam was limited to 0.114° by a parallel slit analyzer (PSA-80).
Peak positions and widths of the Bragg reflections were determined by a self-consistent profile-fitting procedure using Jade 9.1 (Materials Data, Inc., Livermore, CA). The coherent diffraction lengths observed in Zn2+-Aβ40 were estimated by the Scherrer formula from the broadening of the corresponding peaks.
ThT Fluorescence
Triplicate 200-μl samples were examined in plastic clear-bottom 96-well plates (Nunc 96F Maxisorp, Thermo Fisher Scientific, Roskilde, Denmark). The plates were incubated at 25 °C for 72 h without agitation in a Synergy HT multi-mode microplate reader (Bio-Tek Instruments, Winooski, VT) with excitation and emission wavelengths/slit widths of 400 nm/30 nm and 485 nm/20 nm, respectively. To prevent evaporation, plates were tightly sealed with Parafilm (Plastic Packaging, Chicago, IL). The ThT fluorescence data were background-subtracted and plotted using OriginPro 8.0.
Hippocampal Neuron Cultures
Animal experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of the Weizmann Institute and the Israeli national guidelines on animal care. Cell cultures were prepared as described previously (20). Briefly, rat pups were decapitated on the day of birth (P0), and their brains were removed and placed into Petri dishes containing chilled (4 °C), oxygenated Leibovitz L15 medium (Invitrogen) supplemented with 0.6% glucose and gentamicin (20 μg/ml, Sigma). Hippocampi were isolated, incubated with trypsin (0.25% w/v) and DNase (50 μg/ml), triturated, and then transferred to plating medium comprising 5% heat-inactivated horse serum (HS), 5% fetal bovine serum, and B-27 (1 μl/ml) prepared in minimum essential medium (MEM) (Invitrogen) supplemented with 0.6% glucose, 20 μg/ml gentamicin, and 2 mm glutamax (enriched MEM).
Using 24-well plates, ∼105 hippocampal neurons were plated in 1 ml of medium/well onto a hippocampal glial cell feeder layer. The feeder layer was grown on poly-lysine-coated glass cover slips for 2 weeks before transferring the neurons. On day 3, the medium was changed to enriched MEM containing 10% HS and a mixture of 5′-fluoro-2-deoxyuridine/uridine (20 μg/ml and 50 μg/ml, respectively, Sigma) to block glial cell proliferation. On day 7, the medium was changed to MEM-containing 10% HS with no further modifications until cells were used for experiments.
Intracellular Ca2+ Imaging
Postnatal cultures 2–3 weeks after plating were used for Ca2+ imaging. First, cells were washed in serum-free cell-culture media containing 129 mm NaCl, 4 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 10 mm glucose, and 10 mm HEPES (pH 7.4) with osmolarity adjusted to 320 mOsm.
Cells were incubated for 1 h at 23 °C in the dark in the same serum-free media containing 2 μm Fluo-4AM (Invitrogen/Molecular Probes). The cells were then extensively washed for 5 min and imaged using an upright Zeiss PASCAL confocal microscope with an Olympus ×63 water immersion lens (0.9 numerical aperture) and ×1–2 scan zoom in bidirectional scan mode. Laser power, pinhole diameter, and detector gains were adjusted and standardized to avoid photo damage and pixel saturation. Each field was recorded for 5 min without signs of cellular photo toxicity or changes in spontaneous firing rates.
As all Aβ40 preparations initially were prepared in MOPS buffer, we tested their effects on the spontaneous activity of cultured neurons in 10 mm MOPS (pH 6.9 ± 0.1). We found that MOPS, which served as a control, did not affect the rate or the amplitude of Ca2+ activity or [Ca2+]I (p > 0.05, Student's t test). Sequential images were taken at t = 0, 5, 30, 60, 90, and 120 min for each Aβ40 preparation, followed by a 30-min wash step.
Cell Viability Analysis
In initial experiments, we added 10 μm of each Aβ40 preparation (Zn2+-free Aβ40, Aβ40 fibrils or Zn2+-Aβ40 oligomers), MOPS, or Zn2+ to hippocampal neurons and measured neuronal viability after 48 h incubation. The incubation conditions were serum-containing culture media (MEM containing 10% HS) at 37 °C and 5% CO2. Under these conditions, no toxicity was observed for any of the Aβ40 preparations. Comparison of Zn2+-Aβ40 oligomers and control conditions is shown in supplemental Fig. S6.
In a modified protocol, four equal aliquots of Zn2+-free Aβ40, Zn2+-Aβ40 oligomers, or Aβ40 fibrils were added to primary hippocampal neurons every 12 h without changing the incubation conditions (1 ml MEM containing 10% HS at 37 °C, 5% CO2). The final Aβ40 concentration was 10 μm in each case. Cell survival was evaluated 72 h after the last application by assaying neuron-specific enolase or directly by phase-contrast microscopy.
Immunochemistry
Glass cover slips with treated cells were removed from the wells and washed briefly with the standard recording medium. The cells were fixed in 4% paraformaldehyde and 4% sucrose in 0.1 m PBS (pH 7.4) for 20 min and then washed thoroughly with PBS, incubated for 1 h in PBS containing 10% normal goat serum and 0.1% Triton X-100 to reduce nonspecific reactivity, and incubated with rabbit anti-neuron-specific enolase at 4 °C for 24 h. The cells were then washed, incubated with Alexa Fluor 568-labeled caprine anti-rabbit secondary antibody (1:200, Molecular Probes, Eugene, OR) for 1 h, and washed. The coverslips were transferred onto glass slides and mounted in an anti-fading mounting medium comprising 90% glycerol and 0.1% p-phenylenediamine (Sigma) in 20 mm PBS (pH 8.5) for visualization. Confocal image stacks were recorded using a Zeiss LSM 510 laser-scanning microscope with a Zeiss ×40 oil-immersion objective (1.4 numerical aperture) and ×1 scan zoom. Detector gain and amplifier were initially set to obtain pixel densities within a linear range.
Quantification, Statistics, and Digital Images
Time lapse, immunostaining, and phase contrast images were analyzed using Zeiss LSM 510 or Zeiss PASCAL software (Carl Zeiss). The fluorescence signal was measured by producing regions of interest related to the shapes of cell bodies. Alternatively, profiles of fluorescence intensity were generated using ImageJ. Cells were measured and counted using a double-blinded procedure by an independent observer. [Ca2+]i events were counted automatically using a home-made Matlab program (The MathWorks, Inc., Asheboro, NC) and controlled manually.
Statistical analyses were performed by Student's t tests or analysis of variance using Matlab or KaleidaGraph (Synergy Software, Reading, PA). In each experiment, 4–6 cultures were used per group.
Figures were prepared using Photoshop CS2 (Adobe, San Jose, CA). Image brightness and contrast were adjusted uniformly across the entire image.
RESULTS
Morphology of Zn2+-Aβ40 Oligomers in Serum-free Culture Media
The rapid interaction of Zn2+ ions with Aβ40 induced aggregation mainly into quasi-spherical morphologies as observed previously in MOPS buffer (14). The experimental conditions used to assess Aβ morphologies by TEM and AFM were similar to conditions used for neuron cultures. Aβ morphologies were assessed at room temperature in serum-free culture media. Under these conditions, our preparations contained ∼5 μm of unbound Zn2+. In fresh Zn2+-Aβ40 preparations, a distribution of predominantly quasi-spherical particles with 7–15-nm diameter was observed by TEM (Fig. 1, a and b). These Zn2+-Aβ40 assemblies resembled those of toxic spherical oligomers of Zn2+-free Aβ40 (22) stabilized at 4 °C for 30–60 h, although the latter had larger diameters (15–35 nm). The majority of the Zn2+-Aβ40 complexes appeared to have stable size and morphology for at least 2 h at room temperature, whereas ∼20% of these structures increased in size after 2 h (supplemental Fig. S3). In contrast, the morphology and diameter/length of structures observed in Zn2+-free Aβ40 preparations (supplemental Fig. S1, a and b) increased substantially in the first 2 h of incubation. Thus, morphological changes in Zn2+-free Aβ40 occur considerably faster than in Zn2+- stabilized oligomers.
FIGURE 1.

Morphological characterization of freshly prepared metastable Zn2+-Aβ40 oligomers in serum-free culture medium. a, TEM image of Zn2+-Aβ40. b, diameter distribution of Zn2+-Aβ40 derived from TEM images. c, AFM image of Zn2+-Aβ40 in air. d, height distribution of Zn2+-Aβ40 derived from AFM images.
Height analyses of the Zn2+-Aβ40 oligomers by AFM revealed distinct populations with heights of 1, 2, or 4 nm (Fig. 1, c and d). This suggests that aggregates are arranged in mono-, bi-, or tetralayers, assuming ∼1-nm-thick monolayers within which Aβ40 molecules have a hairpin conformation (14).
Height-diameter correlation analysis of native neurotoxic amylospheroids (23) and Zn2+-free cross-linked Aβ40 (24) yielded correlation coefficients of r2 = 0.87 and r2 > 0.95, respectively. A similar height-diameter correlation analysis of Zn2+-Aβ40 populations gave a correlation coefficient of r2 = 0.055, demonstrating that heights and diameters were not correlated. Further analysis showed that the low correlation coefficient resulted from the existence of a relatively narrow population of diameters and three distinct populations of heights (Fig. 1, b–d). This suggests that Zn2+-Aβ40 assembles in defined conformational protein blocks possessing an irregular structural organization in the horizontal plane and a regular organization in the vertical direction. This organization may be induced by stable hairpin conformation of Aβ40 and nonspecific intermolecular Zn2+ coordination (14, 25).
Secondary and Tertiary Structures of Zn2+-Aβ40
The secondary and tertiary structures of Zn2+-Aβ40 oligomers were analyzed by far-UV CD spectroscopy and x-ray powder diffraction (Fig. 2). At t = 0, the CD spectrum of Zn2+-Aβ40 oligomers measured in serum-free media exhibited a minimum at 214 nm, indicating a high β-sheet content which was stable during 2 h of incubation at room temperature (Fig. 2a). Similar CD experiments assessing Zn2+-free Aβ40 preparations at t = 0 or t = 2 h also showed the presence of β-sheets (supplemental Fig. S1c). Yet, the β-sheet content in these preparations increased substantially during the first 2 h of incubation, in agreement with the TEM data discussed above.
FIGURE 2.

Secondary and tertiary structure analyses of Zn2+-Aβ40 oligomers in serum-free culture medium. a, CD spectra of Zn2+-Aβ40. b, x-ray powder diffraction pattern of Zn2+-Aβ40. The intensity in b is given in arbitrary units (a.u.).
X-ray powder diffraction was used to elucidate the long-range order of metastable Zn2+-Aβ40 oligomers (Fig. 2b). Fresh preparations of Zn2+-Aβ40 in MOPS buffer were rapidly concentrated prior to x-ray measurements (see “Experimental Procedures”). The diffraction pattern collected in reflection mode exhibited three peaks with d spacings at 10.25, 4.67, and 3.7 Å, characteristic of amyloid fibrils (26–30) (Fig. 2b). This result was intriguing in view of the non-fibrillar, quasi-spherical morphology observed for the metastable Zn2+-Aβ40 oligomers in both MOPS (14) and in serum-free culture media (Fig. 1). The most pronounced reflection peak corresponded to a distance of 4.67 Å and was attributed to intermolecular hydrogen bond distances. The coherence length of this reflection is ∼20 Å, corresponding to approximately four molecules of Aβ40 in a crystallite along the axis of hydrogen bonding. The reflection referring to 10.25 Å was attributed to the mean distance between peptides in neighboring sheets of Aβ40 molecules with a U-shaped hairpin conformation. This distance is in good agreement with the 1-nm height for monolayer arrangements of Zn2+-Aβ40 oligomers measured by AFM (Fig. 1, c and d). The calculated coherence length for the 10.25 Å diffraction peak is ∼42 Å, corresponding to a tetra-layer of Aβ40, also in agreement with our AFM height analysis, which indicates a height of 4 nm (Fig. 1d). The low-intensity, broad reflection peak (> 6) corresponding to ∼3.7 Å indicates a low degree of order of the pleated β-sheets. Similar peaks were observed in fibrils of glucagon (3.77 Å) (26), Aβ1–28 (3.8 Å) (27), and Aβ40 (3.8 Å) (28) and was interpreted by Glenner et al. (26) as average spacing between Cα atoms in neighboring polypeptide chains. Our CD and x-ray powder diffraction data indicate that binding of Zn2+ to Aβ40 peptide does not interfere with the hairpin conformation observed in Aβ40 fibrils yet induces non-fibrillar morphologies, possibly via intermolecular Zn2+ coordination.
Seeding Aβ40 with Zn2+-Aβ40 Oligomers Inhibits Fibril Growth
Our results indicate that Zn2+-Aβ40 oligomers possess structural characteristics similar to those found in fibrils but do not have fibrillar morphology (14, 17, 18). To explore the overall effect of Zn2+–Aβ40 oligomers on Aβ40 fibril formation, we analyzed their nucleation power by conducting time-dependent ThT fluorescence experiments in serum-free media. The ThT fluorescence assay quantitatively measures formation of cross-β-sheet structures (31) and is commonly used for monitoring Aβ assembly kinetics (32). The ThT fluorescence signal was recorded over 72 h at 25 °C without agitation in Zn2+-free Aβ40 (comprising monomers and low-molecular-weight oligomers (19), Zn2+-Aβ40 oligomers, or mixtures of the two preparations at 9:1, or 4:1 concentration ratio (Fig. 3). Ten μm Zn2+-free Aβ40 displayed a rapid increase in ThT fluorescence in serum-free culture medium. The linear part of the fluorescence curve had a slope of 0.81 ± 0.08 arbitrary units/h. Such a fast increase in ThT fluorescence relative to measurements in hypotonic buffers is typically observed in serum-free medium because of the relatively high concentration of Na+ and Ca2+ salts, which are known to accelerate Aβ aggregation and fibril formation (33–36).
FIGURE 3.
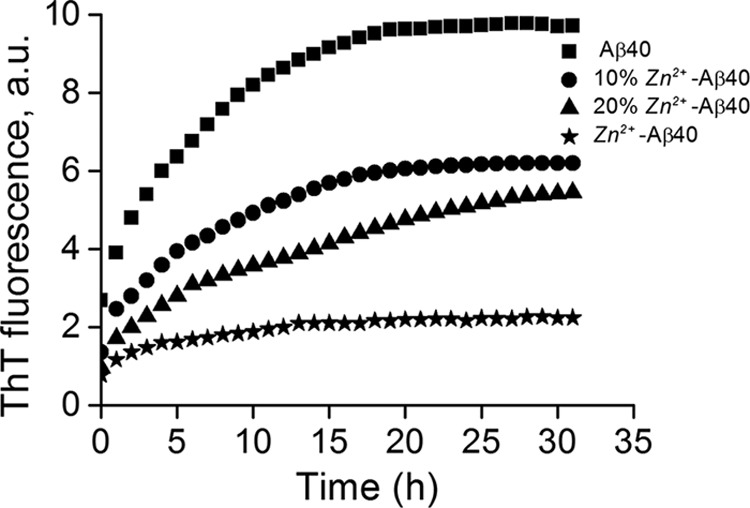
Effects of metastable Zn2+-Aβ40 oligomers on Aβ40 nucleation and fibril formation. β-sheet formation was assessed in solutions of 10 μm Zn2+- free Aβ40, 10 μm Zn2+-Aβ40 oligomers, or mixtures of these two preparations at 9:1 or 4:1 concentration ratios, respectively, by measuring the change in ThT fluorescence over time. ThT fluorescence is given in arbitrary units (a.u.).
Seeding Zn2+-free Aβ40 with 10 or 20% Zn2+-Aβ40 oligomers affected both the slope and the plateau value of the ThT fluorescence. Specifically, the slopes decreased to 0.44 ± 0.05 arbitrary units/h for 10% and 0.33 ± 0.03 arbitrary units/h in the presence of 20% Zn2+-Aβ40 oligomers, respectively. The ThT fluorescence at the plateau decreased by ∼35% and ∼45% relative to the plateau signal of Aβ40 in the presence of 10 and 20% Zn2+-Aβ40 oligomers, respectively, suggesting a reduction of aligned cross-β-sheet content. The sample containing 10 μm Zn2+-Aβ40 oligomers showed little increase in ThT fluorescence relative to Zn2+-free Aβ40. These data suggest that Zn2+-Aβ40 oligomers attenuate the nucleation step of Aβ40 fibrillization and interfere with fibril growth. The ThT results are in agreement with TEM images presented in supplemental Figs. S3 and S4. Thus, our results indicate that not only Zn2+ but also Zn2+-Aβ40 oligomers interfere with Aβ40 assembly.
Metastable Zn2+-Aβ40 oligomers Irreversibly Suppress Spontaneous Neuronal Activity
The most common forms of spontaneous neuronal activity in vivo and in vitro are synchronous intracellular calcium ([Ca2+]i) transients and bursts of action potentials in large populations of neurons involved in local circuits. Typically, in serum-free media, synchronous [Ca2+]i bursts can be recorded at stable rates and amplitudes for 2–3 h at room temperature (21, 37). To evaluate how fresh Zn2+-Aβ40 oligomers affect neuronal function, we added them to primary rat hippocampal neurons in serum-free culture media and measured spontaneous Ca2+ activity using the fluorescent Ca2+-binding dye Fluo-4AM (38). The control conditions included freshly prepared Zn2+-free Aβ40, Aβ40 fibrils, and Zn2+. An additional control was MOPS (pH 6.9) diluted in serum-free cell-culture medium at the same concentration used for preparing Zn2+-Aβ40 oligomers. We found that MOPS did not significantly affect the rate or the amplitude of Ca2+ activity or basal [Ca2+]i (data not shown).
The effect of Zn2+-Aβ40 oligomers on Ca2+ transients in a typical field of cultured hippocampal neurons is shown in Fig. 4, a and b. Initially, the fluorescence intensity of resting [Ca2+]i was consistently below 10 relative fluorescence units. On average, 5–8 transient Ca2+ events per minute were measured, reaching up to 80 relative fluorescence units. After 60 min of incubation with 10 μm Zn2+-Aβ40 oligomers, the mean Ca2+ firing rate gradually decreased to ∼3–5 min−1 (Fig. 4, a and b, and supplemental Fig. S5, a and b), and the net number of Ca2+ events was almost completely suppressed by 120 min (Fig. 4c and supplemental Fig. S5). An early indicator of neuronal damage is a gradual rise in basal [Ca2+]i. In our experiments, an increase in [Ca2+]i was observed ∼30 min after adding freshly prepared Zn2+-Aβ40 oligomers (Fig. 4, a, b, and d) with further increase by a factor of 4–5 by 120 min. Extensively washing out the Zn2+-Aβ40 oligomers did not reverse the change in basal [Ca2+]I, nor did it affect the steady increase in the basal [Ca2+]i signal (Fig. 4d). This suggests that Zn2+-Aβ40-induced alterations of cell physiology were irreversible for at least 60 min after washing.
FIGURE 4.

Effect of metastable Zn2+-Aβ40 oligomers on primary hippocampal neurons. a, top panels, gradual rise in basal [Ca2+]i in cultured neurons. Bottom panels, Ca2+-influx peaks. b, Ca2+-imaging of the same fields after background subtraction. The y axis represents Fluo-4AM fluorescence under carefully standardized experimental conditions, including dye-loading time, temperature, light-source intensity, detector offset, and gain. c, net change of spontaneous [Ca2+]i bursts in the presence of Aβ40 monomers (green), Aβ40 fibrils (blue), Zn2+ (purple), or metastable Zn2+-Aβ40 oligomers (red). Negative values are relative to the initial [Ca2+]i burst rate. d, resting [Ca2+]i in the same population of neurons. The y axis represents Fluo-4AM fluorescence under the same standardized conditions as in b and corresponds to the values of the net [Ca2+]i changes obtained after subtracting initial [Ca2+]i. e, viability of neurons at t = 72 h measured by immunostaining for neuron-specific enolase. The data are presented as mean ± S.E. for three independent experiments compared with the control. N.S., non-significant.
Of all the treatment conditions, only Zn2+-Aβ40 oligomers induced a marked decrease in firing rates and in resting [Ca2+]i fluorescence. Treatment with Zn2+-free Aβ40 induced a slight, transient increase in the neuronal firing rate at ∼10 min (Fig. 4c) that became statistically insignificant by 30 min and completely waned after 60 min. Importantly, when the same preparations of Zn2+-Aβ40 oligomers were stored for at least 16 h at room temperature before application, they did not affect spontaneous Ca2+ activity. TEM images of these Zn2+-Aβ40 oligomer preparations revealed large amorphous aggregates extending more than 100 nm (supplemental Fig. S4), which likely formed by association of the small quasi-spherical oligomers observed at earlier time points (Fig. 1a).
Two other Ca2+-binding dyes, Fura-2AM and Oregon Green BAPTA-2AM yielded very similar results (data not shown), suggesting that the observations were unrelated to the use of Fluo-4AM.
Metastable Zn2+–Aβ40 Oligomers Cause Neuronal Death
Alterations in neuronal activity and basal [Ca2+]i following treatment with Zn2+-Aβ40 oligomers indicated neuronal dysfunction, which might lead to neuronal death. To test this prediction, we applied Zn2+-Aβ40 oligomers, freshly prepared Zn2+-free Aβ40, or fibrillar Aβ40 to primary hippocampal neurons and measured their viability by immunostaining for neuron-specific enolase, a standard assay for specifically detecting neuronal, rather than glial, cell death. The cell viability experiments were performed in serum-containing medium at 37 °C. We could not assess the morphology of Aβ40 under these conditions because they were indistinguishable from those of serum proteins.
In initial experiments, we added 10 μm of each Aβ40 preparation, 10 mm MOPS, or 20 μm Zn2+ to the neurons and measured neuronal viability following incubation for 48 h. Under these conditions, we observed little or no toxicity induced by the Zn2+-Aβ40 oligomers (supplemental Fig. S6) or any of the other conditions (data not shown).
This result was inconsistent with the toxic effect of the Zn2+-Aβ40 oligomers on spontaneous Ca2+ activity and basal [Ca2+]i levels. A potential explanation for this apparent discrepancy was rapid self-association of the metastable Zn2+-Aβ40 oligomers in serum-containing medium into large, non-toxic aggregates, similar to those shown in supplemental Fig. S4. In contrast to serum-free medium, this medium has a complex composition, including serum proteins and growth factors, which likely affect the rate of Aβ40 aggregation.
To test this hypothesis, we changed our protocol. Instead of adding 10 μm of any Aβ40 preparation in one portion at t = 0, the same total amount of each preparation was added in four freshly prepared aliquots (2.5 μm each) at t = 0, 12, 24, and 36 h. Remarkably, under these conditions, the Zn2+-Aβ40 oligomers reduced neuronal survival by 85% ± 3% (Fig. 4e). In contrast, no statistically significant difference was observed between the viability of untreated neurons and those treated with MOPS buffer, Aβ40 fibrils, or 20 μm Zn2+. 10 μm Aβ40 caused 20 ± 8% decrease in neuronal viability (p < 0.05), in agreement with many previous observations. Overall, our results indicate that freshly prepared Zn2+-Aβ40 oligomers are highly toxic to cultured neurons, induce changes in normal cellular physiology within minutes, and cause neuronal death within hours. Importantly, the apparent toxicity of Zn2+-Aβ40 strongly depends on the assembly state of the metastable oligomers, giving weight to their structure-toxicity interplay (Fig. 4 and supplemental Figs. S3, S4, and S6). Although the quasi-spherical Zn2+-Aβ40 assemblies (∅ = 7–15 nm, Fig. 1) were highly toxic, their large aggregates (∅ ≥ 100 nm, supplemental Fig. S4) were benign.
DISCUSSION
The reported results provide a detailed structure-toxicity study of early-forming, metastable, toxic Zn2+-Aβ40 oligomers. Our experiments in primary hippocampal cultures showed that the small Zn2+-Aβ40 oligomers inhibited spontaneous neuronal activity and caused neuronal death. Destabilization of neuronal network activity is thought to cause the cognitive impairment associated with AD (39, 40). Dysregulation of [Ca2+]i is a prominent feature of AD. It is involved both in neuronal excitotoxicity and in apoptosis (41). Our experiments in primary hippocampal neurons showed that the Zn2+-Aβ40 oligomers inhibited spontaneous neuronal activity (Fig. 4, a–c), induced a time-dependent elevation in basal Ca2+ levels (Fig. 4d), and, at later time points, caused neuronal death (Fig. 4e). The formation of Zn2+-Aβ40 oligomers may disrupt Ca2+ metabolism and synaptic communication, and persistent insults may contribute to neuronal apoptosis.
The small (7–15 nm) Zn2+-Aβ40 assemblies have unique structural characteristics. Our TEM, AFM, CD, and x-ray diffraction data indicate that these oligomers are organized in a cross-β arrangement typical of mature amyloid fibrils (42), yet they exhibit quasi-spherical morphologies characteristic of Aβ oligomers (43). Furthermore, we show that the small quasi-spherical oligomers decrease the rate of fibril growth and, thus, may increase the steady-state concentration of toxic Zn2+-free Aβ forms. Our detailed structure-toxicity characterization of the Zn2+-Aβ40 oligomers indicates that only the early-forming small (7–15 nm) assemblies are toxic. Upon incubation (≥16 h), the initial Zn2+-Aβ40 oligomers coalesce into larger structures (supplemental Fig. S4) and concomitantly lose their toxicity (supplemental Fig. S6). Our data provide mechanistic insights into the work of Deshpande et al. (11), who highlighted the role of Zn2+ in the formation and accumulation of toxic Aβ oligomers, and of Cuajungco et al. (44), who observed loss of Aβ toxicity upon prolonged incubation with Zn2+. Though apparently, Zn2+ binding prevents Aβ fibrillogenesis, the toxic behavior of the Zn2+-Aβ40 oligomers prepared here is akin to that of Zn2+-free Aβ oligomers, which become less toxic upon transformation into fibrils (45, 46).
Similarly to native amylospheroids (23), toxic Zn2+-Aβ40 oligomers may be considered off-pathway with regard to fibril formation (14). Other quasi-spherical toxic oligomers exhibiting β-sheet-rich structures have recently been reported for Aβ40 oligomers in the absence of Zn2+ (22). These oligomers formed following 30–60 h incubation at 4 °C. In contrast, the Zn2+-Aβ40 oligomers reported here were obtained within seconds at room temperature and retained their quasi-spherical morphologies for at least 2 h in serum-free medium. It is tempting to hypothesize that Zn2+ binding accelerates formation of oligomers similar to those reported by Chimon et al. (22), although testing this hypothesis will require detailed, side-by-side comparison of the two species.
On the basis of the data obtained here, we propose a structure-kinetic model for Zn2+-Aβ40 assembly spanning from the earliest metastable toxic oligomers to aged, benign morphologies (Fig. 5). Fig. 5a presents putative arrangements of Zn2+-Aβ40 in mono- to multilayer assemblies rich in β-sheet and cross-β assemblies as depicted by our CD, ThT, and x-ray diffraction analyses (Figs. 2 and 3). X-ray diffraction analysis also implies that the major conformation of Aβ40 in the aggregates resembles a hairpin, similar to the one described in Aβ fibrils by Petkova et al. (47, 48). However, our data cannot reveal the super-structural organization of molecules (i.e. symmetry of organization or registry of Aβ40) in multilayered Aβ40 assemblies (Fig. 5a). Combined AFM and x-ray diffraction analyses showed mostly abundant monolayers as well as bilayers and tetralayers but not trilayer arrangements. We propose that monolayers have kinetic and/or thermodynamic preference for arrangement in bilayers, ultimately forming tetralayers, which may be mediated by intermolecular coordination of Zn2+ ions (14, 25). This may result in enhanced Aβ40 C-terminal dynamics, as reported recently (49).
FIGURE 5.

Model of Zn2+-Aβ40 oligomer structure and assembly. a, Aβ40 packing in Zn2+-Aβ40 viewed along the intermolecular H-bond direction. Aβ40 adopts a hairpin conformation and packs in mono-, bi-, or tetralayers. b, hypothetical arrangement of Zn2+ and Aβ40 in a toxic assembly shown perpendicular to the plane of H-bond direction. c, oligomerization pathways of Aβ40 in the absence (top panels) or presence of Zn2+ (bottom panels).
We estimate a total number of 20 molecules of Aβ40 in Zn2+-Aβ40 monomeric oligomers (110 Å/4.67 Å) (Fig. 5b), where 110 Å is the average diameter per oligomer derived from TEM (Fig. 1, a and b) and 4.67 Å is the cross-β-sheet distance measured by x-ray powder diffraction (Fig. 2b). Each oligomer is composed of small crystalline assemblies of ∼4 Aβ40 molecules, as indicated by the x-ray diffraction analysis. These Zn2+-Aβ40 oligomers are distinct in their three-dimensional structures from analogous, toxic oligomers formed in the absence of Zn2+ (43). Fig. 5c presents a comparative scheme of Aβ40 oligomerization pathways in the presence or absence of Zn2+. Metastable Zn2+-Aβ40 oligomers form quasi-spherical mono-, bi-, or tetralayer toxic structures that strongly interfere with fibril formation. Aged Zn2+-Aβ40 oligomers exhibit benign activity, similar to Zn2+-free Aβ40 fibrils, but differ in their morphologies.
In conclusion, our results provide quantitative structural, spectroscopic, and functional analyses of metastable and toxic Zn2+-Aβ40 oligomers in cultured hippocampal neurons. We show that binding of stoichiometric Zn2+ concentrations modulates Aβ40 neurotoxicity via structural organization mechanisms mediated by coordination chemistry. Hence, carefully targeted, Zn2+-specific chelators may be beneficial for treatment of diseases associated with Aβ oligomerization (50–52).
Acknowledgments
The electron microscopy studies were conducted at the Irving and Cherna Moskowitz Center for Nano and Bio-Nano Imaging at the Weizmann Institute of Science.
This work was supported in part by National Institutes of Health Grants AG027818 (to G. B.) and CA098799 (to I. Sagi). This work was also supported by United States-Israel Binational Science Foundation Grant 2007187 (to G. B. and I. Sagi), by a Human Frontier Science Program cross-disciplinary fellowship and a Dean of Faculty fellowship of the Weizmann Institute (to B. B.), and by the Israel Science Foundation, the Kimmelman Center at the Weizmann Institute, and the Ambach family fund (to I. Sagi).

This article contains supplemental Figs. S1–S6.
- AD
- Alzheimer disease
- Aβ
- amyloid β-protein
- APP
- amyloid β-protein precursor
- TEM
- transmission electron microscopy
- AFM
- atomic force microscopy
- ThT
- thioflavin T
- MEM
- minimal essential medium
- HS
- horse serum.
REFERENCES
- 1. Wolfe M. S. (2003) The secretases of Alzheimer's disease. Curr. Top. Dev. Biol. 54, 233–261 [DOI] [PubMed] [Google Scholar]
- 2. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease. Progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 3. Selkoe D. J. (2008) Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav. Brain. Res. 192, 106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heininger K. (2000) A unifying hypothesis of Alzheimer's disease. IV. Causation and sequence of events. Rev. Neurosci. 11, 213–328 [DOI] [PubMed] [Google Scholar]
- 5. Klein W. L., Stine W. B., Jr., Teplow D. B. (2004) Small assemblies of unmodified amyloid β-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiol. Aging 25, 569–580 [DOI] [PubMed] [Google Scholar]
- 6. Lovell M. A., Robertson J. D., Teesdale W. J., Campbell J. L., Markesbery W. R. (1998) Copper, iron and zinc in Alzheimer's disease senile plaques. J. Neurol. Sci. 158, 47–52 [DOI] [PubMed] [Google Scholar]
- 7. Dong J., Atwood C. S., Anderson V. E., Siedlak S. L., Smith M. A., Perry G., Carey P. R. (2003) Metal binding and oxidation of amyloid-β within isolated senile plaque cores. Raman microscopic evidence. Biochemistry 42, 2768–2773 [DOI] [PubMed] [Google Scholar]
- 8. Miller L. M., Wang Q., Telivala T. P., Smith R. J., Lanzirotti A., Miklossy J. (2006) Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with β-amyloid deposits in Alzheimer's disease. J. Struct. Biol. 155, 30–37 [DOI] [PubMed] [Google Scholar]
- 9. Stoltenberg M., Bush A. I., Bach, Smidt G. K., Larsen A., Rungby J., Lund S., Doering P., Danscher G. (2007) Amyloid plaques arise from zinc-enriched cortical layers in APP/PS1 transgenic mice and are paradoxically enlarged with dietary zinc deficiency. J. Neurosci. 150, 357–369 [DOI] [PubMed] [Google Scholar]
- 10. Frederickson C. J., Koh J. Y., Bush A. I. (2005) The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 6, 449–462 [DOI] [PubMed] [Google Scholar]
- 11. Deshpande A., Kawai H., Metherate R., Glabe C. G., Busciglio J. (2009) A role for synaptic zinc in activity-dependent Aβ oligomer formation and accumulation at excitatory synapses. J. Neurosci. 29, 4004–4015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee J. Y., Cole T. B., Palmiter R. D., Suh S. W., Koh J. Y. (2002) Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 99, 7705–7710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bush A. I., Pettingell W. H., Multhaup G., d Paradis M., Vonsattel J. P., Gusella J. F., Beyreuther K., Masters C. L., Tanzi R. E. (1994) Rapid induction of Alzheimer A β amyloid formation by zinc. Science 265, 1464–1467 [DOI] [PubMed] [Google Scholar]
- 14. Noy D., Solomonov I., Sinkevich O., Arad T., Kjaer K., Sagi I. (2008) Zinc-amyloid β interactions on a millisecond time-scale stabilize non-fibrillar Alzheimer-related species. J. Am. Chem. Soc. 130, 1376–1383 [DOI] [PubMed] [Google Scholar]
- 15. Garai K., Sengupta P., Sahoo B., Maiti S. (2006) Selective destabilization of soluble amyloid β oligomers by divalent metal ions. Biochem. Biophys. Res. Commun. 345, 210–215 [DOI] [PubMed] [Google Scholar]
- 16. Huang X., Atwood C. S., Moir R. D., Hartshorn M. A., Vonsattel J. P., Tanzi R. E., Bush A.I. (1997) Zinc-induced Alzheimer's Aβ1–40 aggregation is mediated by conformational factors. J. Biol. Chem. 272, 26464–26470 [DOI] [PubMed] [Google Scholar]
- 17. Kodali R., Williams A. D., Chemuru S., Wetzel R. (2010) Aβ(1–40) forms five distinct amyloid structures whose β-sheet contents and fibril stabilities are correlated. J. Mol. Biol. 401, 503–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen W. T., Liao Y. H., Yu H. M., Cheng I. H., Chen Y. R. (2011) Distinct effects of Zn2+, Cu2+, Fe3+, and Al3+ on amyloid-β stability, oligomerization, and aggregation. Amyloid-β destabilization promotes annular protofibril formation. J. Biol. Chem. 286, 9646–9656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bitan G., Vollers S. S., Teplow D. B. (2003) Elucidation of primary structure elements controlling early amyloid β-protein oligomerization. J. Biol. Chem. 278, 34882–34889 [DOI] [PubMed] [Google Scholar]
- 20. Papa M., Bundman M. C., Greenberger V., Segal M. (1995) Morphological analysis of dendritic spine development in primary cultures of hippocampal neurons. J. Neurosci. 15, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eilers J., Schneggenburger R., Konnerth A. (1995) in Single Channel Recording (Sakmann B., Neher E., eds) pp. 213–229, Plenum Press, New York [Google Scholar]
- 22. Chimon S., Shaibat M. A., Jones C. R., Calero D. C., Aizezi B., Ishii Y. (2007) Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer's β-amyloid. Nat. Struct. Mol. Biol. 14, 1157–1164 [DOI] [PubMed] [Google Scholar]
- 23. Hoshi M., Sato M., Matsumoto S., Noguchi A., Yasutake K., Yoshida N., Sato K. (2003) Spherical aggregates of β-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β. Proc. Natl. Acad. Sci. U.S.A. 100, 6370–6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ono K., Condron M. M., Teplow D. B. (2009) Structure-neurotoxicity relationships of amyloid β-protein oligomers. Proc. Natl. Acad. Sci. U.S.A. 106, 14745–14750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miller Y., Ma B., Nussinov R. (2010) Zinc ions promote Alzheimer Aβ aggregation via population shift of polymorphic states. Proc. Natl. Acad. Sci. U.S.A. 107, 9490–9495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Glenner G. G., Eanes E. D., Bladen H. A., Linke R. P., Termine J. D. (1974) β-Pleated sheet fibrils. A comparison of native amyloid with synthetic protein fibrils. J. Histochem. Cytochem. 22, 1141–1158 [DOI] [PubMed] [Google Scholar]
- 27. Kirschner D. A., Inouye H., Duffy L. K., Sinclair A., Lind M., Selkoe D. J. (1987) Synthetic peptide homologous to β protein from Alzheimer disease forms amyloid-like fibrils in vitro. Proc. Natl. Acad. Sci. U.S.A. 84, 6953–6957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malinchik S. B., Inouye H., Szumowski K. E., Kirschner D. A. (1998) Structural analysis of Alzheimer's β(1–40) amyloid. Protofilament assembly of tubular fibrils. Biophys. J. 74, 537–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sachse C., Fändrich M., Grigorieff N. (2008) Paired β-sheet structure of an Aβ(1–40) amyloid fibril revealed by electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 105, 7462–7466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jahn T. R., Makin O. S., Morris K. L., Marshall K. E., Tian P., Sikorski P., Serpell L. C. (2010) The common architecture of cross-β amyloid. J. Mol. Biol. 395, 717–727 [DOI] [PubMed] [Google Scholar]
- 31. LeVine H., 3rd (1993) Thioflavine T interaction with synthetic Alzheimer's disease β-amyloid peptides. Detection of amyloid aggregation in solution. Protein Sci. 2, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bernstein S. L., Dupuis N. F., Lazo N. D., Wyttenbach T., Condron M. M., Bitan G., Teplow D. B., Shea J. E., Ruotolo B. T., Robinson C. V., Bowers M. T. (2009) Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nat. Chem. 1, 326–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stine W. B., Jr., Dahlgren K. N., Krafft G. A., LaDu M. J. (2003) In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278, 11612–11622 [DOI] [PubMed] [Google Scholar]
- 34. Nichols M. R., Moss M. A., Reed D. K., Lin W. L., Mukhopadhyay R., Hoh J. H., Rosenberry T. L. (2002) Growth of β-amyloid(1–40) protofibrils by monomer elongation and lateral association. Characterization of distinct products by light scattering and atomic force microscopy. Biochemistry 41, 6115–6127 [DOI] [PubMed] [Google Scholar]
- 35. Isaacs A. M., Senn D. B., Yuan M., Shine J. P., Yankner B. A. (2006) Acceleration of amyloid β-peptide aggregation by physiological concentrations of calcium. J. Biol. Chem. 281, 27916–27923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Itkin A., Dupres V., Dufrêne Y. F., Bechinger B., Ruysschaert J. M., Raussens V. (2011) Calcium ions promote formation of amyloid β-peptide (1–40) oligomers causally implicated in neuronal toxicity of Alzheimer's disease. PloS ONE 6, e18250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Neher E. (2011) in Imaging: A Laboratory Manual (Rafael Y., ed) pp. 417–426, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 38. Khazipov R., Esclapez M., Caillard O., Bernard C., Khalilov I., Tyzio R., Hirsch J., Dzhala V., Berger B., Ben-Ari Y. (2001) Early development of neuronal activity in the primate hippocampus in utero. J. Neurosci. 21, 9770–9781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Palop J. J., Chin J., Mucke L. (2006) A network dysfunction perspective on neurodegenerative diseases. Nature 443, 768–773 [DOI] [PubMed] [Google Scholar]
- 40. Uhlhaas P. J., Singer W. (2006) Neural synchrony in brain disorders. Relevance for cognitive dysfunctions and pathophysiology. Neuron 52, 155–168 [DOI] [PubMed] [Google Scholar]
- 41. LaFerla F. M. (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat. Rev. Neurosci. 3, 862–872 [DOI] [PubMed] [Google Scholar]
- 42. Serpell L. C. (2000) Alzheimer's amyloid fibrils. Structure and assembly. Biochim. Biophys. Acta 1502, 16–30 [DOI] [PubMed] [Google Scholar]
- 43. Rahimi F., Shanmugam A., Bitan G. (2008) Structure-function relationships of pre-fibrillar protein assemblies in Alzheimer's disease and related disorders. Curr. Alzheimer Res. 5, 319–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cuajungco M. P., Goldstein L. E., Nunomura A., Smith M. A., Lim J. T., Atwood C. S., Huang X., Farrag Y. W., Perry G., Bush A. I. (2000) Evidence that the β-Amyloid Plaques of Alzheimer's Disease Represent the Redox-silencing and Entombment of Aβ by Zinc. J. Biol. Chem. 275, 19439–19442 [DOI] [PubMed] [Google Scholar]
- 45. Dahlgren K. N., Manelli A. M., Stine W. B., Jr., Baker L. K., Krafft G. A., LaDu M. J. (2002) Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053 [DOI] [PubMed] [Google Scholar]
- 46. Klein W. L., Krafft G. A., Finch C. E. (2001) Targeting small Aβ oligomers. The solution to an Alzheimer's disease conundrum? Trends Neurosci. 24, 219–224 [DOI] [PubMed] [Google Scholar]
- 47. Petkova A. T., Ishii Y., Balbach J. J., Antzutkin O. N., Leapman R. D., Delaglio F., Tycko R. (2002) A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid-state NMR. Proc. Natl. Acad. Sci. U.S.A. 99, 16742–16747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tycko R. (2011) Solid-state NMR studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 62, 279–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rezaei-Ghaleh N., Giller K., Becker S., Zweckstetter M. (2011) Effect of zinc binding on β-amyloid structure and dynamics: implications for Aβ aggregation. Biophys. J. 101, 1202–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cherny R. A., Legg J. T., McLean C. A., Fairlie D. P., Huang X., Atwood C. S., Beyreuther K., Tanzi R. E., Masters C. L., Bush A. I. (1999) Aqueous dissolution of Alzheimer's disease Aβ amyloid deposits by biometal depletion. J. Biol. Chem. 274, 23223–23228 [DOI] [PubMed] [Google Scholar]
- 51. Adlard P. A., Cherny R. A., Finkelstein D. I., Gautier E., Robb E., Cortes M., Volitakis I., Liu X., Smith J. P., Perez K., Laughton K., Li Q. X., Charman S. A., Nicolazzo J. A., Wilkins S., Deleva K., Lynch T., Kok G., Ritchie C. W., Tanzi R. E., Cappai R., Masters C. L., Barnham K. J., Bush A. I. (2008) Rapid restoration of cognition in Alzheimer's transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Aβ. Neuron 59, 43–55 [DOI] [PubMed] [Google Scholar]
- 52. Faux N. G., Ritchie C. W., Gunn A., Rembach A., Tsatsanis A., Bedo J., Harrison J., Lannfelt L., Blennow K., Zetterberg H., Ingelsson M., Masters C. L., Tanzi R. E., Cummings J. L., Herd C. M., Bush A. I. (2010) PBT2 rapidly improves cognition in Alzheimer's Disease. Additional phase II analyses. J. Alzheimers Dis. 20, 509–516 [DOI] [PubMed] [Google Scholar]