

Background: Protein kinase C-related kinases (PRKs) are involved in the development of cancer and hepatitis C.
Results: N-terminal domains of PRK2 inhibit the interaction with its upstream kinase and are responsible for a dimerization that inhibits PRK2 activity.
Conclusion: PRK2 regulation requires a cross-talk between the N- and C-terminal domains.
Significance: The results provide new perspectives to pharmacologically target PRK2.
Keywords: Allosteric Regulation, Phosphatidylinositol-dependent Kinase-1 (PDK1), Protein Conformation, Protein Kinase C (PKC), Protein Kinases, Protein Phosphorylation, Protein-Protein Interactions
Abstract
Protein kinase C-related protein kinases (PRKs) are effectors of the Rho family of small GTPases and play a role in the development of diseases such as prostate cancer and hepatitis C. Here we examined the mechanism underlying the regulation of PRK2 by its N-terminal region. We show that the N-terminal region of PRK2 prevents the interaction with its upstream kinase, the 3-phosphoinositide-dependent kinase 1 (PDK1), which phosphorylates the activation loop of PRK2. We confirm that the N-terminal region directly inhibits the kinase activity of PRK2. However, in contrast to previous models, our data indicate that this inhibition is mediated in trans through an intermolecular PRK2-PRK2 interaction. Our results also suggest that amino acids 487–501, located in the linker region between the N-terminal domains and the catalytic domain, contribute to the PRK2-PRK2 dimer formation. This dimerization is further supported by other N-terminal domains. Additionally, we provide evidence that the region C-terminal to the catalytic domain intramolecularly activates PRK2. Finally, we discovered that the catalytic domain mediates a cross-talk between the inhibitory N-terminal region and the activating C-terminal region. The results presented here describe a novel mechanism of regulation among AGC kinases and offer new insights into potential approaches to pharmacologically regulate PRK2.
Introduction
Protein kinase C-related protein kinases (PRKs),2 also known as protein kinase N and originally identified as the protease-activated kinase (1–3), are effectors of the Rho family of small GTPases such as Rho and Rac (4, 5). The PRK family comprises three isoforms, PRK1–3. Although PRK1 and PRK2 share high homology in their catalytic domains (87%), they share only ∼48% identity in their N-terminal regulatory domains. With 70% identity between their catalytic domains and 40% in their N-terminal domains, PRK2 and PRK3 share an even lower degree of sequence identity. Recent work has confirmed that all three PRK isoforms can support Rho-dependent cell migration (6) and may be considered to be targets for the development of anti-cancer drugs.
PRK2 is involved in actin cytoskeletal organization (4), keratinocyte cell-cell adhesion (7), and differentiation (8), mainly through activation by Rho GTPases (9). In HeLa cells, PRK2 controls the entry into mitosis and exit from cytokinesis (10). Recently, PRK2 was shown to regulate apical junction formation in human bronchial epithelial cells (11) as well as the migration and invasion of 5637 bladder tumor cells (6). PRK1 and PRK2 were also shown to activate the androgen receptor, which plays an important role in the development of prostate cancer (12, 13). Moreover, PRK2 is the host kinase that phosphorylates the hepatitis C virus RNA polymerase, and PRK2 inhibitors such as Y27632 suppress hepatitis C virus replication (14, 15).
The PRKs belong to the family of AGC protein kinases. These protein kinases possess two conserved structural features involved in the regulation of their activity: (i) a C-terminal extension of the catalytic core that contains a conserved hydrophobic motif (HM), including a phosphorylation site and (ii) the so-called HM/PIF pocket in the catalytic domain. The HM/PIF pocket was originally identified in the AGC kinase 3-phosphoinositide-dependent protein kinase 1 (PDK1) as the binding site for the C-terminal extension of PRK2 (PDK1-interacting fragment, PIF). In contrast to the other AGC kinases, PDK1 does not contain an equivalent C-terminal extension (16, 17).
PDK1 is the activation loop kinase for numerous AGC kinases (18, 19). Many substrates of PDK1, including PRK, PKC, S6K, SGK, and RSK isoforms, interact with PDK1 through the binding of their HM to the PIF pocket of PDK1. It is thought that for several substrates, such as SGK, RSK, and S6K, this docking interaction requires the phosphorylation of the HM site, although the phosphorylation-dependent interaction of S6K and PDK1 is controversial (20). Thus, HM phosphorylation appears to be a mechanism that regulates the docking interaction with PDK1 and the phosphorylation of substrates by PDK1 (21–25). In addition, the phosphorylated HM binds intramolecularly to its own HM/PIF pocket. This interaction stabilizes the active conformation of the kinase domain, explaining the mechanism of activation of AGC kinases by HM phosphorylation (26, 27).
Unlike most substrates of PDK1, PRKs and atypical protein kinases C (PKCs) possess an acidic amino acid that mimics a phosphorylated residue at the position corresponding to the HM phosphorylation site in other AGC kinases. Hence, PRKs and atypical PKCs are not regulated through HM phosphorylation. Recently, we described the regulation of PRK2 by a third phosphorylation site conserved in AGC kinases, the zipper (Z)/turn motif (TM) phosphorylation site (28, 29). Accordingly, phosphorylation of the Z/TM site increases the binding affinity of the C-terminal extension of PRK2 to its catalytic domain, thereby supporting dissociation from PDK1 and maximal PRK2 activation (28).
PRKs were first described to be activated by limited proteolysis (2, 30). In addition, PRK2 is cleaved at amino acid 700 by caspases during apoptosis, and the cleavage product, comprising amino acids 700–984 of PRK2, has pro-apoptotic activity (31, 32). Thus, the N-terminal region of the PRKs plays a role in their regulation. By homology with a consensus phosphorylation site sequence of the PKCs, a potential pseudosubstrate sequence was identified, comprising amino acids 39–53 of PRK1 (33). A peptide derived from this sequence inhibits PRK1 activity. However, there are conflicting results with respect to the identity of the PRK1 autoinhibitory sequence because it has also been reported that PRK1 is only activated upon the deletion of amino acids 455–511 (34). Recently, Shiga et al. (35) reported that a peptide derived from this region comprising amino acids 485–499 inhibits the activity of both PRK1 and PRK2. The N-terminal region of the PRKs also contains three Rho-binding domains (Hr1a, Hr1b, and Hr1c) (36, 37) that mediate interactions with the small GTPases Rho and Rac (38, 39). Another conserved domain in the N-terminal region shows significant similarity to the phospholipid-binding C2 domains of PKCϵ and PKCη (40). However, until now, no function has been described for the C2-like domain in the PRKs, although it could be involved in the activation of PRKs by lipids or the targeting of PRKs to the membrane (9, 11). The C2-like domain and the catalytic domain of PRK2 are separated by a linker of 195 amino acids.
In the present work, we examined in detail the role of the N-terminal region of PRK2 in the mechanism of regulation of its activity. We discovered that the N-terminal region of PRK2 strongly inhibits the interaction with its upstream kinase PDK1. Furthermore, this region supports the formation of PRK2 oligomers that are catalytically inactive. Our results suggest novel approaches for the development of compounds to block or strengthen the oligomeric structure of PRK2 and inhibit or activate PRK2 in cells.
EXPERIMENTAL PROCEDURES
General materials and methods are described in the supplemental “Experimental Procedures.”
Antibodies and Peptides
A phosphospecific antibody recognizing the phosphorylated activation loop of several AGC kinases was purchased from Upstate Biotechnology. Another phosphospecific antibody that recognizes the phosphorylated Z/TM site of PKCβ (T641) was purchased from Abcam. Anti-GST and anti-Myc tag antibodies were obtained from Cell Signaling, and anti-FLAG M2 was obtained from Sigma. An antibody against an epitope in the N terminus of PRK2 was purchased from Cell Signaling, and another antibody against an epitope in the C terminus of PRK2 (C-18) was obtained from Santa Cruz Biotechnology. Fluorescently labeled secondary antibodies were from LiCor (IRDye680 and IRDye800) and Rockland (IRDye800 donkey anti-goat).
The peptides KKCrosstide (KKGRPRTSSFAEG), PIFtide (REPRILSEEEQEMFRDFDYIADWC), and PKLtide (PKLQRQKKIFSKQQG) were synthesized by JPT Peptide Technologies. Peptides K/R-E-PKLtide (PELQEQEEIFSEQQG) and Shuffled PIFtide (DFIREESAWQDRSEFLMDEPYERI) were synthesized by GenScript. PIFtide (Cys → Ser) used for AlphaScreen experiments was synthesized by Pepscan and contained a serine instead of the C-terminal cysteine to avoid the formation of disulfide bridges in the assay.
Expression and Purification of Protein Kinases
The expression and purification of GST-fused proteins were performed essentially as previously described (28, 41). GST-fused protein kinases were expressed from pEBG2T-derived plasmids (12.5 μg plasmid/145 mm dish) by transient transfection into HEK 293 or HEK 293T cells utilizing a polyethylenimine method (42).
His-PRK2 was expressed in Sf9 cells using the Bac-to-Bac system from Invitrogen. Expression and purification using nickel-nitrilotriacetic acid and gel filtration was essentially performed as previously described for His-PDK1 (41) with the modification that the tag was not cleaved from His-PRK2. The fractions containing the His-PRK2 protein peak were pooled and concentrated using Vivaspin concentrators (Sartorius).
Protein Kinase Activity Assay
PRK2 activity was measured in a 20-μl mixture containing 50 mm Tris-HCl, pH 7.4, 0.005 mg/ml of bovine serum albumin, 0.1% (v/v) 2-mercaptoethanol, 10 mm MgCl2, 100 μm [γ-32P]ATP (5–50 cpm/pmol), 0.003% Brij-35, various amounts of PRK2 proteins, and KKCrosstide as the substrate. Specificity of the signal was confirmed by the use of purified GST-protein that did not show any activity to phosphorylate KKCrosstide and in addition by the inhibition of PRK2 by Y27632. A control experiment for the specific inhibition by PKLtide is shown in supplemental Fig. S4. Due to differences in basal activity of the constructs, the inhibition by PRK2 K686M shown in Fig. 5 was obtained at 36 nm GST-PRK2(1–984) and 7 nm GST-PRK2Δ642. Experiments were performed in duplicates or triplicates. The results were similar in at least two separate experiments. One unit is defined as the amount of product (in nmol) that is formed per minute.
FIGURE 5.

Inhibition of the kinase activity of PRK2 by its N-terminal region. The kinase activity of wild-type and N-terminal-truncated PRK2 proteins purified from HEK 293T cells was measured in vitro using the polypeptide KKCrosstide as a substrate. A, effect of the kinase-dead mutant GST-FLAG-PRK2 K686M on the kinase activity of GST-PRK2(1–984) or GST-PRK2Δ642. B, inhibition of the kinase activity of GST-PRK2(1–984) (×), Δ463 (▴), Δ500 (○), and Δ642 (■) by a peptide comprising amino acids 487–501 (PKLQRQKKIFSKQQG; PKLtide). C, effect of mutation of basic residues in the PKL region on the kinase activity of PRK2. Basic residues at positions 488 and 491 (PRK2 KR/AA) or 494, 495, and 498 (PRK2 KKK/AAA) were mutated to alanine; *, p < 0.05 (n = 7). D and E, effect of the isolated N-terminal region with (GST-PRK2(1–656)) or without (GST-PRK2(1–463)) the PKL linker on the kinase activity of GST-FLAG-PRK2(1–984) (D) and GST-PRK2Δ642 (E) (tested at 5 nm). Purified GST was used as negative control.
Protein-Protein Interaction Assay
The interactions between GST-PRK2 and Myc-PDK1 or between GST-PRK2 and PRK2 constructs fused to a FLAG, or HA tag, were evaluated by co-transfection of the appropriate plasmids into HEK 293 cells, pulldown of the GST fusion protein with glutathione-Sepharose 4B, and analysis of the bound proteins by SDS-PAGE followed by immunoblotting as previously described (43). The immune complexes were detected by fluorescently labeled secondary antibodies for multiplex detection on the same membrane. The extent of interaction was quantified using the software MultiGauge version 3.0 (Fujifilm) and normalized over the amount of purified GST-fused protein. In all figures, we show duplicates of independent transfections and independent GST pulldown experiments performed in parallel. Experiments were performed at least twice, although most experiments were repeated multiple times, with similar results.
In Vitro Cross-linking
Cross-linking reactions were performed in HEPES buffer (50 mm HEPES-KOH, pH 7.4, 150 mm NaCl, 0.1 mm EDTA, 0.05% (v/v) β-mercaptoethanol) at room temperature (RT) with the homobifunctional cross-linker BS3 (Thermo Scientific) at 1 mm for 5 or 30 min. The samples were analyzed by SDS-PAGE using 3–8% Tris acetate polyacrylamide gels (Invitrogen) and staining with Coomassie Blue R-250.
Cross-linking in Cell Lysates
HEK 293T cells were transfected with plasmids coding for FLAG-tagged PRK2 proteins. After 48 h, the cells were washed 3 times with 5 ml of PBS, collected in 500 μl of PBS, and centrifuged at 100 × g for 5 min. The cell pellet was resuspended in HEPES lysis buffer (50 mm HEPES, pH 7.0, 100 mm NaCl, 0.2 mm MgSO4, 0.5 mm Na3VO4, 1% Triton X-100, supplemented with reconstituted SIGMAFast protease inhibitor mixture), incubated on ice for 30 min, snap frozen, and stored at −80 °C. After thawing, the lysates were cleared by centrifugation at 17,000 × g for 10 min and directly used for cross-linking with 1 mm BS3 for 30 min at RT. For immunoprecipitation, the samples were diluted to 0.4 mg/ml in HEPES lysis buffer, and 1 ml of sample was incubated with 10 μg of anti-FLAG M2 antibody at 4 °C for 1 h. Ten μl of Protein A/G PLUS-agarose (Santa Cruz Biotechnology) was added, and the samples were incubated for 3 h. After two washes with 50 mm Tris, 100 mm NaCl, 0.1 mm EDTA, 0.1% (v/v) β-mercaptoethanol, the agarose was boiled with 30 μl of 2× SDS loading buffer and analyzed by SDS-PAGE using 3–8% Tris acetate gels followed by transfer onto nitrocellulose membranes and immunostaining.
Cleavage of Purified Proteins with PreScission Protease
GST-fused protein purified from HEK 293T cells and bound to glutathione-Sepharose 4B was washed in cleavage buffer (50 mm Tris, 150 mm NaCl, 1 mm EDTA, 1 mm DTT). PreScission protease (0.5 units; GE Healthcare) was added to 15 μl of affinity matrix in 50 μl of cleavage buffer. After an overnight incubation at 4 °C, samples were centrifuged through SpinX columns (Sigma). The samples not treated with PreScission protease were eluted with 50 mm glutathione, and equal glutathione concentrations were also added to the cleaved protein prior to activity assays.
AlphaScreen Assay
The AlphaScreen assay was performed according to the manufacturer's protocol (PerkinElmer Life Sciences). Reactions were performed in a 25-μl final volume in white 384-well microtiter plates (Greiner). The reaction buffer contained 50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm DTT, 0.01% (v/v) Tween 20, and 0.1% (w/v) BSA. GST-PRK2Δ642 and biotin-PKLtide were mixed with varying concentrations of unlabeled KKCrosstide, protamine sulfate, or PIFtide (Cys → Ser). Subsequently, 5 μl of the bead solution containing anti-GST-coated acceptor beads and streptavidin-coated donor beads (20 μg/ml final concentration) was added to the reaction mixture. The proteins and beads were incubated in the dark for 90 min at RT, and the emission of light from the acceptor beads was measured in the EnVision multilabel reader (PerkinElmer Life Sciences) and analyzed using the EnVision manager software. Results confirming the specificity of the interaction between biotin-PKLtide and GST-PRK2Δ642 are shown in supplemental Fig. S5.
Luciferase Reporter Assay
HEK 293 cells were transfected in a 96-well format using a polyethylenimine method. Per well, 100 ng of mouse mammary tumor virus (MMTV)-LUC reporter was co-transfected with 5 ng of pSGhAR and 5 ng of a PRK2-encoding plasmid (12). Twenty hours prior to analysis, the medium was exchanged for medium containing 100 pm R1881 or dimethyl sulfoxide. Two days post-transfection, the luciferase activity was measured using the Bright-GloTM luciferase assay system (Promega) and the EnVision multilabel reader (PerkinElmer Life Sciences).
Statistical Analysis
All experiments were performed at least twice with similar results. GraphPad Prism was used for the statistical analysis. The values are presented as mean ± S.E. in the respective figure. Significance was calculated by one-way analysis of variance followed by Bonferroni post-hoc testing. A p value < 0.05 was considered statistically significant. The immunoblots shown are representative for the immunoblots of at least two different experiments and the statistical analysis is presented in the supplemental data. For AlphaScreen assays and the concentration-dependent effect of PRK2 K686M, PKLtide, PRK2(1–463), and PRK2(1–656) on the kinase activity of PRK2 proteins, a representative result for at least two independent experiments performed in duplicates or triplicates with less than 10% difference is shown and the values are presented as mean ± S.E. of the duplicates or triplicates.
RESULTS
N-terminal Region of PRK2 Negatively Regulates Its Interaction with PDK1
We first analyzed the role of the N-terminal domains of human PRK2 in the regulation of its interaction with its upstream kinase PDK1. Therefore, we performed PRK2-PDK1 interaction studies using PRK2 deletion constructs successively truncated at the N terminus. To this end, HEK 293 cells were co-transfected with plasmids encoding Myc-PDK1 and either wild-type or N-terminal truncated GST-PRK2 proteins (see Fig. 1A for the wild-type and mutant PRK2 proteins used in this study). After cell lysis, GST-PRK2 was pulled down with glutathione-Sepharose, and the binding of Myc-PDK1 to GST-PRK2 was analyzed by immunoblotting using an anti-Myc antibody. As shown in Fig. 2, deletion of PRK2 Rho-binding domains Hr1a, Hr1b, and Hr1c (GST-PRK2Δ295) increased the interaction of PDK1 and PRK2 by ∼3-fold compared with wild-type PRK2 (GST-PRK2(1–984)). Additional deletion of the linker between the C2-like and the catalytic domain (GST-PRK2Δ500; GST-PRK2Δ642) further increased the binding of PRK2 to PDK1 to ∼5-fold greater than wild-type PRK2 (Fig. 2). Thus, the N-terminal region of PRK2 negatively regulated the interaction with the upstream kinase PDK1.
FIGURE 1.

Schematic representations of PRK2, PRK2-derived proteins, and peptides employed in this study. A, the potential pseudosubstrate region (PS), the Rho-binding domains (Hr1a, Hr1b, Hr1c), the C2-like domain (C2), and the PKL sequence (PKL) are indicated. A close up of the C-terminal region of PRK2(1–984) shows the Z/TM site, the hydrophobic IL patch, and the hydrophobic motif. Proteins were expressed with an N-terminal GST-FLAG tag. Wild-type PRK2(1–984) was also expressed with an N-terminal FLAG tag, an N-terminal GST tag, an N-terminal His tag or a C-terminal HA tag, respectively. Additionally, N-terminal-truncated PRK2 proteins were expressed with an N-terminal FLAG tag. GST-PRK2N-PRK2CD-His contains the cleavage site of PreScission protease (LEVLFQGP). In the kinase-dead mutant PRK2 K686M, the active site lysine was mutated to methionine. The IL/AA mutant carried alanine substitutions at the hydrophobic patch Ile965–Leu966. Two additional mutants used in this study (PRK2 KR/AA and PRK2 KKK/AAA) carried alanine substitutions in the PKL region (amino acids 487–501, PKLQRQKKIFSKQQG). B, list of key peptides used in this study.
FIGURE 2.
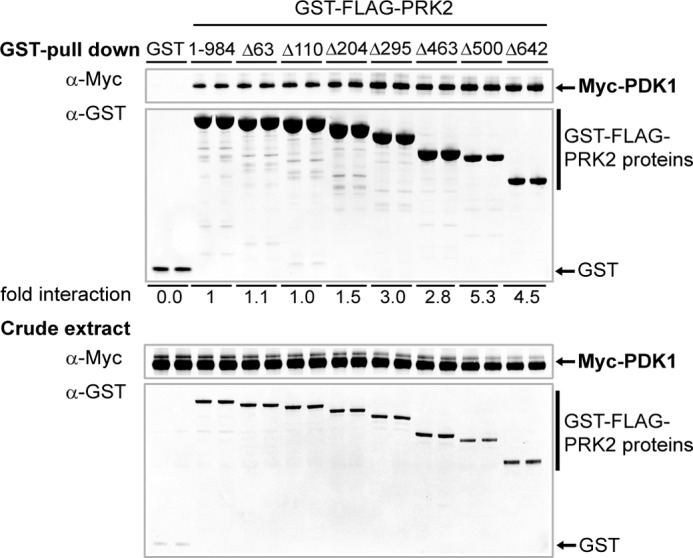
Deletion of the N-terminal domains of PRK2 increases its interaction with PDK1. HEK 293T cells were transiently co-transfected with plasmids encoding Myc-PDK1 and GST-FLAG-PRK2 proteins. After 48 h, the cells were lysed, and the GST-FLAG fusion proteins were purified with glutathione-Sepharose. The bound proteins and aliquots of the cell lysates (crude extract) were analyzed by SDS-PAGE followed by immunoblotting using anti-Myc and anti-GST antibodies, respectively. Duplicates of the transfection of each condition are shown. The immunoblot shown is representative for four experiments and the statistical evaluation is presented in supplemental Table S1.
N-terminal Region of PRK2 Inhibits Its Catalytic Activity
PRKs are reported to be activated by limited proteolysis (2, 30). Kitagawa and colleagues (33) further demonstrated that PRK1 constructs lacking the different N-terminal regions had increased activity. To investigate whether the N-terminal domains of PRK2 directly influence its enzymatic activity, we expressed wild-type PRK2 and N-terminal deletion constructs as GST fusion proteins in HEK 293T cells. After purification with glutathione-Sepharose, the PRK2 proteins were subjected to in vitro kinase activity assays (Fig. 3A). PRK2 lacking the putative pseudosubstrate region (amino acids 49–63; GST-PRK2Δ63) exhibited a similar specific activity as wild-type PRK2. This result argues against a pseudosubstrate function of this region because the deletion of a pseudosubstrate should result in a large increase in kinase activity. Similarly, the specific activity of PRK2 did not considerably change upon the subsequent deletion of amino acids 64–463, including the three Hr1 domains (PRK2Δ110, PRK2Δ204, PRK2Δ295) and the C2-like domain (PRK2Δ463). The further deletion of amino acids 464–500 led to a ∼3-fold increase in specific activity compared with wild-type PRK2, whereas the isolated catalytic domain of PRK2 (GST-PRK2Δ642) showed the highest specific activity, which was ∼9-fold higher than that of wild-type PRK2 (Fig. 3A). Western blotting analysis confirmed that the degree of the activation loop phosphorylation was similar for all PRK2 proteins (Fig. 3B). Thus, differences in the specific activities of the PRK2 truncations were not due to different phosphorylation levels of this site. In contrast, Z/TM site phosphorylation was increased in the deletion constructs, with the highest levels observed in the construct containing only the catalytic domain of PRK2 (PRK2Δ642; Fig. 3C). Because phosphorylation of the Z/TM site is part of the mechanism of activation of PRK2, the increased Z/TM site phosphorylation of GST-PRK2Δ642 could be a reason for its strongly increased specific activity compared with wild-type PRK2 (Fig. 3A). To analyze this possibility, we generated a new PRK2 construct in which we inserted the recognition sequence of the PreScission protease between amino acids 646 and 647 of PRK2 (GST-PRK2N-PRK2CD-His; Fig. 1A). This construct enabled us to eliminate the N-terminal region after purification of the full-length protein. The GST-PRK2N-PRK2CD-His protein was expressed in HEK 293T cells, immobilized on glutathione-Sepharose, cleaved by PreScission protease, and PRK2CD-His comprising the catalytic core was isolated without contamination by the N-terminal region of PRK2. Immunoblot analysis showed that GST-PRK2N-PRK2CD-His and the cleaved product PRK2CD-His were phosphorylated at the Z/TM site to a similar extent (Fig. 3D, left panel). Importantly, the released catalytic domain of PRK2 showed a specific activity that was ∼11-fold increased compared with the uncleaved GST-PRK2N-PRK2CD-His (Fig. 3D, right panel). This demonstrated that the strongly elevated specific activity of the isolated catalytic domain of PRK2 was due not to the increased phosphorylation of the Z/TM site but to deletion of the N-terminal region.
FIGURE 3.

The N-terminal region of PRK2 inhibits its kinase activity. A, the specific activity of wild-type and N-terminal-truncated GST-FLAG-PRK2 proteins purified from HEK 293T cells was measured in vitro; ***, p < 0.001 (n = 9). B and C, levels of activation loop (B) and Z/TM site (C) phosphorylation of N-terminal-truncated PRK2 proteins. Purified GST-FLAG-PRK2 proteins were subjected to SDS-PAGE followed by immunoblotting using phosphospecific antibodies for the activation loop site (T816; B) and the Z/TM site (T958; C). The immunoblot shown is representative for four experiments and the statistical evaluation is presented in supplemental Table S2. D, GST-PRK2N-PRK2CD-His, which contained the PreScission protease cleavage site between amino acids 646 and 647, was cleaved with PreScission protease to release the catalytic domain of PRK2 (amino acids 647–984; PRK2CD-His). Both uncleaved GST-PRK2N-PRK2CD-His and PRK2CD-His were subjected to SDS-PAGE followed by immunoblotting using a phosphospecific Z/TM site antibody or a His tag antibody, respectively, and analyzed as described for B and C. The kinase activity of the respective proteins was measured in vitro using KKCrosstide as a substrate, and the signal was normalized to the protein amount used to measure kinase activity, which was analyzed by SDS-PAGE and quantified by immunoblotting using fluorescently labeled secondary antibodies.
Together, the results described here confirmed that the N-terminal region inhibited the catalytic activity of PRK2. In particular, amino acids 464–642, which do not contain distinct domains, played an important role in the inhibition of PRK2 activity. However, the results did not agree with the model that suggested PRK2 activity is inhibited through the putative pseudosubstrate region located between amino acids 49 and 63.
PRK2 Forms Dimers in Vitro and in Cells
While performing the PRK2 kinase activity measurements described above, we noticed that the specific activity of wild-type PRK2 could not be accurately measured because it was dependent on the protein kinase concentration in the assay, with lower concentrations giving rise to higher specific activities. In contrast, this effect was not observed for a construct comprising only the catalytic domain (PRK2Δ500). Interestingly, when low concentrations (1.8 nm) of PRK2(1–984) were used in the protein kinase activity measurements, the specific activity of PRK2(1–984) was identical to the specific activity of PRK2Δ500. This indicated that inhibition of the activity of wild-type PRK2 depended on the protein concentration. These results were not compatible with a mechanism in which PRK2 activity was inhibited through intramolecular binding of the N-terminal region of PRK2. In contrast, they pointed to an inhibition mediated through intermolecular interactions. Therefore, we analyzed whether PRK2 formed oligomers. As shown in Fig. 4A, PRK2-HA(1–984) significantly bound to GST-PRK2(1–984). This interaction was strongly diminished upon deletion of amino acids 1–500 of PRK2 (GST-PRK2Δ500), demonstrating that the N-terminal region was required for oligomerization of PRK2. To further analyze the intermolecular PRK2 interaction, cross-linking experiments were carried out. For these experiments, His-tagged PRK2(1–984) was recombinantly expressed in Sf9 cells, purified, and incubated with the homobifunctional NHS cross-linker BS3. After 5 min of incubation, oligomerization of purified His-PRK2(1–984) was observed (Fig. 4B). The apparent molecular weights of the oligomers suggested the formation of dimers and tetramers. A similar result was observed when a lysate of HEK 293T cells overexpressing FLAG-PRK2(1–984) was incubated with BS3 (Fig. 4C). The formation of oligomers with the same size as the potential dimer formed by purified His-PRK2(1–984) suggested that the protein complexes did not include other cellular proteins besides PRK2. Most importantly, the results indicated that the complexes observed in vitro reflected an oligomerization that also occurred in the cell.
FIGURE 4.

The N-terminal region of PRK2 mediates an intermolecular PRK2-PRK2 interaction. A, HEK 293 cells were co-transfected with PRK2-HA(1–984) DNA and DNA constructs expressing GST, GST-FLAG-PRK2(1–984), or GST-FLAG-PRK2Δ500, respectively. The interaction was analyzed as described in the legend to Fig. 2 using the indicated antibodies. Duplicates of each transfection condition are shown. B, cross-linking of isolated His-PRK2(1–984) with BS3. The apparent molecular weights of the oligomers formed by His-PRK2(1–984) suggested the formation of dimers and tetramers. C, cross-linking in lysates of HEK 293T cells overexpressing FLAG-PRK2(1–984) or FLAG-PRK2Δ642. D, effect of N-terminal truncation of FLAG-PRK2 on its interaction with GST-PRK2(1–984). HEK 293T cells were co-transfected with GST-PRK2(1–984) and constructs coding for N-terminal truncated FLAG-PRK2 proteins. The interaction was analyzed as described in the legend to Fig. 2 using the indicated antibodies. To compare the level of interaction, duplicates were averaged and a value of 1 was assigned to FLAG-PRK2(1–984). The immunoblot shown is representative for two experiments and the statistical evaluation is presented in supplemental Table S3. A control experiment confirming specificity of the interaction between PRK2 proteins is presented in supplemental Fig. S6.
N-terminal Region of PRK2 Mediates PRK2 Oligomerization and Thereby Inhibits PRK2 Kinase Activity in Trans
Next, we investigated which domain of the N-terminal region participated in the intermolecular PRK2-PRK2 interaction. For this investigation, interaction assays were performed with GST-PRK2(1–984) and FLAG-tagged N-terminal deletion constructs of PRK2 (Fig. 4D). The consecutive deletion of the N-terminal domains, including the Hr1c domain, had only minor effects on the intermolecular PRK2 interaction. However, the interaction between GST-PRK2(1–984) and FLAG-PRK2 constructs was clearly diminished upon deletion of the C2-like domain (FLAG-PRK2Δ463) and was almost abolished when amino acids 464–500 were also deleted (FLAG-PRK2Δ500). Similarly, when FLAG-PRK2Δ642, a construct missing the entire N-terminal region, was transfected, oligomerization was not observed upon the incubation of the cell lysate with the cross-linker BS3 (Fig. 4C).
Taken together, these results confirmed the existence of an intermolecular PRK2-PRK2 interaction that was mediated by the N-terminal domains and demonstrated that, in particular, the interaction required amino acids 464–500 located in the linker between the C2-like domain and the catalytic domain.
We wanted to verify whether the observed oligomerization was indeed the mechanism of inhibition of PRK2 activity. In a kinase activity assay, we measured the influence of a purified kinase-dead mutant of PRK2, in which the lysine necessary for ATP binding was mutated to methionine (PRK2 K686M), on the activity of PRK2. Indeed, we observed a concentration-dependent inhibition of PRK2(1–984) kinase activity in the presence of kinase-dead PRK2 K686M (Fig. 5A). This is in agreement with the previous finding that wild-type PRK2 (PRK2(1–984)) readily formed oligomers. The isolated catalytic domain of PRK2 (PRK2Δ642) was inhibited to a lower degree by PRK2 K686M (Fig. 5A). This was in line with the strongly diminished PRK2-PRK2 interaction observed upon deletion of the N-terminal region (FLAG-PRK2Δ642; Fig. 4D). These results provided evidence that the inhibition of PRK2 by the N-terminal region did not occur intramolecularly but, rather, in trans by the formation of oligomers, in which the N-terminal region of one protein inhibited its interacting partner.
Linker between C2-like Domain and Catalytic Domain Mediates Oligomerization and Inhibition of PRK2 Activity in Trans
Shiga and colleagues (35) recently described a highly basic sequence in the linker between the C2-like and catalytic domain of PRK1 that acts as an inhibitor of PRK1 and PRK2 when used as an isolated peptide in kinase activity assays. This peptide comprised amino acids 485–499 of PRK1 and was shown to compete with the substrate. Similarly, we found that a synthetic peptide with the sequence of the corresponding region in PRK2 (PKLQRQKKIFSKQQG, named “PKLtide”), inhibited the activity of all tested PRK2 proteins in a concentration-dependent manner with IC50 values between 5 and 10 μm (Fig. 5B), indicating a low affinity interaction. The PKL sequence of PRK2 is located in the linker between the C2-like and catalytic domains, hereafter referred to as the PKL linker. To investigate if the PKL region participated in the mechanism of inhibition of PRK2, we prepared PRK2 mutants with alanine substitutions of basic amino acids within the PKL sequence. Kinase activity assays revealed that PRK2 proteins with alanine mutations of Lys488 and Arg491 (PRK2 KR/AA) or lysines 494, 495, and 498 (PRK2 KKK/AAA) had higher specific activities (between 2- and 3-fold) than wild-type PRK2 (PRK2 WT, Fig. 5C). However, the activity of the PRK2 proteins with mutations in the PKL sequence did not reach the level of activity observed for the isolated catalytic domain of PRK2 (PRK2Δ642). Mutation of all basic residues in this region did not lead to a further increase of activity (data not shown). Thus, the PKL sequence contributed to inhibition in trans but did not mediate it alone.
Therefore, we investigated whether the inhibitory effect of kinase-dead PRK2 K686M on the activity of PRK2(1–984) could be explained by the additional binding of other N-terminal domains to PRK2. We performed kinase activity assays with GST-PRK2(1–984) and GST-PRK2Δ642 in the presence and absence of the isolated N-terminal domain (GST-PRK2(1–463) and GST-PRK2(1–656); Fig. 5, D and E). GST-PRK2(1–463), which lacks the PKL linker, showed only a weak inhibitory effect on wild-type PRK2 (PRK2(1–984)) and no effect on the isolated catalytic domain (PRK2Δ642). In contrast, GST-PRK2(1–656), which contains the PKL linker, inhibited the kinase activity of the wild-type PRK2 and the PRK2 catalytic domain (Fig. 5, D and E). The inhibition of the PRK2 catalytic domain by GST-PRK2(1–656) (IC50 = 15 nm) indicated a high affinity of the N-terminal domain to the catalytic domain and was consistent with a tight-binding inhibition because the concentration of PRK2 in the assay was only three times lower than the IC50. Furthermore, the PRK2(1–656) construct comprising all the N-terminal domains including the PKL sequence was ∼500-fold more potent than the isolated PKL sequence (PKLtide; IC50 = 5–10 μm). This suggested that the PKL sequence and other N-terminal domains cooperatively provided the high affinity interaction that inhibited PRK2 upon oligomerization.
To further characterize the PRK2-PRK2 interaction, we performed interaction assays by co-transfecting cells with plasmids encoding the isolated N terminus of PRK2 including (GST-PRK2(1–656)) or excluding (GST-PRK2(1–463)) the PKL linker along with the N-terminal deleted FLAG-tagged PRK2 proteins (supplemental Fig. S1). We concluded that the results can be explained by a model in which PRK2Δ463 is able to oligomerize and the N-terminal region can recognize the dimer but not the monomeric form of the catalytic domain (PRK2Δ642). A schematic model for a possible mechanism that explains these results is presented in supplemental Fig. S1B.
C-terminal PIF Region of PRK2 Is Required for Its Catalytic Activity and Affects Binding of PKLtide
The region C-terminal to the catalytic core has a low but significant homology within most of the protein kinases from the AGC group. In all crystal structures of active AGC kinases solved so far, the C-terminal region interacts with the small lobe of the catalytic domain (supplemental Fig. S2). However, it was still not known if the binding of the C-terminal region of PRK2 to the catalytic domain would also be required for PRK2 activity. To test if this was the case, we used an antibody generated to the C-terminal region of PRK2 (PRK2 C-18) that did not recognize PRK2 when the Ile-Leu hydrophobic patch within the C-terminal sequence, REPRILSEEEQEMFRDFDYIADWC (amino acids 961–984, PIF sequence), was mutated to Ala-Ala (supplemental Fig. S3). The addition of the PRK2 C-18 antibody in kinase activity assays inhibited the activity of PRK2(1–984) and also PRK2Δ500 lacking most of the regions N-terminal to the catalytic domain (Fig. 6, A and B). This inhibition was abolished upon mutation of Ile965 and Leu966 to Ala (PRK2 IL/AA; Fig. 6A) confirming that the inhibition was due to a direct interaction of the antibody with the C-terminal PIF sequence of PRK2. The inclusion of a peptide comprising the nonmutated PIF sequence of PRK2 (PIFtide) in the assay also abolished the inhibition (Fig. 6B). As a further control, the addition of PIFtide mutated at the Ile-Leu site (PIFtide IL/AA) did not affect the inhibition mediated by the antibody (Fig. 6B). These data showed that sequestration of the C-terminal PIF sequence by the PRK2 C-18 antibody inhibited the activity of PRK2, demonstrating that interaction of the C-terminal region with the catalytic domain was required for activity. In addition, the same effect was also observed for a construct containing the catalytic domain of PRK2 (Δ500) that is essentially monomeric and therefore not inhibited by another PRK2 molecule. Based on the above results and on the role of the equivalent C-terminal region in other AGC kinases (supplemental Fig. S2), we suggest that the C-terminal region of PRK2 containing the PIF sequence is intramolecularly bound to the HM/PIF pocket on the catalytic domain in the active monomeric structure of this protein.
FIGURE 6.

The communication between N-terminal and C-terminal regions of PRK2. A and B, the kinase activity of GST-PRK2 proteins purified from HEK 293 cells was measured in vitro using the polypeptide KKCrosstide as a substrate in the presence or absence of the PRK2 C-18 antibody directed to the C terminus of PRK2. A, effect of the PRK2 C-18 antibody on the kinase activity of PRK2 WT and PRK2 with alanine substitutions of Ile965 and Leu966 (PRK2 IL/AA). B, the addition of PIFtide (amino acids 961–984 of PRK2, REPRILSEEEQEMFRDFDYIADWC) but not PIFtide IL/AA abolished the inhibition of the kinase activity of PRK2(1–984) (left) and PRK2Δ500 (right) mediated by the 2 (PRK2 C-18) antibody. C–E, analysis of the effect of the substrate peptide KKCrosstide (C), protamine sulfate (D), PIFtide and Shuffled PIFtide (DFIREESAWQDRSEFLMDEPYERI) (E) on the interaction between GST-PRK2Δ642 and biotin-PKLtide using AlphaScreen technology. Similar to Shuffled PIFtide, a PIFtide mutant (REPRAASGGGQEMRRDRAYIADWS) also did not affect the binding of GST-PRK2Δ642 and biotin-PKLtide (data not shown).
The results described here showed that the N-terminal region blocked the interaction of PRK2 with PDK1 (Fig. 2), whereas previous reports indicate that the C-terminal region is required for the PRK2-PDK1 docking interaction (24). Furthermore, the N-terminal region inhibited the enzymatic activity of PRK2 (Fig. 3), whereas the PIF sequence (Fig. 6B) and phosphorylation of the Z/TM site on the C-terminal segment (28) participated directly in the activation of PRK2. Thus, the results indicated that the N-terminal and C-terminal regions of PRK2 had opposite effects on the activity and on the interaction with PDK1. Therefore, it was of interest to investigate whether there was a direct cross-talk between the N- and C-terminal regions of PRK2.
To this end, we set up an AlphaScreen interaction assay with biotinylated PKLtide and the isolated catalytic domain of PRK2 (GST-PRK2Δ642) using streptavidin (donor) beads and anti-GST (acceptor) beads. Biotin-PKLtide readily interacted with GST-PRK2Δ642, as verified by the emission of light (Fig. 6, C–E). In addition, in agreement with PKLtide being a competitive inhibitor with respect to the substrate, the peptide substrate KKCrosstide (Km = 100 μm) as well as protamine sulfate, a PRK2 substrate with more favorable kinetics of phosphorylation (44), disrupted the binding of PKLtide (Fig. 6, C and D). Surprisingly, PIFtide, expected to bind to the HM/PIF pocket, a site distinct from the substrate-binding site, potently disrupted the interaction between biotin-PKLtide and GST-PRK2Δ642 (Fig. 6E). This result indicated that PIFtide and PKLtide could not bind simultaneously to PRK2, suggesting that the peptides bind to different conformations of the catalytic domain. Because the binding of the PIF sequence to the catalytic domain of PRK2 is required for activity, we deduce that this binding stabilizes the active conformation of PRK2. Therefore, we suggest that PKLtide may bind to the substrate-binding site in an inactive conformation rather than binding as a pseudosubstrate to the active conformation.
PKL Linker of PRK2 Is Required for Inhibition of PRK2 Activity in Cells
PRK2 mediates androgen receptor-dependent gene transcription (12), which can be tested in transfected cells using a luciferase readout. We used this approach to study the effect of different PRK2 constructs on a physiologically relevant signaling system. For this study, wild-type PRK2 and constructs with N-terminal deletions were co-transfected with plasmids coding for the androgen receptor and a luciferase reporter regulated by the androgen- and glucocorticoid-inducible hormone response element found in the MMTV long terminal repeats (45). The effect of the PRK2 proteins on the activity of the androgen receptor was analyzed with and without androgen receptor stimulation using the androgen R1881 (Fig. 7). GST-PRK2Δ500 and GST-PRK2Δ642 prompted increased activity of luciferase in the absence of the antagonist. In particular, PRK2Δ642 induced a similar level of luciferase activity as observed in cells stimulated with R1881 in the absence of plasmids encoding PRK2. On the other hand, all GST-PRK2 constructs containing the PKL sequence were essentially inactive in cells. The results are in agreement with our in vitro data, which showed that the constructs containing the isolated catalytic domain were constitutively active, whereas the presence of the PKL sequence prompted oligomerization and the inhibition of activity. Taken together, the effects of the N-terminal deletion mutants in a relevant PRK2 signaling system in cells correlate well with the results derived from in vitro experiments.
FIGURE 7.
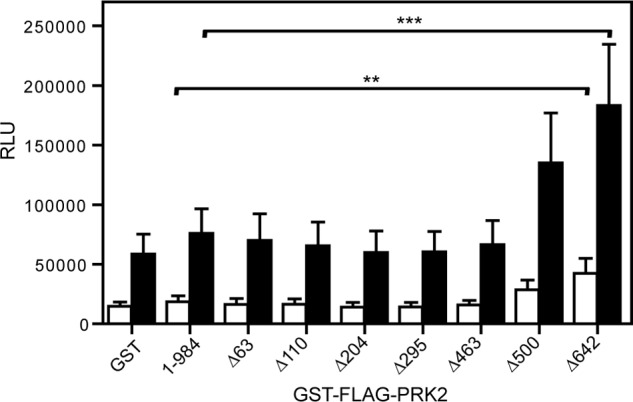
Deletion of the N-terminal region of PRK2 leads to elevated androgen receptor activation. HEK 293 cells were co-transfected with plasmids coding for the androgen receptor, pSGhAR, the MMTV-LUC reporter plasmid and different PRK2-encoding plasmids in triplicates in a 96-well format. For androgen receptor activation, the cells were stimulated by the addition of 100 pm of the agonist R1881 (filled bars) or dimethyl sulfoxide (empty bars) as a control. Luciferase activity was measured using the EnVision multilabel reader. Cells transfected with only pSGhAR and MMTV-LUC or cells co-transfected with empty plasmid (GST) were used as controls (n = 5); *, p < 0.05; **, p < 0.01. RLU, relative luciferase response units.
DISCUSSION
PRKs are potential targets for the treatment of cancer and hepatitis C infection. In the present work, we discovered essential aspects of the mechanism of regulation of PRK2, which had remained elusive over the years: 1) the N-terminal region inhibits the interaction with the upstream kinase PDK1, 2) PRK2 forms oligomers in vitro and within cells, 3) the intermolecular interaction between PRK2 molecules in an oligomer inhibits the catalytic activity of PRK2, and 4) the activating C-terminal PIF sequence displaces the binding of the inhibitory N-terminal PKL sequence. This is the first detailed description of the regulation of AGC kinases by oligomerization. The results further suggest that the catalytic domain of PRK2 is subject to conformational changes and, as a consequence, offer novel insights into potential approaches to pharmacologically regulate PRK2.
The phosphorylation of a number of PDK1 substrates is regulated by the modulation of the ability of the substrates to bind to PDK1 (21–25). In this study, we showed that deletion of the N-terminal region of PRK2 greatly enhanced the ability of PRK2 to interact with PDK1. Thus, these domains actively inhibit the ability of PRK2 to bind to PDK1, which supports the idea that the interaction between PRKs and PDK1 is indeed regulated.
PRKs share a number of characteristics with the group of PKC protein kinases that are inhibited intramolecularly by their N-terminal domains. However, in contrast to previous models of PKC or PRK regulation, our results suggested that the inhibition of PRK2 by its N-terminal domain was not mediated by an intramolecular interaction. Indeed, we provide several lines of evidence indicating that PRK2 forms oligomers, that oligomer formation is mediated by the N-terminal region, and that the inhibition is achieved in trans by the interacting partner in the oligomeric structure. These lines of evidence include the following: 1) the dilution of PRK2 preparations enhanced PRK2 catalytic activity, 2) GST- and FLAG-tagged PRK2 proteins interacted whereas the deletion of the N-terminal region decreased the interaction, 3) the cross-linking of purified wild-type PRK2 produced dimers and, to a lesser degree, tetramers, 4) cross-linking in extracts from cells expressing recombinant wild-type PRK2 produced dimers, whereas the overexpressed catalytic domain of PRK2 did not, 5) wild-type PRK2 was inhibited by the addition of a kinase-dead mutant PRK2, 6) the N-terminal region of PRK2 inhibited the activity in trans, and 7) mutation of the PKL sequence important for dimerization produced PRK2 proteins with higher basal activity. Therefore, we suggest that PRK2 forms dimers and, potentially, higher order oligomers that keep PRK2 inactive. Because we also detected PRK2 tetramers, we cannot discount the possibility that the tetramers are physiologically relevant as well.
The formation of dimers is a mechanism of regulation widely found within receptor tyrosine kinases. The molecular details of the dimer formation shows that they interact through a largely hydrophobic interface (46, 47) that is comparable with the interaction between PDK1 and its substrates through the HM/PIF pocket (19). Homodimers of AGC kinases have also been described in the literature for PKB/Akt, mediated by the PH domain (48, 49), and for PKCα, mediated by the C1 and C2 domains (50). Recent work using fluorescence lifetime imaging microscopy deduced that PDK1 forms homodimers in cells (51). Altogether, the oligomerization mechanisms described for tyrosine kinases and suggested for AGC kinases PKB/Akt, PDK1, and PKCα seem to be different from the mechanism we propose for PRK2. In particular, the mechanism of PRK2 regulation appears very different from the models currently accepted for the regulation of PKCs. We propose that the PRK2-PRK2 interaction is mediated mainly by two binding sites on the catalytic domain (Fig. 8A): the PKL sequence-binding site (binding site 1) and a binding site for other N-terminal domains (binding site 2).
FIGURE 8.

Potential mechanisms for PRK2 dimerization. A, a schematic representation of the active PRK2 monomer. The catalytic domain contains two binding sites for N-terminal domains (binding site 1 and binding site 2, boxed). The N-terminal region contains the PKL linker and additional sites that potentially bind to the catalytic domain. For simplification, the C-terminal extension of PRK2 is not shown and the N-terminal region is minimized. B, a possible model for the dimerization of PRK2 molecules is based on the existence of pre-formed binding sites (binding site 2) already present in the monomer. In this model, the PKL region of one molecule interacts with the dimer partner, whereas the other N-terminal sites interact with both molecules of the dimer.
Consistent with the idea that the PKL sequence binds to the substrate-binding site, the PKL sequence requires positively charged residues, like prototypical substrates of PRK2. In addition, PKLtide is displaced from its binding site by the PRK2 substrates KKCrosstide and protamine sulfate, but not by (K/R)E-PKLtide. The crystal structure of the catalytic subunit of PKA in complex with the pseudosubstrate inhibitor (PKI; see supplemental Fig. S2) shows that the peptide inhibitor stabilizes the close-active structure of the catalytic domain and positions all residues for catalysis (52). By analogy, we suggest that the PKL-binding site (binding site 1) corresponds to the PRK2 peptide-substrate binding groove, contiguous to the ATP-binding site between the small and large lobes of the kinase domain. On the other hand, we currently lack information on the location of the binding region for other N-terminal domains (binding site 2) that support the formation of oligomers of PRK2. Our observations are consistent with a model in which the other N-terminal domains recognize the dimer of PRK2. This finding could be explained if the N-terminal domains recognized the dimer by having recognition sites on both molecules within the dimer (Fig. 8B). Alternatively, the N-terminal domains could recognize a specific conformation of the kinase that is triggered by the formation of the inactive dimer. Interestingly, our present work revealed the possible existence of at least two distinct conformations of the PRK2 kinase domain in the PKL-bound (inactive) and PIF-bound (active) forms of PRK2. It is, therefore, possible that binding of the PKL region to binding site 1 induces a conformational change that creates binding site 2 (Fig. 8).
Except for PDK1, AGC kinases possess a rather conserved extension C-terminal to the catalytic core. The crystal structures of active AGC kinases show that this C-terminal region extends over the small lobe with the hydrophobic motif Phe-Xaa-Xaa-Phe occupying the HM/PIF pocket (supplemental Fig. S2). In previous work we showed that the phosphorylation of this C-terminal region at the Z/TM site of PRK2 triggers a direct interaction with a phosphate-binding site on the small lobe of the catalytic domain. We now further show that the antibodies that bind to the IL hydrophobic patch, located further C-terminal within the PIF sequence, inhibit PRK2 activity, thereby confirming the important role of the C-terminal region on the active structure of PRK2. Because the binding of the C terminus was necessary for activity, the mechanism appears similar to that found in other AGC kinases, where the C-terminal extension stabilizes the catalytic domain in an active conformation. In addition, the binding site for the PIF sequence of PRK2 is not expected to overlap with the substrate-binding site because an interaction with the substrate-binding site would not support but inhibit the activity of PRK2. Notably, we observed that PIFtide, the peptide derived from the PIF region on the C terminus of PRK2, disrupted the interaction between PRK2 and the inhibitory PKLtide. This revealed that a simultaneous binding of PIFtide and PKLtide to PRK2 was not possible, suggesting that either (a) they compete for the same binding site on the small lobe of the catalytic domain or (b) there is an allosteric cross-talk between the PIFtide- and PKLtide-binding sites on PRK2. Both scenarios, however, imply that the inhibitory N-terminal domains and the activating C-terminal region communicate at the level of the catalytic core to regulate PRK2 activity. Although our results cannot fully discard any possibilities, our accumulating evidence suggests that PKLtide binds to the substrate-binding site, but stabilizing an inactive conformation of the catalytic core, which is not suitable for the binding of PIFtide. Thus, it is tempting to speculate that binding the PKL sequence would affect the shape of the HM/PIF pocket, thereby preventing the binding of the C-terminal PIF sequence to the catalytic domain. In any case, however, one should predict that the C-terminal region of the dimeric inactive form of PRK2 would be exposed and, because the PIF sequence has high affinity for PDK1 (∼50 nm), this would readily trigger the interaction with PDK1. However, this was not the case, because wild-type PRK2 had greatly decreased affinity for PDK1 compared with the isolated catalytic domain. Only upon deletion of the N-terminal domains of PRK2 was its interaction with PDK1 enhanced. Therefore, we must hypothesize that, in the inactive structure of PRK2, the C terminus is neither bound to the HM/PIF pocket nor freely available for interaction with PDK1. It is therefore possible that, in the inactive oligomeric structures of PRK2, the N-terminal domains interact directly or indirectly with the C-terminal PIF region of PRK2, preventing its ability to interact with PDK1.
Combining our results described here and previous information on the mechanism of regulation of PRKs, we suggest the following extended model for PRK2 regulation. PRK2 forms dimers and possibly higher order oligomers in cells, which represent the inhibited form of PRK2. The binding of the PKL sequence to the active site of the dimer partner affects the conformation of the catalytic domain, thereby allosterically preventing the binding of the C terminus to the HM/PIF pocket. The activation of PRK2 requires the binding of Rho-GTP to the Hr1 domains of PRK2. Upon binding of Rho to the Hr1 domains and the action of at least one more cellular partner, the PIF motif would be released. The exposed PIF motif would then mediate the docking interaction of PRK2 with PDK1, thereby leading to activation loop phosphorylation of PRK2. Subsequently, the Z/TM site is phosphorylated by a yet unknown protein kinase. Phosphorylation of the activation loop and Z/TM site supports the dissociation of PRK2 from PDK1, and promotes the intramolecular binding of the PIF sequence to the HM/PIF pocket, resulting in stabilization of the active conformation of PRK2. In turn, stabilization of the active conformation would trigger the dissociation of the inhibitory PKL sequence from the active site and, eventually, the release of active monomers of PRK2. Flynn et al. (53) suggest that PRK1 is dephosphorylated and inactive under basal conditions and that it becomes phosphorylated by PDK1 and thus activated upon stimulation. Our data are compatible with such a model but also show that phosphorylated PRK2 can maintain a low basal activity and be activated by regulating the oligomeric structure of the kinase. It is, therefore, possible that both mechanisms co-exist or that distinct mechanisms regulate PRK1 and PRK2 activity in different signaling situations.
Understanding the molecular details of PRK2 regulation opens the possibility to modulate the activity of PRKs in cells. For example, small compounds that stabilize the oligomeric structure will inhibit PRK2 activity, whereas compounds supporting the dissociation of the oligomer will activate PRK2. Over the last years, we provided evidence that the HM/PIF pocket can be targeted with small compounds leading to the development of allosteric activators or allosteric inhibitors of AGC kinases (41, 43, 54–56). Our present results indicate that the HM/PIF pocket also has an allosteric role in the regulation of PRK2 by affecting the binding of the PKL sequence to the substrate-binding site. Small compounds that bind to the HM/PIF pocket in the presence of the PKL sequence would, according to the model, stabilize the inhibition by the N-terminal region. In contrast, compounds mimicking PIFtide would promote the dissociation of PRK2 molecules and thus promote their activation. Compounds with such a novel mode of action could serve as selective pharmacological tools to further define the function of PRKs in cells and organisms. Importantly, because of the increased selectivity of allosteric inhibitors compared with ATP-competitive inhibitors, such compounds would open the possibility for the development of selective anticancer and antiviral drugs.
Acknowledgments
We thank Dr. Eric Metzger for providing the pSGhAR and MMTV-LUC plasmids. We are grateful to Iris Adrian for initial experiments within this project and Dr. Laura A. Lopez-Garcia for preliminary molecular biology work. We thank Tje Lin Chung for help with the statistical evaluations. We are also grateful to Dr. Jörg Schulze for careful reading of the manuscript and comments on the work.
This work was supported by the Bundesministerium für Bildung und Forschung GO-Bio and Deutsche Forschungsgemeinschaft Grant BI 1044/5-1.

This article contains supplemental Experimental Procedures, Tables S1–S3, and Figs. S1–S6.
- PRK
- protein kinase C-related protein kinase (also termed PAK or PKN)
- PKC
- protein kinase C
- PDK1
- 3-phosphoinositide-dependent protein kinase-1
- PKB
- protein kinase B (also termed Akt)
- RSK
- p90 ribosomal S6 kinase
- S6K
- p70 ribosomal S6 kinase
- SGK
- serum and glucocorticoid-induced protein kinase
- AGC kinases
- group of protein kinases comprising PKA, PKG, and PKC
- HM
- hydrophobic motif
- Z/TM phosphorylation site
- zipper/turn motif phosphorylation site
- PIF
- PDK1-interacting fragment
- HCV
- hepatitis C virus
- MMTV
- mouse mammary tumor virus.
REFERENCES
- 1. Palmer R. H., Ridden J., Parker P. J. (1994) Identification of multiple, novel, protein kinase C-related gene products. FEBS Lett. 356, 5–8 [DOI] [PubMed] [Google Scholar]
- 2. Gabrielli B., Wettenhall R. E., Kemp B. E., Quinn M., Bizonova L. (1984) Phosphorylation of ribosomal protein S6 and a peptide analogue of S6 by a protease-activated kinase isolated from rat liver. FEBS Lett. 175, 219–226 [DOI] [PubMed] [Google Scholar]
- 3. Mukai H., Ono Y. (1994) A novel protein kinase with leucine zipper-like sequences. Its catalytic domain is highly homologous to that of protein kinase C. Biochem. Biophys. Res. Commun. 199, 897–904 [DOI] [PubMed] [Google Scholar]
- 4. Vincent S., Settleman J. (1997) The PRK2 kinase is a potential effector target of both Rho and Rac GTPases and regulates actin cytoskeletal organization. Mol. Cell Biol. 17, 2247–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Quilliam L. A., Lambert Q. T., Mickelson-Young L. A., Westwick J. K., Sparks A. B., Kay B. K., Jenkins N. A., Gilbert D. J., Copeland N. G., Der C. J. (1996) Isolation of a NCK-associated kinase, PRK2, an SH3-binding protein and potential effector of Rho protein signaling. J. Biol. Chem. 271, 28772–28776 [DOI] [PubMed] [Google Scholar]
- 6. Lachmann S., Jevons A., De Rycker M., Casamassima A., Radtke S., Collazos A., Parker P. J. (2011) Regulatory domain selectivity in the cell-type specific PKN dependence of cell migration. PLoS One 6, e21732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Calautti E., Grossi M., Mammucari C., Aoyama Y., Pirro M., Ono Y., Li J., Dotto G. P. (2002) Fyn tyrosine kinase is a downstream mediator of Rho/PRK2 function in keratinocyte cell-cell adhesion. J. Cell Biol. 156, 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bourguignon L. Y., Singleton P. A., Diedrich F. (2004) Hyaluronan-CD44 interaction with Rac1-dependent protein kinase Nγ promotes phospholipase Cγ1 activation, Ca2+ signaling, and cortactin-cytoskeleton function leading to keratinocyte adhesion and differentiation. J. Biol. Chem. 279, 29654–29669 [DOI] [PubMed] [Google Scholar]
- 9. Mukai H. (2003) The structure and function of PKN, a protein kinase having a catalytic domain homologous to that of PKC. J. Biochem. 133, 17–27 [DOI] [PubMed] [Google Scholar]
- 10. Schmidt A., Durgan J., Magalhaes A., Hall A. (2007) Rho GTPases regulate PRK2/PKN2 to control entry into mitosis and exit from cytokinesis. EMBO J. 26, 1624–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wallace S. W., Magalhaes A., Hall A. (2011) The Rho target PRK2 regulates apical junction formation in human bronchial epithelial cells. Mol. Cell. Biol. 31, 81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Metzger E., Müller J. M., Ferrari S., Buettner R., Schüle R. (2003) A novel inducible transactivation domain in the androgen receptor. Implications for PRK in prostate cancer. EMBO J. 22, 270–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Metzger E., Yin N., Wissmann M., Kunowska N., Fischer K., Friedrichs N., Patnaik D., Higgins J. M., Potier N., Scheidtmann K. H., Buettner R., Schüle R. (2008) Phosphorylation of histone H3 at threonine 11 establishes a novel chromatin mark for transcriptional regulation. Nat. Cell Biol. 10, 53–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim S. J., Kim J. H., Kim Y. G., Lim H. S., Oh J. W. (2004) Protein kinase C-related kinase 2 regulates hepatitis C virus RNA polymerase function by phosphorylation. J. Biol. Chem. 279, 50031–50041 [DOI] [PubMed] [Google Scholar]
- 15. Kim S. J., Kim J. H., Sun J. M., Kim M. G., Oh J. W. (2009) Suppression of hepatitis C virus replication by protein kinase C-related kinase 2 inhibitors that block phosphorylation of viral RNA polymerase. J. Viral Hepat. 16, 697–704 [DOI] [PubMed] [Google Scholar]
- 16. Vanhaesebroeck B., Alessi D. R. (2000) The PI3K-PDK1 connection. More than just a road to PKB. Biochem. J. 346, 561–576 [PMC free article] [PubMed] [Google Scholar]
- 17. Leslie N. R., Biondi R. M., Alessi D. R. (2001) Phosphoinositide-regulated kinases and phosphoinositide phosphatases. Chem. Rev. 101, 2365–2380 [DOI] [PubMed] [Google Scholar]
- 18. Belham C., Wu S., Avruch J. (1999) Intracellular signalling. PDK1, a kinase at the hub of things. Curr. Biol. 9, R93–96 [DOI] [PubMed] [Google Scholar]
- 19. Biondi R. M., Cheung P. C., Casamayor A., Deak M., Currie R. A., Alessi D. R. (2000) Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J. 19, 979–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Keshwani M. M., von Daake S., Newton A. C., Harris T. K., Taylor S. S. (2011) Hydrophobic motif phosphorylation is not required for activation loop phosphorylation of p70 ribosomal protein S6 kinase 1 (S6K1). J. Biol. Chem. 286, 23552–23558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Biondi R. M., Komander D., Thomas C. C., Lizcano J. M., Deak M., Alessi D. R., van Aalten D. M. (2002) High resolution crystal structure of the human PDK1 catalytic domain defines the regulatory phosphopeptide docking site. EMBO J. 21, 4219–4228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frödin M., Jensen C. J., Merienne K., Gammeltoft S. (2000) A phosphoserine-regulated docking site in the protein kinase RSK2 that recruits and activates PDK1. EMBO J. 19, 2924–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Biondi R. M., Kieloch A., Currie R. A., Deak M., Alessi D. R. (2001) The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 20, 4380–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Balendran A., Biondi R. M., Cheung P. C., Casamayor A., Deak M., Alessi D. R. (2000) A 3-phosphoinositide-dependent protein kinase-1 (PDK1) docking site is required for the phosphorylation of protein kinase Cζ (PKCζ) and PKC-related kinase 2 by PDK1. J. Biol. Chem. 275, 20806–20813 [DOI] [PubMed] [Google Scholar]
- 25. Gao T., Toker A., Newton A. C. (2001) The carboxyl terminus of protein kinase C provides a switch to regulate its interaction with the phosphoinositide-dependent kinase, PDK-1. J. Biol. Chem. 276, 19588–19596 [DOI] [PubMed] [Google Scholar]
- 26. Frödin M., Antal T. L., Dümmler B. A., Jensen C. J., Deak M., Gammeltoft S., Biondi R. M. (2002) A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J. 21, 5396–5407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang J., Cron P., Thompson V., Good V. M., Hess D., Hemmings B. A., Barford D. (2002) Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol. Cell 9, 1227–1240 [DOI] [PubMed] [Google Scholar]
- 28. Dettori R., Sonzogni S., Meyer L., Lopez-Garcia L. A., Morrice N. A., Zeuzem S., Engel M., Piiper A., Neimanis S., Frödin M., Biondi R. M. (2009) Regulation of the interaction between protein kinase C-related protein kinase 2 (PRK2) and its upstream kinase, 3-phosphoinositide-dependent protein kinase 1 (PDK1). J. Biol. Chem. 284, 30318–30327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hauge C., Antal T. L., Hirschberg D., Doehn U., Thorup K., Idrissova L., Hansen K., Jensen O. N., Jørgensen T. J., Biondi R. M., Frödin M. (2007) Mechanism for activation of the growth factor-activated AGC kinases by turn motif phosphorylation. EMBO J. 26, 2251–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mukai H., Kitagawa M., Shibata H., Takanaga H., Mori K., Shimakawa M., Miyahara M., Hirao K., Ono Y. (1994) Activation of PKN, a novel 120-kDa protein kinase with leucine zipper-like sequences, by unsaturated fatty acids and by limited proteolysis. Biochem. Biophys. Res. Commun. 204, 348–356 [DOI] [PubMed] [Google Scholar]
- 31. Takahashi M., Mukai H., Toshimori M., Miyamoto M., Ono Y. (1998) Proteolytic activation of PKN by caspase-3 or related protease during apoptosis. Proc. Natl. Acad. Sci. U.S.A. 95, 11566–11571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koh H., Lee K. H., Kim D., Kim S., Kim J. W., Chung J. (2000) Inhibition of Akt and its anti-apoptotic activities by tumor necrosis factor-induced protein kinase C-related kinase 2 (PRK2) cleavage. J. Biol. Chem. 275, 34451–34458 [DOI] [PubMed] [Google Scholar]
- 33. Kitagawa M., Shibata H., Toshimori M., Mukai H., Ono Y. (1996) The role of the unique motifs in the amino-terminal region of PKN on its enzymatic activity. Biochem. Biophys. Res. Commun. 220, 963–968 [DOI] [PubMed] [Google Scholar]
- 34. Yoshinaga C., Mukai H., Toshimori M., Miyamoto M., Ono Y. (1999) Mutational analysis of the regulatory mechanism of PKN. The regulatory region of PKN contains an arachidonic acid-sensitive autoinhibitory domain. J. Biochem. 126, 475–484 [DOI] [PubMed] [Google Scholar]
- 35. Shiga K., Takayama K., Futaki S., Hutti J. E., Cantley L. C., Ueki K., Ono Y., Mukai H. (2010) Development of an intracellularly acting inhibitory peptide selective for PKN. Biochem. J. 425, 445–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Palmer R. H., Ridden J., Parker P. J. (1995) Cloning and expression patterns of two members of a novel protein kinase-C-related kinase family. Eur. J. Biochem. 227, 344–351 [DOI] [PubMed] [Google Scholar]
- 37. Maesaki R., Ihara K., Shimizu T., Kuroda S., Kaibuchi K., Hakoshima T. (1999) The structural basis of Rho effector recognition revealed by the crystal structure of human RhoA complexed with the effector domain of PKN/PRK1. Mol. Cell 4, 793–803 [DOI] [PubMed] [Google Scholar]
- 38. Amano M., Mukai H., Ono Y., Chihara K., Matsui T., Hamajima Y., Okawa K., Iwamatsu A., Kaibuchi K. (1996) Identification of a putative target for Rho as the serine-threonine kinase protein kinase N. Science 271, 648–650 [DOI] [PubMed] [Google Scholar]
- 39. Watanabe G., Saito Y., Madaule P., Ishizaki T., Fujisawa K., Morii N., Mukai H., Ono Y., Kakizuka A., Narumiya S. (1996) Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase Rho. Science 271, 645–648 [DOI] [PubMed] [Google Scholar]
- 40. Palmer R. H., Parker P. J. (1995) Expression, purification, and characterization of the ubiquitous protein kinase C-related kinase 1. Biochem. J. 309, 315–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Engel M., Hindie V., Lopez-Garcia L. A., Stroba A., Schaeffer F., Adrian I., Imig J., Idrissova L., Nastainczyk W., Zeuzem S., Alzari P. M., Hartmann R. W., Piiper A., Biondi R. M. (2006) Allosteric activation of the protein kinase PDK1 with low molecular weight compounds. EMBO J. 25, 5469–5480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Durocher Y., Perret S., Kamen A. (2002) High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 30, E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lopez-Garcia L. A., Schulze J. O., Fröhner W., Zhang H., Süss E., Weber N., Navratil J., Amon S., Hindie V., Zeuzem S., Jørgensen T. J., Alzari P. M., Neimanis S., Engel M., Biondi R. M. (2011) Allosteric regulation of protein kinase PKCζ by the N-terminal C1 domain and small compounds to the PIF pocket. Chem. Biol. 18, 1463–1473 [DOI] [PubMed] [Google Scholar]
- 44. Yu W., Liu J., Morrice N. A., Wettenhall R. E. (1997) Isolation and characterization of a structural homologue of human PRK2 from rat liver. Distinguishing substrate and lipid activator specificities. J. Biol. Chem. 272, 10030–10034 [DOI] [PubMed] [Google Scholar]
- 45. Cato A. C., Henderson D., Ponta H. (1987) The hormone response element of the mouse mammary tumor virus DNA mediates the progestin and androgen induction of transcription in the proviral long terminal repeat region. EMBO J. 6, 363–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang X., Gureasko J., Shen K., Cole P. A., Kuriyan J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 [DOI] [PubMed] [Google Scholar]
- 47. Jura N., Zhang X., Endres N. F., Seeliger M. A., Schindler T., Kuriyan J. (2011) Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell 42, 9–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Datta K., Franke T. F., Chan T. O., Makris A., Yang S. I., Kaplan D. R., Morrison D. K., Golemis E. A., Tsichlis P. N. (1995) AH/PH domain-mediated interaction between Akt molecules and its potential role in Akt regulation. Mol. Cell. Biol. 15, 2304–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Franke T. F., Kaplan D. R., Cantley L. C., Toker A. (1997) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol 3,4-bisphosphate. Science 275, 665–668 [DOI] [PubMed] [Google Scholar]
- 50. Slater S. J., Seiz J. L., Cook A. C., Buzas C. J., Malinowski S. A., Kershner J. L., Stagliano B. A., Stubbs C. D. (2002) Regulation of PKCα activity by C1-C2 domain interactions. J. Biol. Chem. 277, 15277–15285 [DOI] [PubMed] [Google Scholar]
- 51. Masters T. A., Calleja V., Armoogum D. A., Marsh R. J., Applebee C. J., Laguerre M., Bain A. J., Larijani B. (2010) Regulation of 3-phosphoinositide-dependent protein kinase 1 activity by homodimerization in live cells. Sci. Signal. 3, 13. [DOI] [PubMed] [Google Scholar]
- 52. Knighton D. R., Zheng J. H., Ten Eyck L. F., Xuong N. H., Taylor S. S., Sowadski J. M. (1991) Structure of a peptide inhibitor bound to the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science 253, 414–420 [DOI] [PubMed] [Google Scholar]
- 53. Flynn P., Mellor H., Casamassima A., Parker P. J. (2000) Rho GTPase control of protein kinase C-related protein kinase activation by 3-phosphoinositide-dependent protein kinase. J. Biol. Chem. 275, 11064–11070 [DOI] [PubMed] [Google Scholar]
- 54. Hindie V., Stroba A., Zhang H., Lopez-Garcia L. A., Idrissova L., Zeuzem S., Hirschberg D., Schaeffer F., Jørgensen T. J., Engel M., Alzari P. M., Biondi R. M. (2009) Structure and allosteric effects of low molecular weight activators on the protein kinase PDK1. Nat. Chem. Biol. 5, 758–764 [DOI] [PubMed] [Google Scholar]
- 55. Stroba A., Schaeffer F., Hindie V., Lopez-Garcia L., Adrian I., Fröhner W., Hartmann R. W., Biondi R. M., Engel M. (2009) 3,5-Diphenylpent-2-enoic acids as allosteric activators of the protein kinase PDK1. Structure-activity relationships and thermodynamic characterization of binding as paradigms for PIF-binding pocket-targeting compounds. J. Med. Chem. 52, 4683–4693 [DOI] [PubMed] [Google Scholar]
- 56. Fröhner W., Lopez-Garcia L. A., Neimanis S., Weber N., Navratil J., Maurer F., Stroba A., Zhang H., Biondi R. M., Engel M. (2011) 4-Benzimidazolyl-3-phenylbutanoic acids as novel PIF-pocket-targeting allosteric inhibitors of protein kinase PKCζ. J. Med. Chem. 54, 6714–6723 [DOI] [PubMed] [Google Scholar]