

Abstract
The characterization of protein folding dynamics in terms of secondary and tertiary structures is important in elucidating the features of intraprotein interactions that lead to specific folded structures. Apomyoglobin (apoMb), possessing seven helices termed A–E, G, and H in the native state, has a folding intermediate composed of the A, G, and H helices, whose formation in the submillisecond time domain has not been clearly characterized. In this study, we used a rapid-mixing device combined with circular dichroism and small-angle x-ray scattering to observe the submillisecond folding dynamics of apoMb in terms of helical content (fH) and radius of gyration (Rg), respectively. The folding of apoMb from the acid-unfolded state at pH 2.2 was initiated by a pH jump to 6.0. A significant collapse, corresponding to ≈50% of the overall change in Rg from the unfolded to native conformation, was observed within 300 μs after the pH jump. The collapsed intermediate has a fH of 33% and a globular shape that involves >80% of all its atoms. Subsequently, a stepwise helix formation was detected, which was interpreted to be associated with a conformational search for the correct tertiary contacts. The characterized folding dynamics of apoMb indicates the importance of the initial collapse event, which is suggested to facilitate the subsequent conformational search and the helix formation leading to the native structure.
Proteins can spontaneously and quickly form their folded structures from randomly unfolded conformations. It was proposed that fast folding proteins possess smooth potential surfaces that are strongly biased toward the folded conformations (1, 2). This proposal is largely consistent with the folding dynamics of small proteins characterized based on Φ-value analysis (3, 4). It is unknown, however, how the smooth and biased potential of a protein, regarded as a funnel around the specific folded conformation, is organized from the complex and numerous intraprotein interactions among the main chains and side chains. Two important classes of intraprotein interactions contribute to the stabilization of protein structures. The hydrogen bonding interactions between the amide groups of main chains mainly stabilize the secondary structure (5, 6). In contrast, the hydrophobic interactions between side chains are mainly responsible for the stabilization of the tertiary structure (7). The characterizations of protein folding dynamics based on kinetic observations of secondary and tertiary structures, and analyses of observed dynamics along with the properties of primary sequences are important to understand the funnel-shaped potentials in terms of the realistic intraprotein interactions.
In this study, we chose horse apomyoglobin (apoMb) as representative of α-helical proteins and characterized its submillisecond folding dynamics. At neutral pH, apoMb possesses seven helices labeled A–E, G, and H (8), which amount to 55% of α-helical content (fH, see Materials and Methods). The helices assemble into a globular shape with a radius of gyration (Rg) of 18.2 Å through tertiary contacts between hydrophobic residues located inside the protein. The acid-unfolded state of apoMb is observed at pH 2.2, and possesses small fH (5%) and large Rg (29.7 Å). The protein forms a static intermediate at pH 4.2 with a significant fH (33%). Hydrogen/deuterium exchange experiments indicated that the intermediate possesses protected A, G, and H helices (9). Although there are no data on Rg for the static intermediate at pH 4.2, the specific tertiary contacts between the N- and C-terminal helices suggest that the intermediate possesses a compact domain composed of the A, G, and H helices (10, 11). The three-state folding scheme observed in equilibrium was also demonstrated in kinetic folding experiments in the millisecond time domain. After a pH jump from 2.2 to 6.0, the unfolded apoMb forms a kinetic intermediate within the dead time (≈5 ms) of stopped-flow devices, which subsequently converts to the native conformation with a time constant of several hundred milliseconds (12). The kinetic intermediate possesses protected A, G, and H helices (fH = 35%) and a collapsed conformation (Rg = 23 Å) (12, 13). Because its pattern of protection is similar to that of the static intermediate, the kinetic intermediate observed after 5 ms is considered to be identical to the static intermediate stabilized at pH 4.2 (12).
Deviations in the folding dynamics of apoMb from the three-state scheme and the presence of several kinetic intermediates have been suggested based on recent experimental investigations (14–19) and theoretical considerations (20). Temperature-jump experiments suggested a partial helix formation before the tertiary contacts formed between the N and C termini, although α-helical content was not directly monitored (17, 18). Theoretical modeling of the folding of apoMb assumes that the formation of helices precedes that of tertiary contacts as described in the diffusion collision model (20). Such suggestions imply the importance of local hydrogen bonding in the initial stage of the folding. However, the observation that no peptide fragments of the helix regions of apoMb except for the H helix can maintain helical conformations (21) suggests a collapse of the main chain before the formation of the secondary and tertiary structures as assumed in the hydrophobic collapse model (22). The difficulty in observing secondary and tertiary structures quantitatively with a submillisecond time resolution precludes us from resolving and analyzing these processes.
We recently developed an experimental strategy to explore the submillisecond folding dynamics of proteins based on the combined use of a solution mixing device, CD, and small-angle x-ray scattering (SAXS) (23–25). The CD signal monitored at 222 nm indicates α-helical content (fH). The SAXS method is useful to monitor the compactness (Rg) and shape of the scattering molecule, both of which are sensitive to the formations of tertiary structures. We used the developed strategy to induce horse apoMb folding upon a pH-jump from pH 2.2 to 6.0, and to observe its folding behaviors in the time domain from 300 μs to 1 s.
Materials and Methods
Preparation of apoMb. Horse skeletal muscle myoglobin was purchased from Sigma. Other chemicals were of the highest grade available and used without further purification. All procedures were performed at 4°C. Following the method of Teale (26), heme was extracted from holomyoglobin and the apoprotein was dialyzed against deionized water overnight. Unexpectedly, our preliminary SAXS and dynamic light scattering measurements showed that this procedure produces a minor species of apoMb that induces aggregation upon refolding. We found that the following protocol was effective in removing the minor component. To the dialyzed sample containing ≈100 μM of apoMb, 1 M HCl was added until the pH was 2.2. The unfolded sample was refolded by dilution (0.2-fold) with 150 mM citrate buffer to a final pH of 6.0 (80 μM apoMb). The resultant solution was incubated for 90 min and centrifuged at 40,000 × g to remove precipitates. The supernatant was dialyzed again with deionized water overnight. Finally, unfolded apoMb was prepared by adding appropriate amounts of 1 M HCl and 5 M NaCl and by adjusting the pH and chloride iron concentration to 2.2 and 25 mM, respectively. Unfolded apoMb was then frozen and stored at –80°C.
The folding reactions were initiated by mixing the unfolded protein solution at pH 2.2 and 150 mM citrate buffer at a volume ratio of 5:1 to a final pH of 6.0. The final protein concentrations determined by using ε280 = 14,300 M–1·cm–1 (27) were 20 and 80 μM for CD and SAXS measurements, respectively. The final concentrations of sodium citrate and chloride ion were 25 and 20 mM, respectively. All kinetic and static measurements were performed at 26 ± 1°C.
Continuous-Flow Device. The basic design of the mixer used in this study was the same as that of the T-shaped plate mixer reported previously (25). We used a stainless mixing plate with a 1-mm thickness and a T-shaped flow channel. The width of the observation channel was 200 μm. The mixing plate was placed between two quartz plates 10 mm thick for the CD measurements, and between kapton sheets 50 μm and 25 μm thick for the SAXS experiments. The rate of flow of the mixed sample in the observation channel was adjusted from 2.6 to 0.84 mm/ms by changing the velocity at which the sample was supplied. Because the mixing efficiency was affected by the flow speed, we confirmed that complete mixing occurred at each speed by examining the absorbance at 222 nm in a dilution experiment with holomyoglobin. At the fastest flow speed (2.6 mm/ms), 98% mixing is achieved at 130 μs. The width (800 μm) of the incident light for the kinetic CD measurements corresponds to ±150 μs of the time width. Similarly, the time width for the kinetic SAXS measurements was ±80 μs.
CD Measurements. The refolding kinetics of apoMb in the time frame from 280 μs to 17 ms was monitored with the continuous-flow equipment. The kinetic traces in the time domain after 20 ms were collected by using a stopped-flow instrument (Unisoku, Osaka, Japan). The reproducibility of the CD traces obtained with the continuous-flow device was confirmed five times under the same experimental conditions. At least 100 mixing experiments were performed and averaged to obtain the stopped-flow traces. The effect of the optical retardation of the flow-cell on the CD spectra was corrected by following a procedure described previously (24). The helical content (fH) was determined from the mean molar ellipticity at 222 nm ([θ]222) according to Eq. 1 (28):
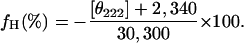 |
[1] |
SAXS Measurements. All SAXS measurements were performed at beamline BL45XU of SPring-8 (29). The basic procedure for the SAXS measurements was described previously (25). Following the recent report by Arai et al. (30), we attached a fan-shaped mask in front of the charge-coupled device detector to correct the contrast reduction. The refolding kinetics for the time domain from 300 μs to 10 ms was monitored with the continuous-flow device. The x-ray wavelength and the camera length were 1.5 Å and 588 mm, respectively. The data collection time was varied from 8 to 12 s depending on the status of the beam. The refolding kinetics for the time domain after 22 ms was monitored with the stopped-flow mixer (Unisoku) (25). The x-ray wavelength and the camera length were 0.9 Å and 655 mm, respectively. The static scattering profiles for the acid-unfolded state at pH 2.2 and for the native state at pH 6.0 were measured by using a standard cell with a path length of 3 mm. The x-ray wavelength and the camera length were 0.9 Å and 587 mm, respectively.
After subtracting the buffer profile, the sample profile was scaled by using the data collection time, the x-ray intensity, and the protein concentration. We estimated the radius of gyration and the zero-angle scattering intensity based on the Guinier approximation (Eq. 2):
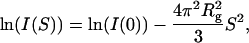 |
[2] |
where S = (2sinθ)/λ; 2θ is the scattering angle; λ is the wavelength of the incident x-ray; Rg is the radius of gyration; I(S) is the scattering intensity at S; and I(0) is the zero-angle scattering intensity. In this study, the maximum S value for the Guinier fitting region was determined by the criterion 2πRgS < 1.5. The Rg value for the native conformation at infinite dilution was estimated by using the Guinier approximation for the calculated scattering profile at infinite dilution that was obtained by extrapolating the scattering intensities at each S observed at seven concentrations between 1.3 and 0.6 mg·ml–1. The estimated value (18.2 ± 0.2 Å) was slightly smaller than the reported value (19.7 ± 0.6 Å) (31). The Rg value for the unfolded structure at infinite dilution (29.7 ± 1.7 Å) was similarly estimated by using the Debye approximation for random-coil chains (32) based on the scattering profiles at seven concentrations between 1.9 and 0.7 mg·ml–1, and was consistent with the reported value (30.2 ± 2.5 Å) (31). The intensities of the observed scattering profiles were normalized relative to the value obtained for a standard solution of lysozyme at each observation point of the flow cell.
Pair distribution functions, P(r)s, were calculated by using the gnom package (33). In brief, a series of trial P(r)s was calculated with various input values of the maximum r (Dmax) at 1 Å resolution for each scattering profile whose S range was limited from 0.0048 to 0.032 Å–1. The selected P(r) possessed the smallest Dmax value and root-mean square difference between the original scattering profile and the reproduced profile from the P(r) function (34, 35). The reliability of the selected P(r) was also confirmed by the coincidence between the Rg value estimated based on the Guinier fitting of the original scattering and the Rg value calculated from the P(r) function (34). The errors in the ordinate values of P(r) were within 2% at each data point.
Results
Stepwise Helix Formation Identified by CD Spectroscopy. To estimate the time-dependent changes in α-helical content during the folding of apoMb, the mean residue ellipticity at 222 nm ([θ]222) was followed after a pH-jump from 2.2 to 6.0 (Fig. 1). We used the continuous-flow and stopped-flow mixing devices for the observations from 280 μs to 17 ms and from 20 ms to 1s, respectively. The well connected data points certify the reliability of the current results. The time-dependent changes in [θ]222 indicate a burst phase within the dead time of the device (280 μs) and two observable phases from 280 μs to 1 s. The observable phases can be well fitted by two exponential decays with time constants of 5 ± 0.5 ms and 49 ± 8 ms (Fig. 1). Extrapolating the fitting to t = 0 leads to a [θ]222 of –11,500 ± 330 deg·cm2·dmol–1, which is distinct from the value for the initial unfolded form (–4,300 deg·cm2·dmol–1) and the difference corresponds to the burst phase amplitude. [θ]222 at 1 s coincides with that of the native conformation (–18,500 deg·cm2·dmol–1). We confirmed that the kinetic CD traces after 20 ms are independent of the protein concentrations from 10 to 40 μM (data not shown), indicating that the final phase is not caused by the possible accumulation of a dimeric intermediate (36). Thus, we identified three kinetic phases in the α-helix formation of apoMb.
Fig. 1.

The time course of the change in mean residue ellipticity at 222 nm ([θ]222) for the folding of apoMb initiated by the pH-jump from 2.2 to 6.0 at 26°C (open circle). The filled square and circle represent [θ]222 for the unfolded and native states, respectively. The dotted line shows [θ]222 at pH 4.2. The line indicates a double exponential fitting (5 ± 0.5 ms and 49 ± 8 ms) of the kinetic data. The amplitudes of the burst, first, and second kinetic phases estimated from the fittings are –11,500 ± 330, –4,230 ± 230, and –2,420 ± 230 (deg·cm2·dmol–1), respectively. The residuals of the single and double exponential fittings are indicated in the upper graphs. The right axis represents helical content (fH) calculated from Eq. 1.
Changes in Size and Shape Determined with SAXS Measurements. To investigate the formation of the tertiary structure, we next conducted time-resolved SAXS measurements by using the same pH-jump process, and estimated Rg from the Guinier analyses of the data obtained. The changes of Rg in Fig. 2 Upper were well fitted by a single exponential decay with a time constant of 56 ms (line in Fig. 2 Upper). In contrast to the time-resolved CD results that indicate two observable phases, the SAXS data denote only a single observable phase whose rate constant (56 ms) is comparable to that of the final phase (49 ms) observed in the CD results. The extrapolation of the fitting to t = 0 leads to an Rg of 23.7 ± 0.9 Å, which is similar to the result (23 ± 2 Å) for the kinetic intermediate characterized at 100 ms and 4°C (13). Thus, an ≈6 Å contraction of the main chain from the unfolded conformation (29.7 Å) was demonstrated to occur within a surprisingly short period (<300 μs) after initiation of the folding.
Fig. 2.

Time-dependent changes in the radius of gyration (Rg) and the zero-angle scattering intensity (I (0)) obtained from the Guinier analysis of the SAXS profiles during apoMb folding. (Upper) The time course of the change in Rg for the folding of apoMb initiated by the pH-jump from 2.2 to 6.0 at 26°C (open circle). The filled square represents Rg for the unfolded conformation at infinite dilution (29.7 ± 1.7 Å). The Rg value for the native structure at infinite dilution was 18.2 ± 0.2 Å. The line indicates a single exponential fitting (56 ± 10 ms) of the kinetic data from 300 μs to 1 s. The extrapolation of the fitting to a longer time period leads to an Rg of 18.7 Å.(Lower) The time course of the change in I (0) for the folding apoMb (open circle). The line indicates a single exponential fitting (27 ± 3.8 ms) of the data.
Time-resolved changes in the molecular conformation can also be deduced from the zero-angle scattering intensity, I(0), estimated from the Guinier analyses. The I(0) values are constant in the time domain from 300 μs to 10 ms, but decrease by ≈33% in the subsequent time domain (Fig. 2 Lower). The changes in I(0) are usually explained either by the presence of a dimeric intermediate (36, 37) or by the difference in hydration structures between the folding intermediates and the native state (30, 38, 39). To explain the observed change in I(0) with the dissociation of the putative dimer into the native monomer, ≈40% of apoMb must be in the dimeric state in the time domain from 300 μs to 10 ms. Because the reported dimeric intermediate has an Rg of ≈32 Å (36), the apparent Rg in the time domain should become ≈27 Å, which is larger than the current observation (23.7 Å). Therefore, the participation of the dimeric intermediate is not consistent with the collapsed Rg detected in the current study.
To explore the shapes of the folding intermediates and to confirm the sole formation of the monomeric intermediate, we next calculated Kratky plots from the scattering profiles. Although a plateau in the moderate-angle region of the Kratky plots indicates a random coil conformation, a peak in the plots indicates the globularity of proteins. The peak position, S, corresponds to the size of the globular part of the molecule as indicated in Eq. 3 (40):
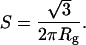 |
[3] |
The Kratky plots calculated for the time domain from 300 μs to 9 ms are very similar and possess a single peak at ≈0.013 Å–1 in S (Fig. 3). According to Eq. 3, the peak position corresponds to an Rg of ≈21 Å, which is comparable to that obtained in the Guinier analysis (23.7 Å). We note that the putative dimeric intermediate with an Rg of 32 ± 3 Å (36) should exhibit a shoulder at ≈0.0086 ± 0.0007 Å–1 in S, which is absent from the plots. Thus, the compact and globular intermediate composed of monomeric apoMb is formed within 300 μs, and does not significantly change its shape in the time domain from 300 μs to 9 ms.
Fig. 3.

Time-resolved Kratky profiles during the folding of apoMb initiated by the pH-jump from 2.2 to 6.0. For clarification, four representative profiles are displayed, which were obtained for the static native conformation (black) and for the time-resolved data at 300 μs (blue), 600 μs, (red), and 9 ms (green) after initiating the folding. Assuming the sequential folding scheme (Scheme 1), 9 ms is close to the time point when I2 is maximal.
Finally, we calculated the pair distribution functions, P(r), to obtain quantitative estimates for the domain sizes of the kinetic intermediates (Fig. 4). The P(r) functions are the Fourier transformations of the scattering profiles and indicate distribution functions of linear distances between every pair of atoms. The P(r) function for apoMb in the native state shows a symmetric peak at ≈21 Å, which is indicative of a compact globular shape. On the other hand, the P(r) functions at 300 μs, 600μs, and 9 ms are composed of a main symmetrical peak near 24 Å and of a weak shoulder near 62 Å. These features suggest that transient species in the time scale from 300 μs to 9 ms comprise a main globular domain and a small extended part. The ratio of the globular domain and the extended part does not change significantly in this time scale within the experimental errors. Fitting the main symmetrical peak using a G function, we estimated that >80% of atoms are involved in the main globular domain.
Fig. 4.

The pair distribution functions, P(r), calculated from the time-resolved scattering profiles of apoMb folding. The P(r) functions were calculated by using the gnom package (33) from the four scattering profiles indicated in Fig. 3. The blue, red, and green lines correspond to the functions for 300 μs, 600 μs, and 9 ms after initiating the folding, respectively. The black line corresponds to the native conformation. The maximum r value, Dmax, for the protein in its native state is 55 Å. All Dmax values for the transient species at 300 μs, 600 μs, and 9 ms are 75 Å. To clarify the differences between P(r) functions, they were normalized to have a constant integral area.
Discussion
Possible Folding Schemes. We identified three kinetic phases, including a burst phase, in the process by which helices form in horse apoMb, which cannot be explained by the three-state folding scheme with a single intermediate. To explain the three phases, one needs to assume that at least two folding intermediates other than the native and unfolded conformation exist. In fact, only two folding schemes are possible with two intermediates. The first is a sequential folding scheme with linearly connected intermediates (Scheme 1), where the formation of I1, I2, and N corresponds to the burst, first (5 ms), and second (49 ms) phases, respectively. The first phase corresponds to a lag phase before the appearance of I2, which is absent in the kinetic Rg trace. We interpret that the absence reflects the similarity in protein size between I1 and I2. Thus, the two intermediates possess the same Rg (23.7 Å) and different fH values (30 and 44% for I1 and I2, respectively). This scheme resembles the four-state scheme proposed by Baldwin and his colleagues (U ↔ Ia ↔ Ib ↔N) to explain the folding of sperm whale apoMb (14, 15). They reported that the formation of Ia from the unfolded structure occurs with a time constant of 50 μs at 20°C (15). Furthermore, the kinetic equilibria between Ia and Ib, and between Ib and N take place with time constants of ≈30 and ≈300 ms at 4°C, respectively (15). Because the current experiments were conducted at 26°C, it is possible that the observed I1 and I2 conformations correspond to Ia and Ib, respectively. Although the fH reported for Ib (33% at pH 4.2) (14) is different from that of the current study (44% at pH 6.0), the difference might be caused by differences in the sample species and the solution condition. The sequential folding scheme suggested here is therefore consistent with previous observations.
Scheme 1.

The second possibility is a parallel folding scheme as indicated in Scheme 2. If we consider that the amplitudes of the first and second phases observed in [θ]222 correspond to the amounts of IP1 and IP2 with the same [θ]222 of –11,500 deg·cm2·dmol–1, ≈65% of the protein in its native state should already be accumulated at 10 ms. However, Rg values (23.7 Å) are constant from 300 μs to 10 ms, which indicates an absence of the native conformation at 10 ms. As suggested in the double jump experiment for sperm whale apoMb (14), the parallel folding scheme cannot explain the current observation.
Scheme 2.

The sequential folding scheme with the two intermediates (Scheme 1) is therefore the simplest model to explain the current observations. A recent pulsed hydrogen/deuterium exchange study, however, detected inhomogeneous components in the early phase of the folding of apoMb from sperm whale (41). A temperature jump infrared study for horse apoMb also reported inhomogeneous dynamics in the early core formation (42). Although the current data do not support this inhomogeneity, it might be possible to accommodate inhomogeneity into Scheme 1 by modifying the I1 conformation into several subcomponents. In the following discussion, however, we will adopt a simple Scheme 1 and describe properties of apoMb folding that do not depend on possible inhomogeneity of I1.
Molecular Mechanism of apoMb Folding. The first remarkable finding of this study is the rapid and significant collapse and helix formation that occurs within the dead time of the experiments (<300 μs). The amount of collapsed domain estimated from the P(r) function (>80%) cannot be explained only by the A, G, and H helices (≈40%), indicating that the regions outside these helices should be collapsed in both the I1 and I2 states. The interpretation seems to contradict the previous consensus based mainly on hydrogen/deuterium exchange experiments on the static intermediate at pH 4.2 (9), i.e., that the collapsed domain is composed of only the A, G, and H helices. However, recent heteronuclear NMR investigations demonstrated that, besides the A, G, and H helices, B, C, D, and E helices show a partially helical conformation at pH 4.2 (43, 44). We conclude that the initial collapse of apoMb involves not only the terminal helices but also the central unprotected helices.
To explain the rapid and large-scale collapse observed in the early phase of the folding, we propose that the collapse is promoted by the central hydrophobic region of apoMb. The hydrophobicity plot of apoMb sequences shows a conserved maximum in the middle of the E helix (Fig. 6, which is published as supporting information on the PNAS web site). Furthermore, it has been indicated that the central region of apoMb is important for the stability of the static and kinetic intermediates. For example, apo-miniMb (residues 32–139) lacking the A, half of the B, and part of the H helix still has an equilibrium intermediate with a significant fH value (31%) (45), indicating that not only the tertiary contacts between the N and C termini but also the central part of the apoMb sequence is important for the formation of the intermediate. A mutant apoMb, in which a hydrophilic His-64 located at the E helix is changed to a hydrophobic Phe (H64F), folds faster than the wild type and forms a kinetic intermediate having larger numbers of protected amide protons (46), demonstrating the importance of the E helix hydrophobicity for the early folding phase of apoMb. We suggest that the E-helix region located in the middle of apoMb attracts other hydrophobic residues and drives the fast and large-scale collapse.
The second important observation of this study is the processes after the I1 state, which can be described as the stepwise helix formations within the collapsed conformation. Interestingly, several previous reports on the folding of apoMb suggested changes in the tertiary contacts in this time domain. Baldwin et al. (14) observed that the initial folding intermediate (Ia) possesses the red shifted Trp fluorescence than that of the next intermediate (Ib), suggesting that Trp residues in the A helix of Ia are more exposed to solvents than those of Ib. Haruta and Kitagawa (16) used UV resonance Raman spectroscopy and found that the A helix Trp residues are in a hydrophilic environment at 250 μs, hydrophobic environment at ≈1 ms, and partially hydrophobic environment at ≈5 ms in the folding of horse apoMb. The reported time constant (3 ms) for the last phase is similar to that observed in the current CD experiment (5 ms). The changes in the surroundings of the Trp-residues demonstrate the rearrangements of the tertiary contacts around the A helix. The folding after the initial collapse can therefore be interpreted as a stepwise process in which apoMb searches for the correct tertiary contacts within the collapsed conformation to stabilize the helical structures.
Finally, we will comment on I2 that appears as the final intermediate. The intermediate accumulates at 10 ms and possesses a fH of 44%. Furthermore, a reduction in both Rg and I(0) was observed in the process leading from I2 to the native conformation. The I2 structure has not been resolved in previous kinetic CD studies, probably because of the limited time resolution of the stopped-flow apparatus (12, 46); however, the structured intermediate was frequently observed. The equilibrium conformation of apoMb stabilized in the presence of trichloroacetate was reported to have a fH of 45% and Rg of 23.1 Å (31, 47, 48) (shown in Fig. 5). The kinetic intermediate, in which the B helix, in addition to the A, G, and H helices, is protected, was also suggested in a pulse-label NMR study (12). Both observations imply the presence of an intermediate with increased amounts of protected helices, which might correspond to I2. Although the fH of I2 was comparable to that of the native conformation, the I(0) value for I2 was demonstrated to be 33% larger. An increase in I(0) for folding intermediates is usually interpreted to mean that the number of hydrated water molecules is larger for the intermediates than for the protein in its native state (30, 38, 39). Thus, one intriguing explanation for the current observations is that the collapsed domain of I2 possesses near native helices and many hydrated water molecules. The reduction in I(0) observed in the conversion from I2 to the native conformation implies that the major changes in the hydration structures occurs at the rate determining step in the folding of apoMb (30, 38).
Fig. 5.

Comparison of the folding trajectories for apoMb and cyt c in the two-dimensional conformational space defined by α-helical content (fH) and the radius of gyration (Rg). The filled circle and square represent the unfolded (U) and native (N) conformations of apoMb, respectively. Open circles correspond to the kinetic intermediates (I1 and I2). The open diamond represents apoMb in the trichloroacetate stabilized state (31, 47, 48). The open triangles indicate the folding trajectory for cyt c determined previously (25).
Implications of the Initial Collapse. To clarify both general and specific features of the folding dynamics, we examined the folding trajectories of apoMb and cytochrome c (cyt c) in the conformational space defined by Rg and fH in Fig. 5 (25). These are the only proteins without S–S linkages whose folding dynamics have been characterized in terms of Rg and fH. Both proteins similarly exhibit cooperative acquisitions of fH and Rg after the initial collapse, indicating that the secondary and tertiary structures are largely formed together during the kinetic processes of protein folding. The difference between the two, however, is apparent in the early kinetic phase. The folding of apoMb (153 residues) is characterized by a large-scale collapse, corresponding to ≈50% of the overall change in Rg from the unfolded to native state. This rapid collapse accompanies the formation of the A, G, and H helices, which amounts to ≈60% of the overall change in fH. In contrast, the initial collapse of cyt c (104 residues) corresponds to only ≈35% of the total (25). Interestingly, it is reported that the IgG-binding domain of protein L (62 residue) does not exhibit an initial collapse (38). Thus, there seems to be a tendency for larger proteins to undergo greater hydrophobic collapse in the early stages of folding. In contrast to previous studies that assumed a diffusion–collision pathway for the folding of apoMb (20), comparison of the characterized trajectories suggests the importance of the initial hydrophobic collapse.
Hydrophobic collapse has been speculated to be either favorable or unfavorable to fast protein folding, depending on the nature of the collapse (22, 49–52). If the collapse is purely nonspecific, it might retard the organization of both the main chain and side chains because of nonnative interactions. In contrast, the collapse might facilitate the conformational search of proteins by reducing the number of accessible conformations. The collapse might also facilitate hydrogen bonding, because the dehydration of hydrogen bonds between main chain amides is energetically unfavorable and should be compensated by the hydrophobic interactions between the side chains (6, 53). The current results demonstrate that the initial collapse of apoMb involves unstructured helices besides the structured A, G, and H helices, and might imply the formation of nonnative contacts in the intermediates. Despite the possible nonnative contacts, however, the rate constant of the folding of apoMb [56 ms at 26°C (this work), 1.2 s at 8°C (36)] is of the same order as those of cyt c [14 ms at 27°C (25), 7s at 10°C (54)] and the IgG binding domain (8 s at 5°C) (38). This might indicate that the retardation of apoMb folding due to the putative nonnative contacts is compensated for by the facile conformational search in the collapsed intermediate (I2). We suggest that the initial hydrophobic collapse of larger proteins should be favorable for the subsequent conformational search for and hydrogen bonding of the native structure.
Conclusion
The submillisecond time-resolved CD and SAXS measurements reported here provide detailed information on the time course of the change in helical content, and shape and compactness of the main chain during the folding of apoMb. A fast and large-scale collapse of apoMb in the initial kinetic stage and subsequent stepwise formation of helices were demonstrated. It was suggested that the initial collapse facilitates the subsequent conformational search for the correct tertiary contacts in large proteins such as apoMb.
Supplementary Material
Acknowledgments
This work was supported by Grants-in-Aids for Scientific Research from the Ministry of Education, Science, Sports, and Culture (to S.T., K.I., and I.M.). S.A. is supported by the Special Postdoctoral Researchers Program of RIKEN. T.K. is supported by the fellowship of Japan Society for the Promotion of Science to Young Scientists.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: apoMb, apomyoglobin; SAXS, small-angle x-ray scattering; cyt c, cytochrome c.
References
- 1.Go, N. (1983) Annu. Rev. Biophys. Bioeng. 12, 183–210. [DOI] [PubMed] [Google Scholar]
- 2.Bryngelson, J. D., Onuchic, J. N., Socci, N. D. & Wolynes, P. G. (1995) Proteins 21, 167–195. [DOI] [PubMed] [Google Scholar]
- 3.Daggett, V. & Fersht, A. R. (2003) Trends Biochem. Sci. 28, 18–25. [DOI] [PubMed] [Google Scholar]
- 4.Shoemaker, B. A., Wang, J. & Wolynes, P. G. (1997) Proc. Natl. Acad. Sci. USA 94, 777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aurora, R., Creamer, T. P., Srinivasan, R. & Rose, G. D. (1997) J. Biol. Chem. 272, 1413–1416. [DOI] [PubMed] [Google Scholar]
- 6.Baldwin, R. L. (2003) J. Biol. Chem. 278, 17581–17588. [DOI] [PubMed] [Google Scholar]
- 7.Kauzmann, W. (1959) Adv. Protein Chem. 14, 1–63. [DOI] [PubMed] [Google Scholar]
- 8.Eliezer, D. & Wright, P. E. (1996) J. Mol. Biol. 263, 531–538. [DOI] [PubMed] [Google Scholar]
- 9.Hughson, F. M., Wright, P. E. & Baldwin, R. L. (1990) Science 249, 1544–1548. [DOI] [PubMed] [Google Scholar]
- 10.Kay, M. S. & Baldwin, R. L. (1996) Nat. Struct. Biol. 3, 439–445. [DOI] [PubMed] [Google Scholar]
- 11.Kay, M. S., Ramos, C. H. I. & Baldwin, R. L. (1999) Proc. Natl. Acad. Sci. USA 96, 2007–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jennings, P. A. & Wright, P. E. (1993) Science 262, 892–896. [DOI] [PubMed] [Google Scholar]
- 13.Eliezer, D., Jennings, P. A., Wright, P. E., Doniach, S., Hodgson, K. O. & Tsuruta, H. (1995) Science 270, 487–488. [DOI] [PubMed] [Google Scholar]
- 14.Jamin, M. & Baldwin, R. L. (1998) J. Mol. Biol. 276, 491–504. [DOI] [PubMed] [Google Scholar]
- 15.Jamin, M., Yeh, S. R., Rousseau, D. L. & Baldwin, R. L. (1999) J. Mol. Biol. 292, 731–740. [DOI] [PubMed] [Google Scholar]
- 16.Haruta, N. & Kitagawa, T. (2002) Biochemistry 41, 6595–6604. [DOI] [PubMed] [Google Scholar]
- 17.Ballew, R. M., Sabelko, J. & Gruebele, M. (1996) Nat. Struct. Biol. 3, 923–926. [DOI] [PubMed] [Google Scholar]
- 18.Ballew, R. M., Sabelko, J. & Gruebele, M. (1996) Proc. Natl. Acad. Sci. USA 93, 5759–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilmanshin, R., Callender, R. H. & Dyer, R. B. (1998) Nat. Struct. Biol. 5, 363–365. [DOI] [PubMed] [Google Scholar]
- 20.Pappu, R. V. & Weaver, D. L. (1998) Protein Sci. 7, 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reymond, M. T., Merutka, G., Dyson, H. J. & Wright, P. E. (1997) Protein Sci. 6, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dill, K. A. (1985) Biochemistry 24, 1501–1509. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi, S., Yeh, S. R., Das, T. K., Chan, C. K., Gottfried, D. S. & Rousseau, D. L. (1997) Nat. Struct. Biol. 4, 44–50. [DOI] [PubMed] [Google Scholar]
- 24.Akiyama, S., Takahashi, S., Ishimori, K. & Morishima, I. (2000) Nat. Struct. Biol. 7, 514–520. [DOI] [PubMed] [Google Scholar]
- 25.Akiyama, S., Takahashi, S., Kimura, T., Ishimori, K., Morishima, I., Nishikawa, Y. & Fujisawa, T. (2002) Proc. Natl. Acad. Sci. USA 99, 1329–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teale, F. W. J. (1959) Biochem. Biophys. Acta 35, 543–543. [DOI] [PubMed] [Google Scholar]
- 27.Goto, Y., Calciano, L. J. & Fink, A. L. (1990) Proc. Natl. Acad. Sci. USA 87, 573–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen, Y. H., Yang, J. T. & Martinez, H. M. (1972) Biochemistry 11, 4120–4131. [DOI] [PubMed] [Google Scholar]
- 29.Fujisawa, T., Inoue, K., Oka, T., Iwamoto, H., Uruga, T., Kumasaka, T., Inoko, Y., Yagi, N., Yamamoto, M. & Ueki, T. (2000) J. Appl. Crystallogr. 33, 797–800. [Google Scholar]
- 30.Arai, M., Ito, K., Inobe, T., Nakao, M., Maki, K., Kamagata, K., Kihara, H., Amemiya, Y. & Kuwajima, K. (2002) J. Mol. Biol. 321, 121–132. [DOI] [PubMed] [Google Scholar]
- 31.Kataoka, M., Nishii, I., Fujisawa, T., Ueki, T., Tokunaga, F. & Goto, Y. (1995) J. Mol. Biol. 249, 215–228. [DOI] [PubMed] [Google Scholar]
- 32.Calmettes, P., Durand, D., Desmadril, M., Minard, P., Receveur, V. & Smith, J. C. (1994) Biophys. Chem. 53, 105–113. [DOI] [PubMed] [Google Scholar]
- 33.Semenyuk, A. V. & Svergun, D. I. (1991) J. Appl. Crystallogr. 24, 537–540. [Google Scholar]
- 34.Flanagan, J. M., Kataoka, M., Fujisawa, T. & Engelman, D. M. (1993) Biochemistry 32, 10359–10370. [DOI] [PubMed] [Google Scholar]
- 35.Moore, P. B. (1980) J. Appl. Cryst. 13, 168–175. [Google Scholar]
- 36.Eliezer, D., Chiba, K., Tsuruta, H., Doniach, S., Hodgson, K. O. & Kihara, H. (1993) Biophys. J. 65, 912–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Segel, D. J., Eliezer, D., Uversky, V., Fink, A. L., Hodgson, K. O. & Doniach, S. (1999) Biochemistry 38, 15352–15359. [DOI] [PubMed] [Google Scholar]
- 38.Plaxco, K. W., Millett, I. S., Segel, D. J., Doniach, S. & Baker, D. (1999) Nat. Struct. Biol. 6, 554–556. [DOI] [PubMed] [Google Scholar]
- 39.Chen, L. L., Wildegger, G., Kiefhaber, T., Hodgson, K. O. & Doniach, S. (1998) J. Mol. Biol. 276, 225–237. [DOI] [PubMed] [Google Scholar]
- 40.Semisotnov, G. V., Kihara, H., Kotova, N. V., Kimura, K., Amemiya, Y., Wakabayashi, K., Serdyuk, I. N., Timchenko, A. A., Chiba, K., Nikaido, K., et al. (1996) J. Mol. Biol. 262, 559–574. [DOI] [PubMed] [Google Scholar]
- 41.Nishimura, C., Dyson, H. J. & Wright, P. E. (2002) J. Mol. Biol. 322, 483–489. [DOI] [PubMed] [Google Scholar]
- 42.Gulotta, M., Gilmanshin, R., Buscher, T. C., Callender, R. H. & Dyer, R. B. (2001) Biochemistry 40, 5137–5143. [DOI] [PubMed] [Google Scholar]
- 43.Eliezer, D., Yao, J., Dyson, H. J. & Wright, P. E. (1998) Nat. Struct. Biol. 5, 148–155. [DOI] [PubMed] [Google Scholar]
- 44.Eliezer, D., Chung, J., Dyson, H. J. & Wright, P. E. (2000) Biochemistry 39, 2894–2901. [DOI] [PubMed] [Google Scholar]
- 45.Desanctis, G., Ascoli, F. & Brunori, M. (1994) Proc. Natl. Acad. Sci. USA 91, 11507–11511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia, C., Nishimura, C., Cavagnero, S., Dyson, H. J. & Wright, P. E. (2000) Biochemistry 39, 11227–11237. [DOI] [PubMed] [Google Scholar]
- 47.Nishii, I., Kataoka, M., Tokunaga, F. & Goto, Y. (1994) Biochemistry 33, 4903–4909. [DOI] [PubMed] [Google Scholar]
- 48.Loh, S. N., Kay, M. S. & Baldwin, R. L. (1995) Proc. Natl. Acad. Sci. USA 92, 5446–5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gutin, A. M., Abkevich, V. I. & Shakhnovich, E. I. (1995) Biochemistry 34, 3066–3076. [DOI] [PubMed] [Google Scholar]
- 50.Thirumalai, D. & Klimov, D. K. (1999) Curr. Opin. Struct. Biol. 9, 197–207. [DOI] [PubMed] [Google Scholar]
- 51.Hardin, C., Luthey-Schulten, Z. & Wolynes, P. G. (1999) Proteins Struct. Funct. Genet. 34, 281–294. [PubMed] [Google Scholar]
- 52.Sadqi, M., Lapidus, L. J. & Munoz, V. (2003) Proc. Natl. Acad. Sci. USA 100, 12117–12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fernandez, A., Kardos, J. & Goto, Y. (2003) FEBS Lett. 536, 187–192. [DOI] [PubMed] [Google Scholar]
- 54.Elove, G. A., Bhuyan, A. K. & Roder, H. (1994) Biochemistry 33, 6925–6935. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}