

Background: Hereditary pancreatitis is caused by mutations in human cationic trypsinogen. Chymotrypsin C (CTRC) is a proteolytic regulator of trypsinogen activation.
Results: Clinically relevant trypsinogen mutations increase autoactivation in the presence of CTRC.
Conclusion: Pathological trypsinogen activation in hereditary pancreatitis is dependent on CTRC.
Significance: Therapeutic suppression of trypsinogen autoactivation is warranted in carriers of pancreatitis-causing mutations.
Keywords: Enzyme Degradation, Enzyme Mutation, Genetic Diseases, Pancreas, Protease, Autoactivation, Chronic Pancreatitis, Chymotrypsin, Trypsin, Trypsinogen
Abstract
Mutations in human cationic trypsinogen (PRSS1) cause autosomal dominant hereditary pancreatitis. Increased intrapancreatic autoactivation of trypsinogen mutants has been hypothesized to initiate the disease. Autoactivation of cationic trypsinogen is proteolytically regulated by chymotrypsin C (CTRC), which mitigates the development of trypsin activity by promoting degradation of both trypsinogen and trypsin. Paradoxically, CTRC also increases the rate of autoactivation by processing the trypsinogen activation peptide to a shorter form. The aim of this study was to investigate the effect of CTRC on the autoactivation of clinically relevant trypsinogen mutants. We found that in the presence of CTRC, trypsinogen mutants associated with classic hereditary pancreatitis (N29I, N29T, V39A, R122C, and R122H) autoactivated at increased rates and reached markedly higher active trypsin levels compared with wild-type cationic trypsinogen. The A16V mutant, known for its variable disease penetrance, exhibited a smaller increase in autoactivation. The mechanistic basis of increased activation was mutation-specific and involved resistance to degradation (N29I, N29T, V39A, R122C, and R122H) and/or increased N-terminal processing by CTRC (A16V and N29I). These observations indicate that hereditary pancreatitis is caused by CTRC-dependent dysregulation of cationic trypsinogen autoactivation, which results in elevated trypsin levels in the pancreas.
Introduction
Hereditary chronic pancreatitis is an autosomal dominant genetic disorder characterized by incomplete penetrance and variable expressivity. In approximately half of the affected families, the disease is caused by heterozygous mutations in the PRSS1 (protease, serine, 1) gene, which encodes human cationic trypsinogen (1–4). The human pancreas secretes three isoforms of trypsinogen, and the cationic isoform contributes about two-thirds of the trypsinogen content in the pancreatic juice. In the large majority of hereditary pancreatitis families worldwide, the causative mutation is either R122H (∼70%) or N29I (∼20%). Less frequently, the same amino acid positions are altered by mutations R122C (∼3%) and N29T (<1%), respectively. In addition, a large number of rare PRSS1 mutations have been identified not only in kindreds with hereditary pancreatitis but also in patients with sporadic idiopathic pancreatitis. Many of these most likely represent harmless variants or mutations with variable or low disease penetrance (5).
Functional studies using recombinant cationic trypsinogen mutants demonstrated that mutations N29I, N29T, and R122H increased autoactivation (trypsin-mediated trypsinogen activation), albeit to a modest degree (6–8). Convincing evidence that increased autoactivation is an important mechanism in hereditary pancreatitis came from studies on a subset of rare mutations (D22G, K23R, and K23I_I24insIDK) that affect the trypsinogen activation peptide and cause a dramatic increase in autoactivation (9–11). However, activation peptide mutations did not seem to cause more severe disease, and the apparent lack of correlation between biochemical and clinical phenotypes associated with different trypsinogen mutations remained puzzling. An early study found that mutation R122C caused loss of function due to misfolding; however, this proved to be an artifact of the in vitro refolding procedure used at the time (12). On the other hand, mutant R116C, another cysteine mutant associated with hereditary pancreatitis, was shown to misfold and elicit endoplasmic reticulum stress in HEK293T cells, suggesting that mutation-induced misfolding and consequent endoplasmic reticulum stress may be an alternative disease mechanism, unrelated to trypsinogen activation (13). Whether or not mutant R116C misfolds in acinar cells is still uncertain, and the role of endoplasmic reticulum stress in pancreatitis, although intensely researched, remains speculative.
A number of recent studies from our laboratory demonstrated that cationic trypsinogen and trypsin are under the regulation of chymotrypsin C (CTRC)2 in humans. First, we found that CTRC stimulates autoactivation of cationic trypsinogen by processing the trypsinogen activation peptide to a shorter form, which is more readily cleaved by trypsin (14). The A16V cationic trypsinogen mutation, which changes the N-terminal residue of the activation peptide, increases the rate of N-terminal processing by CTRC. Subsequently, we found that CTRC promotes degradation of cationic trypsin by a mechanism that involves cleavage of the Leu-81–Glu-82 peptide bond in the calcium-binding loop and an autolytic cleavage by trypsin at the Arg-122–Val-123 peptide bond (15). Both cleavages are required, and mutation of either Leu-81 or Arg-122 blocks degradation. CTRC-mediated cleavage at Leu-81 is calcium-dependent, and millimolar concentrations of calcium protect trypsin against degradation. Finally, we and others obtained genetic evidence that loss-of-function variants of CTRC increase the risk for chronic pancreatitis in humans, indicating that CTRC plays an important protective role in the pancreas against premature trypsinogen activation (16–18). The CTRC-mediated effects on trypsinogen and trypsin are highly specific, and other chymotrypsin or elastase isoforms have no such activity.
We also observed that CTRC degrades cationic trypsinogen at a faster rate than cationic trypsin (15), suggesting that CTRC might regulate mainly the activation of trypsinogen to trypsin rather than controlling active trypsin levels through degradation. Autoactivation of cationic trypsinogen and its hereditary pancreatitis-associated mutants has never been studied in the presence of CTRC. In this study, we therefore undertook these experiments based on the hypothesis that pancreatitis-associated mutants may exert their effect in a CTRC-dependent manner. The findings reveal a prominent biochemical phenotype shared by all disease-causing mutants that involves altered regulation by CTRC and, as a consequence, strongly increased trypsinogen activation in the presence of CTRC.
EXPERIMENTAL PROCEDURES
Nomenclature
Amino acid residues in human cationic trypsinogen (PRSS1) are numbered starting with the initiator methionine of the primary translation product according to the recommendations of the Human Genome Variation Society.
Expression Plasmids and Mutagenesis
Construction of the pTrapT7-intein-Hu1, pTrapT7-Hu1-K23Q, and pcDNA3.1(−)-Hu1 expression plasmids harboring the coding sequence for human cationic trypsinogen (Hu1) was reported previously (6, 7, 19, 20). In an earlier publication, mutation K23Q was denoted as K15Q, using the conventional chymotrypsin numbering (20). In the pTrapT7-intein-Hu1 plasmid, the native N terminus of human cationic trypsinogen (Ala-Pro-Phe) is fused to the C terminus of a mini-intein (19). In the pTrapT7-Hu1-K23Q plasmid, the N-terminal sequence of trypsinogen was changed to Met-Ala-Phe-Pro-Val due to cloning manipulations (20). Mutations were introduced using overlap-extension PCR mutagenesis and were verified by DNA sequencing of the entire gene.
Expression, Purification, and Activation of Human CTRC
Construction of the pcDNA3.1(−)-CTRC expression plasmid was reported previously (15). In this study, a His10 affinity tag was engineered onto the C terminus of the protein. CTRC was expressed in HEK293T cells with transient transfection as described (21). Approximately 900 ml of conditioned medium was used for purification on a nickel-nitrilotriacetic acid Superflow cartridge (Qiagen) equilibrated with Buffer NPI-20 (50 mm NaH2PO4, 300 mm NaCl, and 20 mm imidazole (pH 8.0)). The medium was loaded onto the column at a flow rate of 4 ml/min using a loading pump, and the column was washed with Buffer NPI-20 until the absorbance at 280 nm returned to the base line. CTRC was eluted with Buffer NPI-250 (50 mm NaH2PO4, 300 mm NaCl, and 250 mm imidazole (pH 8.0)) at a flow rate of 2 ml/min, and 5-ml fractions were collected. Aliquots (100 μl) of the fractions were analyzed by 15% SDS-PAGE and Coomassie Blue staining, and peak fractions were pooled and dialyzed for 24 h against two changes of 3.5 liters of 50 mm NaH2PO4 buffer (pH 8.0) containing 300 mm NaCl. The dialyzed CTRC proenzyme solution was concentrated using a Vivaspin concentrator (10-kDa molecular mass cutoff). CTRC zymogen was activated with human cationic trypsin, and CTRC concentrations were determined by active site titration with ecotin as described previously (22). The final working CTRC stock solution was diluted to 500 nm in 0.1 m Tris-HCl (pH 8.0) and 0.05% Tween 20.
Expression and Purification of Human Cationic Trypsinogen
Wild-type and mutant trypsinogens were expressed as intein fusion proteins in aminopeptidase P-deficient Escherichia coli strain LG-3 as described previously (19, 23). After spontaneous self-cleavage, the fusion protein gave rise to cationic trypsinogen with a homogeneous authentic N-terminal sequence. K23Q trypsinogen mutants were expressed in E. coli BL21(DE3) cells, with the exception of the A16V/K23Q double mutant, which was made as an intein fusion and expressed in strain LG-3. Trypsinogen was solubilized from the inclusion bodies, refolded in vitro, and purified by ecotin affinity chromatography according to published protocols (6, 7, 19, 23, 24). Mutant R122C, which is prone to misfolding in vitro (12), was expressed in HEK293T cells by transient transfection. The growth medium contained 1 mm benzamidine to prevent autoactivation. Trypsinogen was purified from 400 ml of conditioned medium by ecotin chromatography. The concentrations of trypsinogen preparations were calculated from their UV absorbance at 280 nm using an extinction coefficient of 37,525 m−1 cm−1.
Trypsin Activity Assay
Trypsin activity was measured in 0.1 m Tris-HCl (pH 8.0) containing 1 mm CaCl2 and 0.05% Tween 20 using the chromogenic trypsin substrate N-benzyloxycarbonyl-Gly-Pro-Arg p-nitroanilide at a final concentration of 0.15 mm in a final volume of 200 μl at 22 °C. Typically, a 2-μl sample was mixed with 48 μl of assay buffer, and the reaction was started by the addition of 150 μl of substrate dissolved in the same buffer. The release of the yellow p-nitroaniline was followed for 1 min at 405 nm in a SpectraMax Plus384 microplate reader (Molecular Devices).
Gel Electrophoresis and Densitometry
Trypsinogen samples were precipitated with trichloroacetic acid (10% final concentration), and the precipitate was recovered by centrifugation, dissolved in 20 μl of Laemmli sample buffer containing 100 mm dithiothreitol (final concentration), and heat-denatured at 95 °C for 5 min. Electrophoretic separation was performed on 15% SDS-polyacrylamide mini gels in standard Tris/glycine buffer. Gels were stained for 30 min with 0.5% Brilliant Blue R-250 dissolved in 40% methanol and 10% acetic acid and destained overnight with 30% methanol and 10% acetic acid. Gels were dried between two layers of cellophane and scanned at a resolution of 300 dots/inch in grayscale mode. Quantitation of bands was carried out with the Quantity One 4.6.9 software (Bio-Rad). Rectangles were drawn around each band of interest, and an identical rectangle was used in each lane for background subtraction.
RESULTS
CTRC Regulates Autoactivation of Cationic Trypsinogen
Human cationic trypsinogen exhibits an unusually high propensity for autoactivation relative to other mammalian trypsinogens studied so far (25). When trypsinogen at 1 μm concentration was incubated with 10 nm trypsin at 37 °C at pH 8.0 in the presence of 1 mm Ca2+, conversion to trypsin was essentially complete within 40 min (Fig. 1A), and the final trypsin activity remained stable. The addition of low concentrations (1–25 nm) of human CTRC to this reaction resulted in a slightly increased rate of autoactivation, with reduced final trypsin levels that were inversely proportional to the CTRC concentration present and that slowly decreased over time from their peak value (Fig. 1A). The small increase in the rate of autoactivation was due to N-terminal processing by CTRC (14); suppression of the final trypsin levels was caused by CTRC-mediated trypsinogen degradation, and the slow decrease in trypsin activity was due to trypsin degradation by CTRC (15). Previously, we determined that CTRC-dependent trypsin degradation involves cleavages at Leu-81 (by CTRC) and Arg-122 (by trypsin) (15). To confirm that the same cleavages are required for trypsinogen degradation during autoactivation in the presence of CTRC, mutants L81A and R122A were studied (Fig. 1, B and C). In the absence of CTRC, autoactivation of mutant L81A was identical to that of the wild type, whereas mutant R122A showed a small increase in the rate (cf. Fig. 1, A–C). Thus, as expected, these mutations had no major impact on autoactivation per se. In contrast, when assayed in the presence of 5 or 25 nm CTRC, both mutations protected against CTRC-induced trypsinogen degradation and resulted in rapid autoactivation to high trypsin levels (Fig. 1, B and C). Mutant L81A autoactivated to the same trypsin levels with or without CTRC (Fig. 1B), whereas mutant R122A showed only a small CTRC-dependent decrease in the final trypsin activity (Fig. 1C). Furthermore, due to the lack of degradation, the effect of CTRC-mediated N-terminal processing became much more apparent, i.e. the rates of autoactivation were noticeably increased. These results indicate that CTRC regulates autoactivation of human cationic trypsinogen, and mutations that interfere with these processes can have profound effects on trypsin levels resulting from trypsinogen activation. Consequently, we hypothesized that naturally occurring hereditary pancreatitis-associated cationic trypsinogen mutations might exert their effect through this mechanism.
FIGURE 1.
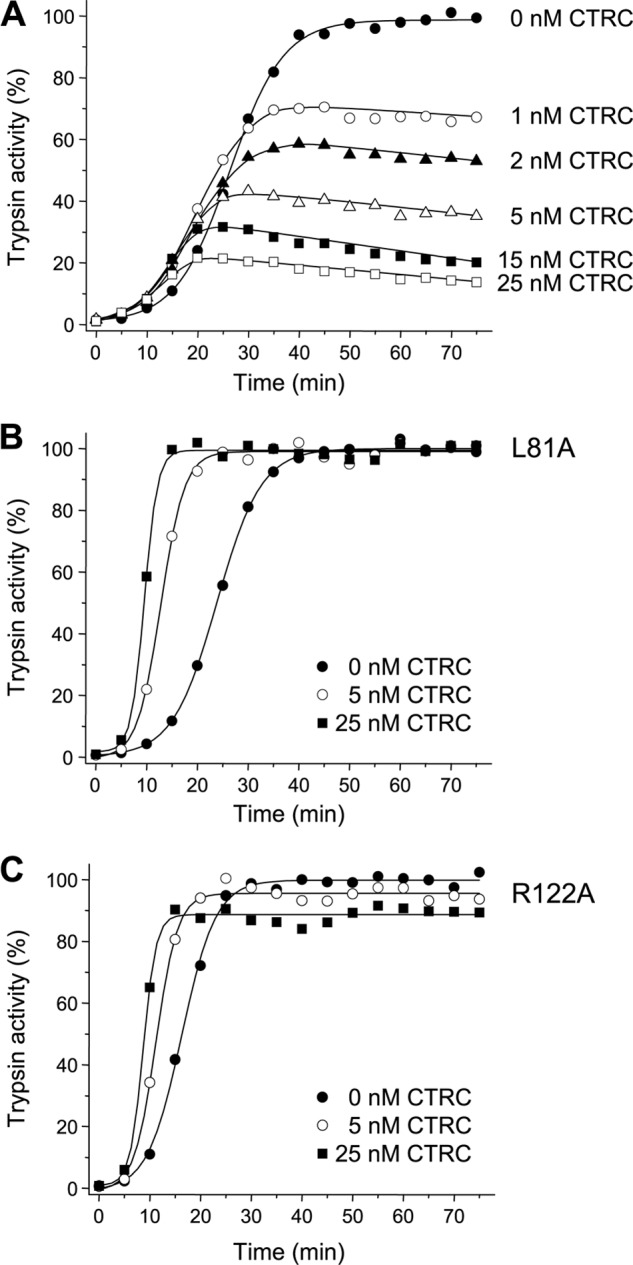
Effect of CTRC on autoactivation of human wild-type cationic trypsinogen (A) and mutants L81A (B) and R122A (C). Wild-type and mutant trypsinogens were incubated at 1 μm with 10 nm initial trypsin in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, and 0.05% Tween 20 (final concentrations) in the absence or presence of the indicated CTRC concentrations at 37 °C. At given times, 2-μl aliquots were removed, and trypsin activity was measured as described under “Experimental Procedures.” Trypsin activity is expressed as a percentage of the maximal activity in the absence of CTRC. Representative experiments from two replicates are shown.
Autoactivation of Cationic Trypsinogen Mutants in Presence of CTRC
Mutations of Arg-122 (R122C and R122H) are the clinically most frequent pancreatitis-causing genetic variants. Because both mutations block the trypsin-mediated cleavage of the Arg-122–Val-123 peptide bond, CTRC-dependent trypsinogen degradation should be impaired, resulting in increased autoactivation in a manner that was already observed with the R122A non-natural mutation in Fig. 1C. Because mutant R122C tends to misfold during the in vitro refolding procedure used (12), this mutant was expressed in transiently transfected HEK293T cells and purified from the conditioned medium. As an appropriate control, a wild-type cationic trypsinogen preparation was also produced by the same method. When tested in the absence of CTRC, both mutations R122C and R122H increased the rate of autoactivation of cationic trypsinogen to a modest extent, 1.2- and 1.5-fold, respectively (Fig. 2A). In contrast, autoactivation of mutants R122C and R122H was dramatically increased in the presence of CTRC relative to the wild type (Fig. 2, B and C). Trypsin levels remained stable over the time course studied, consistent with previous observations that trypsin mutants R122C and R122H are resistant to degradation by CTRC (15). In the absence of CTRC, rates of autoactivation can be estimated assuming that the reaction proceeds as “trypsinogen + trypsin = 2 trypsin” and degradation is negligible, but the same formula cannot be used to describe autoactivation in a quantitative manner once CTRC is present in the system. From a biological perspective, the peak trypsin value and the time to reach the maximal amplitude seem intuitively important. Therefore, we compared the area under the curve between the wild type and mutants from time 0 until the time the wild type reached its peak trypsin activity. We found that mutants R122C and R122H showed 3.3- and 3.5-fold higher values, respectively, compared with wild type in the presence of 5 nm CTRC, and these values increased to 5.0- and 4.8-fold, respectively, in the presence of 25 nm CTRC. The dependence of these values on the CTRC concentration suggests that the differences between wild-type and mutant trypsinogens may be even larger under physiological conditions, where CTRC concentrations are higher than used experimentally (see “Discussion”).
FIGURE 2.
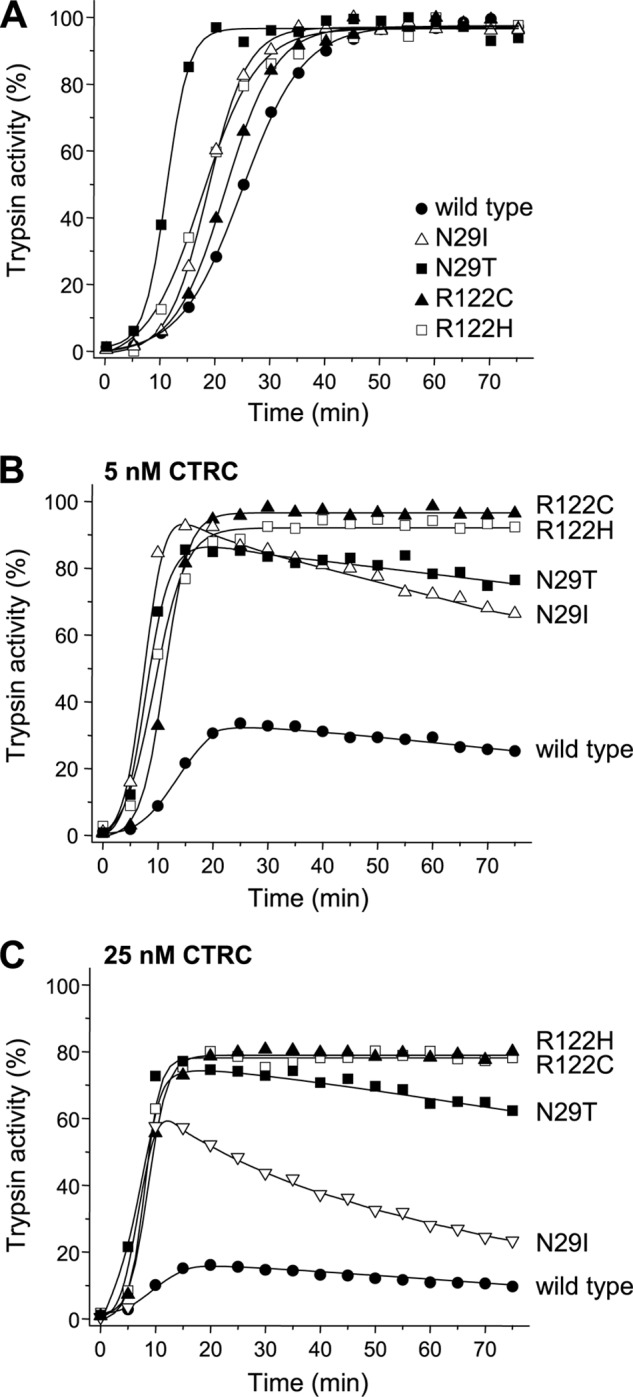
Autoactivation of pancreatitis-associated cationic trypsinogen mutants N29I, N29T, R122C, and R122H in presence of CTRC. Wild-type and mutant trypsinogens were incubated at 1 μm with 10 nm initial trypsin in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, and 0.05% Tween 20 at 37 °C in the absence (A) or presence of CTRC at 5 nm (B) or 25 nm (C) (final concentrations). At the indicated times, 2-μl aliquots were removed, and trypsin activity was measured as described under “Experimental Procedures.” Trypsin activity is expressed as a percentage of the maximal activity in the absence of CTRC. Representative experiments from two or three replicates are shown.
Mutations of Asn-29 (N29I and N29T) are the second most prevalent hereditary pancreatitis-associated changes in cationic trypsinogen. In the absence of CTRC, mutation N29I slightly (1.5-fold) increased the rate of autoactivation, whereas mutation N29T had a somewhat more pronounced (2.5-fold) stimulating effect (Fig. 2A). In the presence of CTRC, compared with wild-type trypsinogen, autoactivation was strongly accelerated by both mutations, resulting in remarkably higher trypsin levels (Fig. 2, B and C). For the N29I mutant, the values of the area under the curve were 4.1-fold higher than those for the wild type in the presence of both CTRC concentrations tested. For the N29T mutant, the same values were 3.7- and 5.2-fold higher compared with the wild type at 5 and 25 nm CTRC, respectively. In contrast to mutants R122C and R122H, trypsin mutants N29I and N29T were prone to degradation by CTRC, and trypsin levels steadily decreased over time. Interestingly, trypsin degradation showed mutation-specific differences, as trypsin mutant N29I was more readily degraded by CTRC than trypsin mutant N29T, which had superior stability, particularly at higher CTRC concentrations (Fig. 2C).
The results presented in Fig. 2 indicate that mutations associated with classic autosomal dominant hereditary pancreatitis exhibit a similar biochemical phenotype in the presence of CTRC that is characterized by increased rates of trypsinogen activation with high levels of trypsin generated. To extend these studies, we tested the effect of mutation V39A on trypsinogen autoactivation. This mutation was described in a single hereditary pancreatitis family in Italy, where it showed convincing segregation with the disease in seven family members (26). In the absence of CTRC, the V39A mutation had no significant effect on trypsinogen autoactivation, whereas in the presence of CTRC, a large increase in autoactivation was observed compared with wild-type cationic trypsinogen (Fig. 3). Mutant V39A autoactivated at a slower rate than mutant R122H, and final trypsin levels approached but remained lower than those seen with mutant R122H. The values of the area under the curve were 2.1- and 2.7-fold higher compared with wild type in the presence of 5 and 25 nm CTRC, respectively (cf. mutant R122H, 2.9- and 4.7-fold in the same experiment). These quantitative differences notwithstanding, mutant V39A exhibited a biochemical phenotype similar to the more frequent pancreatitis-associated mutations.
FIGURE 3.
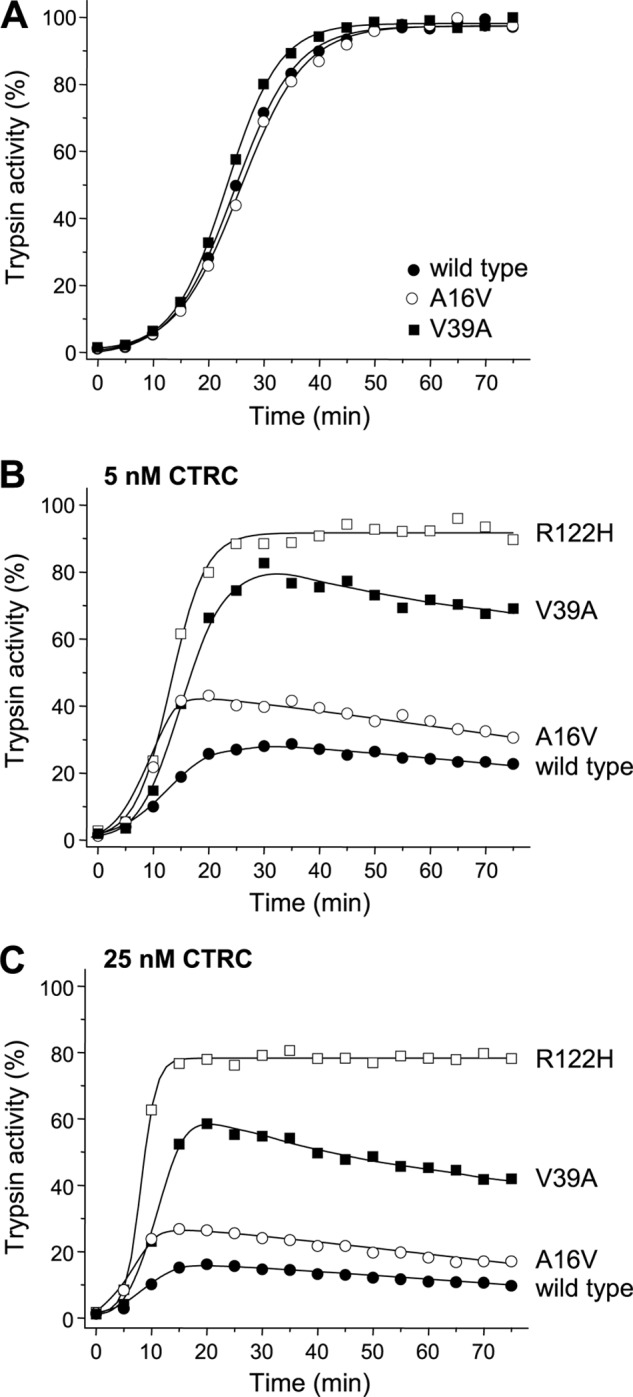
Autoactivation of pancreatitis-associated cationic trypsinogen mutants A16V and V39A in presence of CTRC. For comparison, mutant R122H was also included. Wild-type and mutant trypsinogens were incubated at 1 μm with 10 nm initial trypsin in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, and 0.05% Tween 20 at 37 °C in the absence (A) or presence of CTRC at 5 nm (B) or 25 nm (C) (final concentrations). At the indicated times, 2-μl aliquots were removed, and trypsin activity was measured as described under “Experimental Procedures.” Trypsin activity is expressed as a percentage of the maximal activity in the absence of CTRC. Representative experiments from two replicates are shown.
Mutation A16V is associated with variable disease penetrance and was identified not only in hereditary pancreatitis families but also in sporadic cases with no family history, suggesting a weaker biochemical phenotype (27, 28). The mutation is found in ∼3% of PRSS1 mutation-positive pancreatitis cases, although a recent study from Denmark reported higher regional occurrence (29). The A16V mutation alters the N-terminal amino acid residue of the trypsinogen activation peptide. Amino acid 16 is the first residue of mature trypsinogen, as the secretory signal peptide (amino acids 1–15) is cleaved off during endoplasmic reticulum entry. Because the activation peptide is released during the activation process, mutation A16V is not present in active trypsin and thus cannot alter trypsin function. Previously, we demonstrated that mutation A16V increased the N-terminal processing of the trypsinogen activation peptide by CTRC at the Phe-18–Asp-19 peptide bond by 4-fold, which is expected to result in increased autoactivation (14). Here, we found that in the absence of CTRC, mutation A16V had no effect on trypsinogen autoactivation (Fig. 3A), as demonstrated previously (19). In the presence of CTRC, the rate of autoactivation was increased measurably relative to the wild type, albeit to a lesser degree than observed with the highly disease penetrant mutations (Fig. 3, B and C). The values of the area under the curve were 1.8- and 2.0-fold higher compared with the wild type in the presence of 5 and 25 nm CTRC, respectively. Thus, the variable clinical presentation of the A16V mutation is consistent with its milder biochemical phenotype.
Mechanistic Basis of Increased Autoactivation in Presence of CTRC
The results presented above indicate that proteolytic regulation of cationic trypsinogen mutants by CTRC is altered in such a manner that results in more robust trypsinogen activation. In theory, trypsinogen mutations can cause increased autoactivation by at least four different mechanisms: increased trypsin-mediated cleavage of the activation peptide caused either (i) directly by a mutational effect or (ii) indirectly by increased N-terminal processing at the Phe-18–Asp-19 peptide bond by CTRC; or impaired trypsinogen degradation during autoactivation due to (iii) slower cleavage of the Leu-81–Glu-82 peptide bond by CTRC or (iv) decreased cleavage of the Arg-122–Val-123 peptide bond by trypsin. In the following experiments, these potential mechanisms were explored.
Mechanism i
As presented in Figs. 2A and 3A and reported previously (6, 7, 19), most mutations studied here had little direct effect on autoactivation. Mutations R122H and N29I increased the rate of autoactivation by 1.5-fold, and mutation R122C by 1.2-fold. Mutations A16V and V39A had no measurable stimulating effect. The only exception was mutation N29T, which increased the rate of autoactivation by ∼2.5-fold.
Mechanism ii
N-terminal processing of cationic trypsinogen by CTRC removes the three N-terminal amino acid residues from the activation peptide, which results in a small but detectable shift on SDS-polyacrylamide gels run under nonreducing conditions. Using this assay, we previously found that mutations A16V and N29I increased CTRC-mediated N-terminal processing of cationic trypsinogen by 4- and 1.4-fold, respectively (14). In our current experiments, we confirmed these findings and found even stronger effects after densitometric analysis (Fig. 4). Thus, mutation A16V increased the rate of N-terminal processing by 5.8-fold relative to the wild type, and mutation N29I by 2.6-fold. On the other hand, mutations V39A, R122C, and R122H had no appreciable effect on this CTRC activity. Interestingly, mutant N29T was processed slightly (1.3-fold) slower than wild-type trypsinogen (Fig. 4).
FIGURE 4.
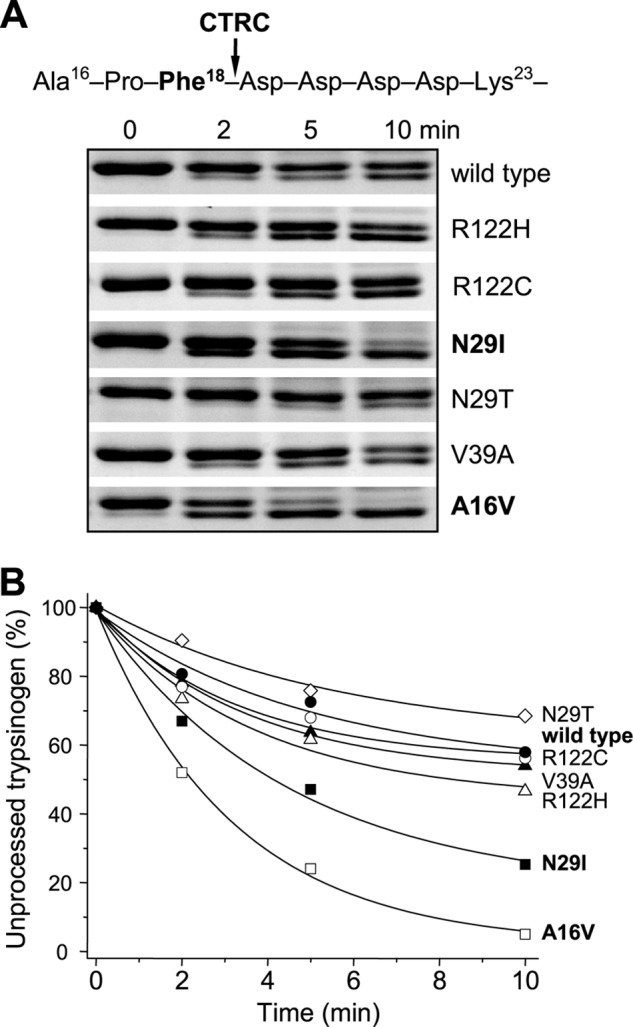
N-terminal processing of human cationic trypsinogen and pancreatitis-associated mutants by CTRC. Wild-type and mutant trypsinogens were incubated at 2 μm with 25 nm CTRC in 0.1 m Tris-HCl (pH 8.0), 1 mm CaCl2, and 20 nm SPINK1 trypsin inhibitor (final concentrations) at 37 °C. The trypsin inhibitor was included to prevent autoactivation. A, at the indicated times, reactions were terminated by precipitation with 10% trichloroacetic acid (final concentration), and samples were analyzed by 15% nonreducing SDS-PAGE and Coomassie Blue staining. Relevant segments of representative gels from two replicates demonstrate the small mobility shift of the trypsinogen band caused by N-terminal processing. B, densitometric analysis of stained gels showing the changes in the intensity of the unprocessed intact trypsinogen band as a percentage of the total intensity of the processed and unprocessed bands. Rates of processing were calculated from linear fits to semilogarithmic plots. Error bars were omitted for clarity. The S.E. was within 10% of the mean.
Mechanism iii
CTRC-mediated degradation of cationic trypsinogen is initiated by cleavage of the Leu-81–Glu-82 peptide bond in the calcium-binding loop (15). This cleavage is rapid when the binding loop is unoccupied, whereas calcium binding inhibits cleavage (KD ∼ 20 μm), with complete inhibition requiring 10 mm or higher concentrations. Experimentally, the reaction is best followed in the absence of calcium when full time courses of degradation can be measured using SDS-PAGE and densitometry. Fig. 5 demonstrates that the rate of cleavage by CTRC at Leu-81 became measurably reduced in mutants N29I (2.5-fold), N29T (3.6-fold), V39A (1.5-fold), R122C (2.1-fold), and R122H (3.1-fold), whereas it was essentially unchanged in mutant A16V.
FIGURE 5.
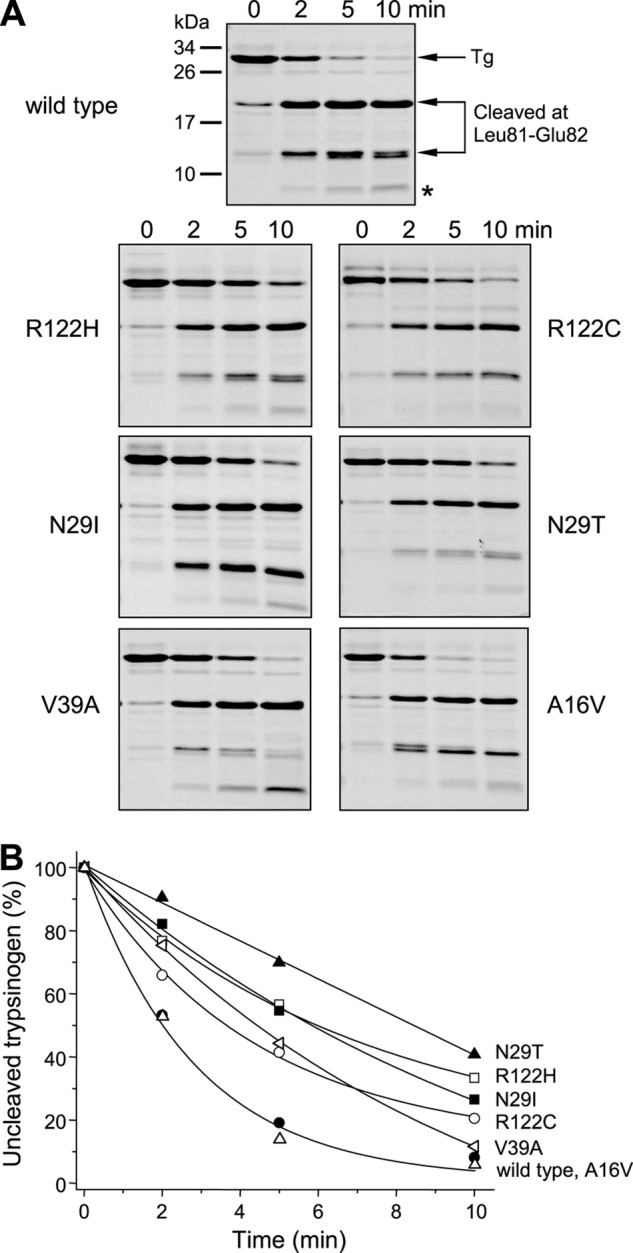
Cleavage of Leu-81–Glu-82 peptide bond in human cationic trypsinogen and pancreatitis-associated mutants by CTRC. Wild-type and mutant trypsinogens were incubated at 2 μm with 25 nm CTRC in 0.1 m Tris-HCl (pH 8.0) and 20 nm SPINK1 trypsin inhibitor (final concentrations) at 37 °C. The trypsin inhibitor was included to prevent autoactivation. A, at the indicated times, reactions were terminated by precipitation with 10% trichloroacetic acid (final concentration), and samples were analyzed by 15% reducing SDS-PAGE and Coomassie Blue staining. The asterisk indicates the faint band migrating at ∼5 kDa, which is the product of a secondary cleavage at Leu-41 by CTRC. Representative gels of two or more experiments are shown. B, densitometric analysis of stained gels showing the changes in the intensity of the unprocessed intact trypsinogen band. Rates of degradation were calculated from linear fits to semilogarithmic plots. Error bars were omitted for clarity. The S.E. was within 12% of the mean.
Mechanism iv
Trypsin-mediated cleavage of the Arg-122–Val-123 peptide bond is required for CTRC-dependent trypsinogen degradation (15). Cleavage of this peptide bond by trypsin does not proceed to completion, but due to trypsin-mediated peptide bond resynthesis, a dynamic equilibrium is reached between the uncleaved and cleaved trypsinogen forms (20). To investigate the effect of pancreatitis-associated mutations on the ability of trypsin to cleave trypsinogen at Arg-122, we used a stable non-activable trypsinogen in which the activating site was mutated (K23Q) (20). Mutations were transferred to the K23Q background, and trypsinogen at 2 μm concentration was digested with 10 nm trypsin in the presence of low calcium concentrations (5 μm), which favor cleavage. Time courses of digestion were analyzed by SDS-PAGE and densitometry (Fig. 6). Because the two products of the cleavage reaction co-migrated as a single band, measurement of the hydrolysis equilibrium between intact and cleaved trypsinogens was straightforward. The equilibrium mixture of wild-type trypsinogen contained 35% intact and 65% cleaved forms. Not surprisingly, mutations R122H and R122C (data not shown) completely blocked cleavage at Arg-122. Mutations N29I and N29T slightly (1.2- and 1.3-fold, respectively) decreased the rate of cleavage and increased the equilibrium levels of uncleaved trypsinogen to 40 and 45%, respectively. Remarkably, mutation V39A decreased the rate of cleavage by at least 3-fold and shifted the cleavage equilibrium toward the uncleaved form (∼50%). Finally, as expected, mutation A16V had no effect on this reaction (data not shown). Taken together, it appears that the mechanistic basis of increased trypsinogen autoactivation in the presence of CTRC is mutation-specific and may involve a combination of relatively smaller effects, as summarized in Table 1.
FIGURE 6.
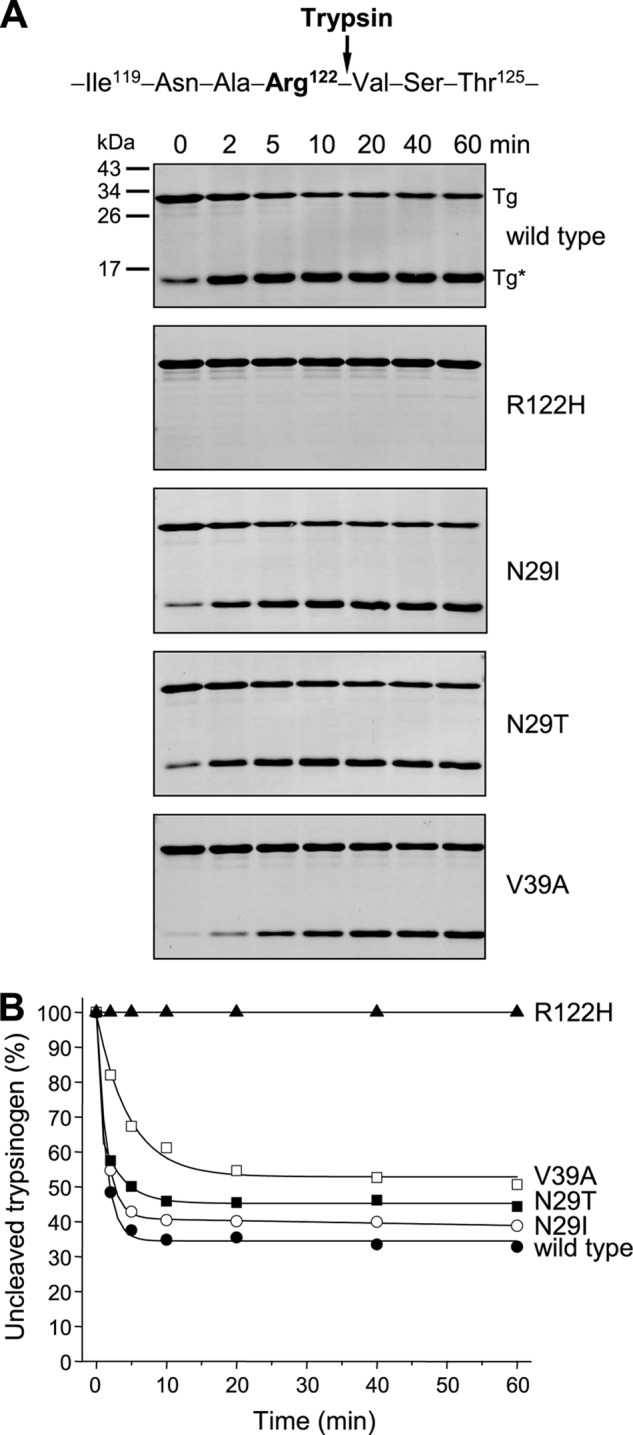
Cleavage of Arg-122–Val-123 peptide bond in human cationic trypsinogen and pancreatitis-associated mutants by cationic trypsin. Wild-type and mutant trypsinogens were incubated at 2 μm with 10 nm human cationic trypsin in 0.1 m Tris-HCl (pH 8.0) and 5 μm CaCl2 (final concentrations) at 37 °C. All trypsinogens contained the K23Q mutation to prevent autoactivation. A, at the indicated times, reactions were terminated by precipitation with 10% trichloroacetic acid (final concentration), and samples were analyzed by 15% reducing SDS-PAGE and Coomassie Blue staining. Note that the two cleavage fragments co-migrated and appeared as a single band at ∼15 kDa. Representative gels of two experiments are shown. B, densitometric analysis of stained gels showing the changes in the intensity of the uncleaved intact trypsinogen band as a percentage of the total intensity of the cleaved (Tg*) and uncleaved (Tg) bands. Error bars were omitted for clarity. The S.E. was within 10% of the mean.
TABLE 1.
Mechanistic basis of increased autoactivation of pancreatitis-associated human cationic trypsinogen mutants
Changes that result in increased autoactivation in the presence of CTRC are shown in boldface. See “Results” for details.
| Mutation | Autoactivation without CTRC | Trypsinogen processing at Phe-18 by CTRC | Trypsinogen cleavage at Arg-122 by trypsin | Trypsinogen cleavage at Leu-81 by CTRC | Trypsin degradation by CTRC |
|---|---|---|---|---|---|
| A16V | Unchanged | Increased 5.8-fold | Unchanged | Unchanged | Unchanged |
| N29I | Increased 1.5-fold | Increased 2.6-fold | Decreased 1.2-fold | Decreased 2.5-fold | Unchanged |
| N29T | Increased 2.5-fold | Decreased 1.3-fold | Decreased 1.3-fold | Decreased 3.6-fold | Decreased 3.3-fold |
| V39A | Unchanged | Unchanged | Decreased 3-fold | Decreased 1.5-fold | Decreased 2.9-fold |
| R122C | Increased 1.2-fold | Unchanged | Blocked | Decreased 2.1-fold | Decreased 11-fold |
| R122H | Increased 1.5-fold | Unchanged | Blocked | Decreased 3.1-fold | Decreased 11-fold |
Effect of Pancreatitis-associated Mutations on Trypsin Stability
Historically, the biochemical effects of pancreatitis-associated trypsinogen mutations used to be interpreted in the context of trypsin stability (1). In light of the present and other recent data, the primary effects of trypsinogen mutations should be considered in terms of trypsinogen autoactivation. Nonetheless, final trypsin levels achieved during trypsinogen autoactivation are also influenced by trypsin stability to a lesser degree. As shown in Fig. 1A, peak trypsin levels slowly declined due to CTRC-mediated trypsin degradation, and mutations N29T, R122C, and R122H seemed to render trypsin more resistant to CTRC-mediated degradation, as indicated by the sustained trypsin plateau phase in Fig. 2 (B and C). To compare the effect of pancreatitis-associated mutations on trypsin degradation in a more quantitative manner, we activated wild-type and mutant trypsinogens with human enteropeptidase and followed the loss of trypsin activity after the addition of 25 nm CTRC. We performed these experiments in the presence of low calcium concentrations (5 μm) to allow the reactions to proceed at a higher rate. Fig. 7 demonstrates that, relative to wild-type trypsin, inactivation of trypsin mutants R122C and R122H proceeded at 11-fold slower rates. Mutants N29T and V39A were degraded ∼3-fold slower than wild-type trypsin, whereas degradation of mutant N29I was comparable with the wild type (see also Table 1). Note that mutation A16V had no effect on trypsin stability, as this mutation is not present in active trypsin.
FIGURE 7.
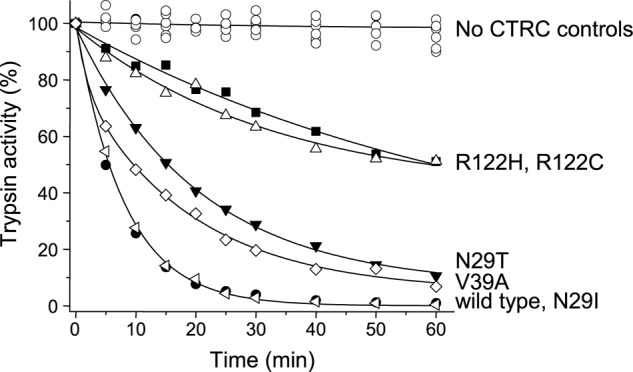
Inactivation of human cationic trypsin and pancreatitis-associated mutants by CTRC. Wild-type and mutant trypsinogens at 1 μm were activated to trypsin with 7 ng/ml (64 pm) human enteropeptidase in 0.1 m Tris-HCl (pH 8.0), 5 μm CaCl2, and 0.05% Tween 20 for 30 min at 37 °C (final concentrations). Initial trypsin activity was determined, and incubations were continued at 37 °C in the absence or presence of 25 nm CTRC. Loss of trypsin activity was followed by withdrawing 2-μl aliquots and measuring trypsin activity as described under “Experimental Procedures.” Trypsin activity is expressed as a percentage of the initial activity. Rates of trypsin inactivation were calculated from linear fits to semilogarithmic plots. Data points represent the average of two or three experiments. Error bars were omitted for clarity. The S.E. was within 10% of the mean.
DISCUSSION
In this study, we have demonstrated that cationic trypsinogen mutations that are associated with classic autosomal dominant hereditary pancreatitis all exhibit markedly increased autoactivation in the presence of CTRC. These findings mark the successful conclusion of a more than decade-long quest for the common biochemical defect caused by hereditary pancreatitis-associated mutations. Increased autoactivation was proposed as a unifying mechanism as early as 2000; however, the clinically common mutations N29I and R122H had a relatively minor, not particularly convincing effect (7) (also see Fig. 2A). Even more confusing was the observation that activation peptide mutations D22G, K23R, and K23I_I24insIDK stimulated autoactivation in a dramatic manner yet still caused essentially the same clinical disease as mutations N29I and R122H (9–11). Thus, there appeared to be no correlation between the biochemical phenotype and the biological effect. The results presented here resolve this apparent contradiction and clearly demonstrate a very similar biochemical phenotype for the highly penetrant, hereditary pancreatitis-associated mutations N29I, N29T, V39A, R122C, and R122H. Furthermore, consistent with its variable disease penetrance, mutant A16V was shown to exhibit a smaller increase in autoactivation than mutants causing hereditary pancreatitis.
Unexpectedly, investigations into the mechanistic basis of increased autoactivation revealed several mutation-specific mechanisms, often acting in concert (see Table 1). (i) Even in the absence of CTRC, mutations N29I, N29T, R122C, and R122H had a direct stimulatory effect on the rate of autoactivation, which ranged from 1.2- to 2.5-fold. These relatively small effects represent increased proteolytic accessibility of the activation peptide caused by the mutations. (ii) Mutations A16V and N29I increased the rate of N-terminal processing of the trypsinogen activation peptide by CTRC by 5.8- and 2.6-fold, respectively. N-terminal processing stimulates trypsin-mediated cleavage of the activation peptide (i.e. autoactivation) by relieving an electrostatic inhibitory interaction between the tetra-aspartate motif in the activation peptide and Asp-218 on trypsin (14). Notably, this is the only biochemical change caused by mutation A16V. (iii) CTRC-dependent degradation of cationic trypsinogen requires cleavages at Leu-81 by CTRC and at Arg-122 by trypsin. With the exception of A16V, all mutations affected both of these cleavages. With respect to cleavage at Leu-81 by CTRC, mutations N29I, N29T, V39A, R122C, and R122H all had a modest inhibitory effect that ranged from 1.5- to 3.6-fold. (iv) With respect to the autolytic cleavage at Arg-122, mutations R122C and R122H blocked this reaction, whereas mutation V39A decreased the rate of cleavage by 3-fold. Mutations N29I and N29T also had a small but measurable negative effect on the rate of cleavage. Furthermore, all mutations shifted the cleavage equilibrium toward uncleaved trypsinogen to various degrees. The altered cleavage at Arg-122 seemed to be the dominant functional effect for mutations R122C, R122H, and possibly V39A. As summarized in Table 1, the synergistic action of these four mechanisms is best seen with mutant N29I, where all four mechanisms increase autoactivation to a relatively small extent, but their cumulative effect becomes striking (see Fig. 2B). Three of the four mechanisms are operational in mutants N29T, R122C, and R122H and two in mutant V39A (Table 1). As pointed out above, the only exception is mutant A16V, where a single mechanism is responsible for the disease-associated phenotype of increased autoactivation. Finally, four mutations (N29T, V39A, R122C, and R122H) also reduced CTRC-mediated trypsin degradation, which may also contribute to the peak trypsin levels achieved during autoactivation. However, one of the key messages of this study is that the pathogenic effects of pancreatitis-associated trypsinogen mutations should be interpreted, first and foremost, in the context of trypsinogen autoactivation and not trypsin stability, as suggested earlier (1).
The structural basis for the decreased cleavage at Leu-81 by CTRC and at Arg-122 by trypsin in the trypsinogen mutants is not readily apparent, but available crystal structures may give some clues. In the bovine trypsinogen x-ray structures (Protein Data Bank codes 2TGD and 1TGN), the side chains of Arg-122 (Arg-117 in chymotrypsin numbering), Asn-84 (Asn-79 in chymotrypsin numbering), and Asn-33 (Asn-25 in chymotrypsin numbering) are within “striking distance” (∼4 Å or less) of each other, suggesting that subtle structural changes altering these interactions may influence cleavage at Arg-122 (Fig. 8). Because Leu-81 in human cationic trypsinogen lies in proximity to Asn-84, the same mechanism may account for altered cleavage at Leu-81. Mutations N29I, N29T, and possibly V39A may affect Asn-33, whereas mutations R122C and R122H may affect both Asn-33 and Asn-84.
FIGURE 8.

Potential interactions between Asn-84, Arg-122, and Asn-33 in trypsinogen. A, ribbon diagram of bovine trypsinogen (Protein Data Bank code 1TGN). Positions 29 and 81 were changed to Asn and Leu, respectively, as found in human cationic trypsinogen. The activation peptide is disordered, and the first N-terminal amino acid residue visible is Val-25. The N-terminal peptide and Asn-33 are colored in blue; the calcium-binding loop and Asn-84 are in green with Leu-81 in black; and the Arg-122 turn is shown in orange with Arg-122 in red. Also shown are the positions of pancreatitis-associated mutations N29I, N29T (with Asn-29 in blue), and V39A (with Val-39 in magenta). The image was rendered using PyMOL 1.3 (Schrödinger, LLC). B, schematic representation of the interactions highlighted in the structural model in A.
The disease-relevant compartment in the pancreas where increased trypsinogen activation might take place in hereditary pancreatitis is unknown, but the pancreatic juice within the excretory ducts seems to be the best candidate. This notion is supported by the observations that mutations in the cystic fibrosis transmembrane conductance regulator, expressed in duct epithelia, or anatomical variations in duct morphology increase the risk for chronic pancreatitis (30). The autoactivation experiments presented here were performed in the presence of 1 mm CaCl2. In human pancreatic juice, total calcium concentrations between 0.2 and 0.9 mm were reported, with higher values found in patients with chronic pancreatitis (31–33). One study reported ionized calcium concentrations at 0.3 mm, with 0.6 mm total calcium (32). We chose to use 1 mm calcium in our experiments because, under these conditions, trypsinogen autoactivation is easy to measure with high trypsin signal even in the presence of CTRC. At slightly lower calcium concentrations that may prevail in the pancreatic juice, autoactivation reactions would proceed with similar kinetics but with a smaller amplitude. Another important factor to consider is the CTRC concentrations used. CTRC is secreted in a zymogen form (chymotrypsinogen C) to the pancreatic juice at levels that are ∼20-fold lower than those of cationic trypsinogen as estimated by immunoblotting using purified proenzymes as standards.3 CTRC has to be activated by trypsin, and this activating cleavage reaction is at least 100-fold faster than trypsin-mediated trypsinogen activation.4 Consequently, small amounts of trypsin generated in the pancreatic juice can preferentially activate CTRC before trypsinogen autoactivation would progress. We performed our experiments at lower CTRC-to-trypsinogen ratios (1:200 and 1:80) than one would expect to find during premature trypsinogen activation in the pancreas. It is likely that in normal pancreatic juice, CTRC-dependent protection against premature autoactivation is more effective and development of trypsin activity is much more dampened than indicated in our autoactivation figures. Under these conditions, the higher autoactivation of mutant trypsinogens seen in our experiments would manifest as increased likelihood of appearance of trypsin activity. Such a scenario is consistent with the clinical course of hereditary pancreatitis, which involves recurrent acute attacks of pancreatitis with symptom-free intervals.
In summary, we have demonstrated that mutations associated with hereditary pancreatitis alter CTRC-dependent proteolytic regulation of human cationic trypsinogen and result in markedly increased autoactivation. The findings suggest that targeted therapy for the management of pancreatitis risk should focus on continuous long-term suppression of trypsinogen autoactivation.
Acknowledgments
Zsófia Nemoda, Zsolt Rónai, Richárd Szmola, and Zoltán Kukor are gratefully acknowledged for initiating some of the experiments in this project. We thank Gábor Pál for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK058088, R01 DK082412, and R01 DK082412-S2 (to M. S.-T.).
Z. Rónai and M. Sahin-Tóth, unpublished data.
A. Geisz and M. Sahin-Tóth, unpublished data.
- CTRC
- chymotrypsin C.
REFERENCES
- 1. Whitcomb D. C., Gorry M. C., Preston R. A., Furey W., Sossenheimer M. J., Ulrich C. D., Martin S. P., Gates L. K., Jr., Amann S. T., Toskes P. P., Liddle R., McGrath K., Uomo G., Post J. C., Ehrlich G. D. (1996) Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat. Genet. 14, 141–145 [DOI] [PubMed] [Google Scholar]
- 2. Teich N., Rosendahl J., Tóth M., Mössner J., Sahin-Tóth M. (2006) Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis. Hum. Mutat. 27, 721–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Howes N., Lerch M. M., Greenhalf W., Stocken D. D., Ellis I., Simon P., Truninger K., Ammann R., Cavallini G., Charnley R. M., Uomo G., Delhaye M., Spicak J., Drumm B., Jansen J., Mountford R., Whitcomb D. C., Neoptolemos J. P., and European Registry of Hereditary Pancreatitis and Pancreatic Cancer (EUROPAC) (2004) Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin. Gastroenterol. Hepatol. 2, 252–261 [DOI] [PubMed] [Google Scholar]
- 4. Rebours V., Boutron-Ruault M. C., Schnee M., Férec C., Le Maréchal C., Hentic O., Maire F., Hammel P., Ruszniewski P., Lévy P. (2009) The natural history of hereditary pancreatitis: a national series. Gut 58, 97–103 [DOI] [PubMed] [Google Scholar]
- 5. Szmola R., Sahin-Tóth M. (2010) Uncertainties in the classification of human cationic trypsinogen (PRSS1) variants as hereditary pancreatitis-associated mutations. J. Med. Genet. 47, 348–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sahin-Tóth M. (2000) Human cationic trypsinogen. Role of Asn-21 in zymogen activation and implications in hereditary pancreatitis. J. Biol. Chem. 275, 22750–22755 [DOI] [PubMed] [Google Scholar]
- 7. Sahin-Tóth M., Tóth M. (2000) Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem. Biophys. Res. Commun. 278, 286–289 [DOI] [PubMed] [Google Scholar]
- 8. Teich N., Nemoda Z., Köhler H., Heinritz W., Mössner J., Keim V., Sahin-Tóth M. (2005) Gene conversion between functional trypsinogen genes PRSS1 and PRSS2 associated with chronic pancreatitis in a 6-year-old girl. Hum. Mutat. 25, 343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Teich N., Ockenga J., Hoffmeister A., Manns M., Mössner J., Keim V. (2000) Chronic pancreatitis associated with an activation peptide mutation that facilitates trypsin activation. Gastroenterology 119, 461–465 [DOI] [PubMed] [Google Scholar]
- 10. Chen J. M., Kukor Z., Le Maréchal C., Tóth M., Tsakiris L., Raguénès O., Férec C., Sahin-Tóth M. (2003) Evolution of trypsinogen activation peptides. Mol. Biol. Evol. 20, 1767–1777 [DOI] [PubMed] [Google Scholar]
- 11. Joergensen M. T., Geisz A., Brusgaard K., Schaffalitzky de Muckadell O. B., Hegyi P., Gerdes A. M., Sahin-Tóth M. (2011) Intragenic duplication: a novel mutational mechanism in hereditary pancreatitis. Pancreas 40, 540–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Simon P., Weiss F. U., Sahin-Tóth M., Parry M., Nayler O., Lenfers B., Schnekenburger J., Mayerle J., Domschke W., Lerch M. M. (2002) Hereditary pancreatitis caused by a novel PRSS1 mutation (Arg-122 → Cys) that alters autoactivation and autodegradation of cationic trypsinogen. J. Biol. Chem. 277, 5404–5410 [DOI] [PubMed] [Google Scholar]
- 13. Kereszturi E., Szmola R., Kukor Z., Simon P., Weiss F. U., Lerch M. M., Sahin-Tóth M. (2009) Hereditary pancreatitis caused by mutation-induced misfolding of human cationic trypsinogen: a novel disease mechanism. Hum. Mutat. 30, 575–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nemoda Z., Sahin-Tóth M. (2006) Chymotrypsin C (caldecrin) stimulates autoactivation of human cationic trypsinogen. J. Biol. Chem. 281, 11879–11886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Szmola R., Sahin-Tóth M. (2007) Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin: identity with Rinderknecht's enzyme Y. Proc. Natl. Acad. Sci. U.S.A. 104, 11227–11232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rosendahl J., Witt H., Szmola R., Bhatia E., Ózsvári B., Landt O., Schulz H. U., Gress T. M., Pfützer R., Löhr M., Kovacs P., Blüher M., Stumvoll M., Choudhuri G., Hegyi P., te Morsche R. H., Drenth J. P., Truninger K., Macek M., Jr., Puhl G., Witt U., Schmidt H., Büning C., Ockenga J., Kage A., Groneberg D. A., Nickel R., Berg T., Wiedenmann B., Bödeker H., Keim V., Mössner J., Teich N., Sahin-Tóth M. (2008) Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat. Genet. 40, 78–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Masson E., Chen J. M., Scotet V., Le Maréchal C., Férec C. (2008) Association of rare chymotrypsinogen C (CTRC) gene variations in patients with idiopathic chronic pancreatitis. Hum. Genet. 123, 83–91 [DOI] [PubMed] [Google Scholar]
- 18. Zhou J., Sahin-Tóth M. (2011) Chymotrypsin C mutations in chronic pancreatitis. J. Gastroenterol. Hepatol. 26, 1238–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Király O., Guan L., Szepessy E., Tóth M., Kukor Z., Sahin-Tóth M. (2006) Expression of human cationic trypsinogen with an authentic N terminus using intein-mediated splicing in aminopeptidase P-deficient Escherichia coli. Protein Expr. Purif. 48, 104–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kukor Z., Tóth M., Pál G., Sahin-Tóth M. (2002) Human cationic trypsinogen. Arg-117 is the reactive site of an inhibitory surface loop that controls spontaneous zymogen activation. J. Biol. Chem. 277, 6111–6117 [DOI] [PubMed] [Google Scholar]
- 21. Bence M., Sahin-Tóth M. (2011) Asparagine-linked glycosylation of human chymotrypsin C is required for folding and secretion but not for enzyme activity. FEBS J. 278, 4338–4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Szabó A., Héja D., Szakács D., Zboray K., Kékesi K. A., Radisky E. S., Sahin-Tóth M., Pál G. (2011) High affinity small protein inhibitors of human chymotrypsin C (CTRC) selected by phage display reveal unusual preference for P4′ acidic residues. J. Biol. Chem. 286, 22535–22545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Király O., Guan L., Sahin-Tóth M. (2011) Expression of recombinant proteins with uniform N termini. Methods Mol. Biol. 705, 175–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lengyel Z., Pál G., Sahin-Tóth M. (1998) Affinity purification of recombinant trypsinogen using immobilized ecotin. Protein Expr. Purif. 12, 291–294 [DOI] [PubMed] [Google Scholar]
- 25. Ózsvári B., Hegyi P., Sahin-Tóth M. (2008) The guinea pig pancreas secretes a single trypsinogen isoform, which is defective in autoactivation. Pancreas 37, 182–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arduino C., Salacone P., Pasini B., Brusco A., Salmin P., Bacillo E., Robecchi A., Cestino L., Cirillo S., Regge D., Cappello N., Gaia E. (2005) Association of a new cationic trypsinogen gene mutation (V39A) with chronic pancreatitis in an Italian family. Gut 54, 1663–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Witt H., Luck W., Becker M. (1999) A signal peptide cleavage site mutation in the cationic trypsinogen gene is strongly associated with chronic pancreatitis. Gastroenterology 117, 7–10 [DOI] [PubMed] [Google Scholar]
- 28. Grocock C. J., Rebours V., Delhaye M. N., Andrén-Sandberg A., Weiss F. U., Mountford R., Harcus M. J., Niemczyck E., Vitone L. J., Dodd S., Jørgensen M. T., Ammann R. W., Schaffalitzky de Muckadell O., Butler J. V., Burgess P., Kerr B., Charnley R., Sutton R., Raraty M. G., Devière J., Whitcomb D. C., Neoptolemos J. P., Lévy P., Lerch M. M., Greenhalf W., and European Registry of Hereditary Pancreatitis and Pancreatic Cancer (2010) The variable phenotype of the p.A16V mutation of cationic trypsinogen (PRSS1) in pancreatitis families. Gut 59, 357–363 [DOI] [PubMed] [Google Scholar]
- 29. Joergensen M. T., Brusgaard K., Crüger D. G., Gerdes A. M., Schaffalitzky de Muckadell O. B. (2010) Genetic, epidemiological, and clinical aspects of hereditary pancreatitis: a population-based cohort study in Denmark. Am. J. Gastroenterol. 105, 1876–1883 [DOI] [PubMed] [Google Scholar]
- 30. Bertin C., Pelletier A. L., Vullierme M. P., Bienvenu T., Rebours V., Hentic O., Maire F., Hammel P., Vilgrain V., Ruszniewski P., Lévy P. (2012) Pancreas divisum is not a cause of pancreatitis by itself but acts as a partner of genetic mutations. Am. J. Gastroenterol. 107, 311–317 [DOI] [PubMed] [Google Scholar]
- 31. Harada H., Takeda M., Yabe H., Hanafusa E., Hayashi T., Kunichika K., Kochi F., Mishima K., Kimura I. (1980) The calcium concentration in human pure pancreatic juice in chronic pancreatitis. Gastroenterol. Jpn. 15, 355–361 [DOI] [PubMed] [Google Scholar]
- 32. Gerolami A., Marteau C., Matteo A., Sahel J., Portugal H., Pauli A. M., Pastor J., Sarles H. (1989) Calcium carbonate saturation in human pancreatic juice: possible role of ductal H+ secretion. Gastroenterology 96, 881–884 [PubMed] [Google Scholar]
- 33. Furui T., Kondoh S., Harada T., Takeuchi K., Shiraishi K., Kaino S., Okuda S., Okita K., Nakamura K. (2000) Calcium concentration and artificial precipitates in human pancreatic juice. Pancreas 21, 257–261 [DOI] [PubMed] [Google Scholar]